

Summary
The ability to selectively enrich or deplete T lymphocytes of specific phenotype and function holds significant promise for application in adoptive immunotherapy protocols. Although CD4+ T cells can have an impact on CD8+ T-cell effector function, memory, and maintenance, a subset of CD4+ T cells, CD25+ regulatory T cells (Treg), can regulate peripheral self tolerance and possess the ability to suppress antitumor responses. The authors report the ability to selectively deplete CD25+ Treg cells from patient leukapheresis samples using a clinical-grade, large-scale immunomagnetic system. Using leukapheresis samples containing up to 1.3 × 1010 white blood cells, efficient depletion of Treg cells was measured by flow cytometric analysis of CD25 expression and FOXP3 expression on post-depletion products. Remnant CD25+ cells could not be detected in CD25-depleted products after short-term culture in IL-2 or enriched following secondary immunomagnetic selection for CD25+ cells, confirming that efficient depletion had occurred. In parallel to efficient enrichment of CD25− cells, immunomagnetic selection resulted in the recovery of Treg cells, since CD25+ lymphocytes removed during depletion were primarily composed of CD4+ T cells that expressed high levels of FOXP3 and possessed suppressive activity against autologous TCR-stimulated CD4+ CD25− T cells in vitro. These results show that selective separation of functional CD25+ Treg cells from large-scale samples can be performed in large scale under clinical-grade conditions with sufficient selection, recovery, viability, ability to expand, and function for potential use in adoptive immunotherapy.
Keywords: human, CD25, regulatory T cells, clinical, depletion
Naturally occurring CD4+ CD25+ T regulatory (Treg) cells are involved in the maintenance of homeostatic peripheral self tolerance by suppressing autoreactive T cells.1 In mice and humans, Treg cells develop in the thymus and represent about 5% to 10% of the peripheral CD4+ T-cell compartment. These regulatory cells are characterized by their constitutive expression of CD25, CTLA-4, glucocorticoid-induced TNFR (GITR), and the transcription factor Forkhead box P3 (Foxp3).1 Compared with CD25−CD4+ T cells, Treg cells exhibit a hypoproliferative capacity and possess the ability to suppress CD8+ and CD25− CD4+ T cell activation in vitro. The mechanism of suppression is cell contact-dependent, but how regulatory T cells induce and maintain self tolerance is unknown.
The importance of Treg cells as key mediators of self tolerance is witnessed in individuals genetically predisposed to the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX), a recessive and often fatal disorder of early childhood caused by loss of function mutations of FOXP3 and the resultant lack of Treg cells.2 In mice, the scurfy strain harbor a similar mutation in Foxp3 resulting in the absence of Treg cells and consequentially the induction of fatal T cell–mediated autoimmunity mediated by unregulated CD4+ T-helper cells directed against self tissue antigens.3 Like scurfy mice, the depletion of Treg cells4 from normal mice results in autoimmune destruction of a number of self tissue organs. Further, transfer of a normal repertoire of CD25− CD4+ T-helper cells to lymphopenic mice results in autoimmune induction, a process inhibited by the co-transfer of Treg cells. 3 In the absence of CD25+ Treg cells, organ-specific destruction of tissue expressing a self antigen can be augmented with self antigen vaccination or through the provision of inflammatory signals.5 Furthermore, depletion of Treg cells can enhance tumor protection to tumor-associated antigens expressed as self antigens and elicit concomitant immunity in tumor-bearing mice.6,7
The influence of CD4+ T-cell subsets on adoptive immunotherapy has recently been evaluated using a well-described murine model of cancer wherein adoptive transfer of tumor/self reactive CD8+ T cells with vaccination and IL-2 leads to regression of large established melanoma in lymphodepleted hosts.8 Although CD4+ T cells can affect CD8+ T-cell effector function, memory, and maintenance, the absence of CD4+ T cells augmented adoptive immunotherapy when using CD8+ T cells directed against a persisting tumor/self antigen.9 However, adoptive transfer of CD25−CD4+ T-helper cells with tumor/self reactive CD8+ T cells and vaccination into CD4+ T cell-deficient recipients resulted in autoimmunity and regression of established tumor. Maintenance of tumor/self CD8+ effector cells was dependent on CD25− CD4+ T helper cells capable of producing IL-2. Transfer of CD25+ Treg cells alone or combined with CD25− CD4+ T-helper cells inhibited effective immunotherapy.
Peripheral blood lymphocytes from melanoma patients contain functional CD4+ CD25+ Treg cells, and melanoma antigen-specific Treg cells have been described.10,11 In addition, CD4+ CD25+ Treg cells are overrepresented in human metastatic melanoma lymph nodes and can inhibit the function of infiltrating T cells.12 In ovarian carcinoma, Treg cells are reported to contribute to the growth of human tumors in vivo by suppressing tumor-specific T-cell immunity and have been reported to be associated with a high death hazard and reduced survival.13 Thus, the negative immunoregulatory effects of Treg cells may explain the poor clinical response rates reported in cancer patients receiving non-immunodepleting immunotherapy, and offers a foundation for novel immunotherapeutic approaches designed to neutralize the suppressive effects of Treg cells in vivo and bolster antitumor immune responses.
METHODS
Cells and Culture Conditions
Donor peripheral blood mononuclear cells (PBMCs) were obtained by leukapheresis. PBMCs were purified on Ficoll-Hypaque step gradients (LSM Lymphocyte Separation Medium; ICN Biochemicals, Aurora, OH). In some cases, cells were cultured for 72 hours in 600 IU/mL rhIL-2 (Chiron Corp, Emeryville, CA) and complete medium (CM) consisting of RPMI 1640 (Invitrogen Corp, Carlsbad, CA) supplemented with 2 mM glutamine (Biofluids, Rockville, MD), 25 mM HEPES buffer (Biofluids), 100 U/mL penicillin (Biofluids), 100 μg/mL streptomycin (Biofluids), 50 μM 2-mercaptoethanol (Invitrogen), and 10% heat-inactivated human AB sera (Gemini Bioproducts, Woodland, CA). CD4+ T-cell enrichments were performed using an indirect magnetic labeling system, the CD4+ T Cell Isolation Kit II (Miltenyi Biotech, Auburn, CA).
Depletion of CD25+ T Cells Ex Vivo
Depletion of CD25+ T cells was performed using the CliniMACSPLUS instrument (Miltenyi Biotech) according to standard user protocol. In brief, the leukapheresis product was washed and resuspended in CliniMACS PBS/EDTA (Miltenyi Biotech) supplemented with human serum albumin (ZLB Bioplasma Inc, Glendale, CA) in a cell preparation bag to which one vial of CliniMACS CD25 MicroBeads (Miltenyi Biotech) was added. Incubation was performed for 30 minutes at room temperature on an orbital shaker. Cells were washed, resuspended, and applied to the CliniMACSPLUS instrument with the depletion 2.1 program selected. Upon completion of the depletion program, CD25-depleted cells and the CD25+ cell fraction were collected in separate collection bags. In other experiments, secondary CD25 enrichment was performed using immunomagnetic selection according to the manufacturer’s instructions (Dynal Biotech, Oslo, Norway).
Suppression Assays
Fresh cell samples were washed and resuspended in CM. Co-cultures containing 5 × 103 CD4+ CD25− T cells with titered numbers of cells from the CD25+ fraction and 1 × 104 autologous CD3-depleted feeder cells (4,000 rad) were established in 96-well U-bottom plates, in triplicate. CD3+ cells were removed by immunomagnetic depletion (Dynal Biotech). To increase the purity of CD25+ cells used in suppression assays, secondary magnetic separation was performed on the CD25+ fraction using a MACS separation column (Miltenyi Biotech). Cells were stimulated with 5 μg/mL soluble OKT3 antibody alone or with 100 CU/mL rhIL-2. Cell cultures were then incubated at 37°C for 3, 5, or 7 days and pulsed with 1 μCi [3H]-thymidine per well for the final 18 hours of incubation. Plates were harvested onto nylon filters using the Betaplate system and radioactivity was quantified using a Betaplate counter. Results are expressed in cpm as the mean of triplicate cultures ± SEM. Percentage suppression was calculated as 1 − ([CD25+ CD25− mixed culture proliferation − CD25+ cell proliferation]/CD25− cell proliferation) × 100.
RNA Isolation and cDNA Synthesis
Total RNA was isolated using RNeasy columns (Qiagen, Valencia, CA) according to the manufacturer’s instructions. RNA was eluted in 50 μL of RNase-free water and used as template for one round of reverse transcription for cDNA synthesis. Total RNA (500 ng) was added to a cocktail (Superscript First-Strand Synthesis system, Invitrogen, Carlsbad, CA) containing 25 ng oligo-dT12–18, 10× RT buffer (200 mM Tris-HCl [pH 8.4], 500 mM KCl), 2.5 mM MgCl2, 10 mM DTT, 0.5 mM dNTPs, DEPC-treated water, and 10 u/μL SuperScript II reverse transcriptase(Invitrogen) in a 20-μL reaction. Reverse transcription was performed at 42°C for 90 minutes, after which 2 units of RNase H (Invitrogen) was added and the reaction incubated at 37°C for 20 minutes. Reactions were subsequently diluted 10-fold with DEPC-treated water and stored at −20°C.
Real-Time PCR
The ABI PRISM 7700 Sequence Detector System (Applied Biosystems, Foster City, CA) was used for quantitative mRNA expression analysis. Primers and probes for β-actin14—forward 5′-GCGAGAAGATGACCCAGGATC-3′, reverse 5′-CCAGTGGTACGGCCAGAGG-3′, TaqMan probe 5′(FAM)-CCAGCCATGTACGTTGCTATCCAGGC-(TAMRA)3′—were synthesized by Applied Biosystems. For analysis of Foxp3, Assay on Demand (Applied Biosystems) primers and probes were used with TaqMan Universal Master Mix (Applied Biosystems). The conditions used for PCR were 50°C for 2 minutes, 95°C for 10 minutes, and then 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. For each experiment a no-template control reaction was performed. Each cDNA sample was tested in duplicate and the mean values were calculated. Copy number quantitation was based on the TaqMan principle, depending on the number of cycles required for threshold detection of the fluorescence signal (cycle threshold, Ct) as described.15,16 Briefly, 10-fold serial dilutions of plasmid constructs containing Foxp3 or β-actin sequences of known concentrations were prepared for development of standard curves for quantitation of test samples. Plasmids were linearized and made in the presence of pCDNA plasmid carrier.
Monoclonal Antibodies and Flow Cytometric Immunofluorescence Analysis
FITC-conjugated anti-human CD4, CD8, CD20, and CD16 antibodies were obtained from BD Biosciences (San Jose, CA). PE-conjugated and biotinylated CD25 antibodies (4A3 clone; Miltenyi Biotech) were used. Biotin-labeled antibody was detected with PE-labeled anti-biotin antibody (Miltenyi Biotech). PE-conjugated CD3 or APC-labeled CD3 antibody was used in some analyses. Fresh cells were resuspended in FACS buffer consisting of PBS with 2% FBS (Gemini Bioproducts, Woodland, CA) at 107 cells/mL and blocked with 10% normal mouse Ig (Caltag Labs, Burlingame, CA) for 10 minutes on ice. 106 cells in 100 μL were stained with fluorochrome conjugated mAbs at 4°C for 40 minutes in the dark. Cells were washed twice, briefly stained with propidium iodide (PI) for nonviable cell exclusion, and subsequently analyzed in a FACSCalibur (Becton Dickinson). For CD25 staining, cells were first stained with biotinylated anti-CD25 antibody, washed and treated with PE-labeled anti-biotin antibody, washed twice, and analyzed.
RESULTS
Cell Counts After Clinical-Scale Depletion of CD25+ Cells
In our murine model studies, CD4+ CD25+ Treg cells have been shown to suppress cell transfer-based anti-melanoma immunotherapies.9 Alternatively, co-transfer of CD4+ CD25− T-helper cells with CD8+ effector T cells augmented antitumor efficacy. Based on these findings, we sought to selectively deplete CD25+ Treg cells and enrich for CD25− T-helper cells from patient pheresis samples for potential application in the clinical setting. To selectively deplete CD25+ T cells from patient pheresis samples, a clinical-grade, large-scale magnetic bead separation device, the CliniMACSPLUS instrument, was used. CliniMACS cell separation technology has been shown to support the isolation of CD34+ hemopoietic stem cells for cancer patients receiving high-dose therapy.17–19 The approach uses iron colloid-labeled antibody binding to marker-positive cells, followed by application of the cells to a magnetic column in a closed-chamber system. For CD25 depletion, one vial of CliniMACS CD25 reagent (anti-CD25 antibody conjugated to an iron-dextran colloid) is dosed to separate up to 6 × 109 CD25+ cells out of a total not exceeding 40 × 109 white blood cells.
As an initial attempt, a donor leukapheresis product (donor 1) consisting of 1.3 × 1010 cells was labeled with CD25 reagent, washed, and applied to the CliniMACSPLUS instrument for automated selection of labeled cells. Depletion of the magnetically labeled cells from the pheresis resulted in the isolation of 5.4 × 109 CD25-depleted cells, accounting for 42% of the starting cell number (Table 1). The CD25+ cell fraction contained 2.3 × 109 cells (18%). Sixty-two percent of the starting pheresis sample was accounted for in the positive selection, negative selection, and waste bags combined. Upon reevaluation, it was noted that cell counts had been performed prior to cell washing but not immediately before application to the CliniMACSPLUS device, which may account in part for the calculated 38% cell loss in the post-separation products. A second test depletion was performed to validate the capability of the CliniMACSPLUS system to remove CD25+ T cells from a patient pheresis sample. A second pheresis (donor 2) of 4.4 × 109 cells was labeled with CD25 reagent, washed, and counted immediately before application to the CliniMACSPLUS instrument. Following magnetic selection, the CD25-depleted cell fraction contained 71% (3.1 × 109 cells) of the starting pheresis sample and the CD25+ cell product contained 27% of the pheresis. Nearly 100% of the starting pheresis cell number was accounted for in cell counts from the positive, negative, and waste collection bags combined. In both attempts to deplete CD25+ cells from pheresis products, CD25-depleted cells were isolated in numbers sufficient for potential application in adoptive cell transfer therapy.
TABLE 1.
Enumeration of Post-CD25 Separation Cell Products
|
Post-Separation Cell Products |
||||||
|---|---|---|---|---|---|---|
| Donor | Leukapheresis | CD25−Cell Fraction | CD25+Cell Fraction | Waste Bag | Total Yield | |
| 1 | Cell number (×106) | 13,000* | 5,440 | 2,300 | 368 | 8,108 |
| Percentage of leukapheresis | N/A | 41.8 | 17.6 | 2.8 | 62.2 | |
| 2 | Cell number (×106) | 4,360 | 3,100 | 1,170 | 88 | 4,358 |
| Percentage of leukapheresis | N/A | 71.1 | 26.8 | 2.0 | 99.9 | |
Values represent the total number of cells from the indicated sample determined as cell concentration multiplied by volume. Cell counts were taken before and after CD25 depletion.
Cell counting was performed before addition of CD25 reagent and washing, but not immediately before depletion and therefore may underestimate the actual number of cells applied to the CliniMACSPLUS instrument.
Flow Cytometric Analysis of Pre- and Post-Depletion Cell Products
Following magnetic selection, flow cytometric analysis was performed on pre- and post-depletion cell products to evaluate the efficiency of the CD25 depletion. Pre-depletion pheresis samples and CD25-depleted cells showed normal forward and side scatter profiles, with lymphocyte and monocyte populations evident (Fig. 1A). CD25+ cell fractions showed a light scattering consistent with the size and granularity of lymphocytes and comprised primarily CD3+ cells (Table 2). Despite the selective depletion of CD3+ lymphocytes, the CD25-depleted fractions showed only minimal reductions in CD3 frequency compared with pre-separation samples. Small increases in CD8+ cell frequency were offset by decreases in CD4 lymphocytes in depleted fractions. In contrast, CD25+ fractions were largely made up of CD4+ T cells. CD25+ fractions were also devoid of CD19+ B and CD16+ NK cells (not shown).
FIGURE 1.
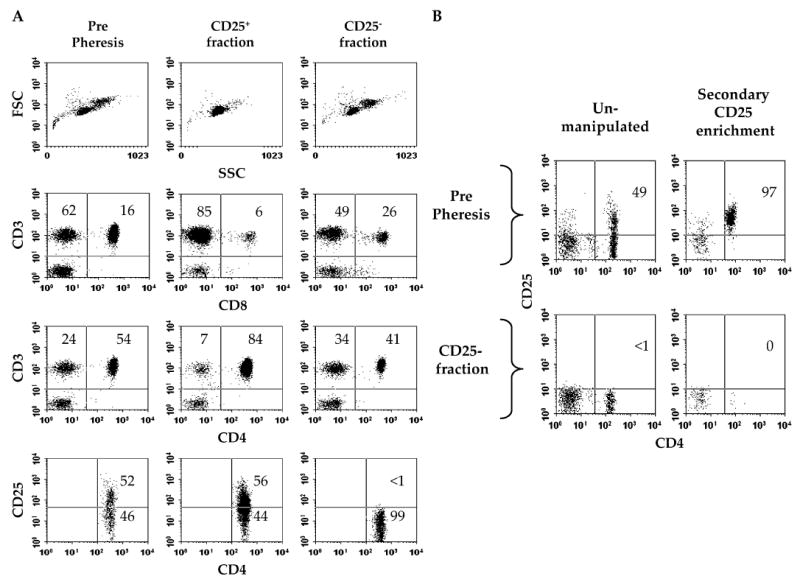
Cells expressing high levels of CD25 are effectively depleted from leukapheresis products by CliniMACS-based separation. Representative flow cytometric data from donor 1 leukapheresis are shown. A, CD4 T cells do not express CD25 after clinical-scale depletion. Forward (FSC) and side scatter (SSC) analysis was performed on the leukapheresis products. For CD3/CD8 and CD3/CD4 analysis, cells were gated through a viable (PI−) lymphocyte gate. For CD4/CD25 analysis, cells were further gated through a viable CD4+ gating. Values represent the percentage of total gated cells contained within the indicated quadrants. B, Remnant CD25-expressing cells cannot be enriched from the CD25− cell fraction. Pre-separation pheresis cells and cells from the CD25− fraction were further enriched for CD25-expressing cells or unmanipulated and analyzed for CD4 and CD25 expression. Values indicate the percentage of CD4+ T cells expressing CD25 before or after immunomagnetic capture.
TABLE 2.
Phenotypic Analysis of Pheresis Products After CD25 Depletion
|
Lymphocyte-Gated Cells |
||||||
|---|---|---|---|---|---|---|
| Donor | %CD3+of Total | % CD3+ | % CD8+of CD3 | % CD4+of CD3 | % CD4+CD25+ | |
| 1 | Pre-separation | 51 | 78 | 22 | 70 | 52 |
| Post-CD25+ fraction | 84 | 91 | 7 | 91 | 56 | |
| Post-CD25− fraction | 45 | 75 | 36 | 55 | <1 | |
| 2 | Pre-separation | 55 | 77 | 65 | 33 | 61 |
| Post-CD25+ fraction | 83 | 88 | 16 | 82 | 67 | |
| Post-CD25− fraction | 51 | 72 | 79 | 17 | <1 | |
Pre- and post-CD25 depletion donor 1 and 2 pheresis samples were blocked with normal mouse immunoglobulin before staining with mouse anti-CD3, CD4, CD8, or CD25 antibodies and viable cells selected through PI gating. For CD25 staining, primary biotin-labeled antibody staining was followed by secondary anti-biotin antibody staining. Values correspond to the percentage of positive events within the total cell population or lymphocyte-gated population as indicated.
Flow cytometric analysis performed on pre- and post-depletion cell products showed the efficient depletion of CD25-expressing cells from the CD25-depleted fractions. Less than 1% of CD4 lymphocytes expressed high levels of CD25 following immunomagnetic separation. CD4 T cells in the CD25+ cell fraction, however, expressed CD25 at frequencies slightly higher than detected in pre-separation phereses, demonstrating selective enrichment of CD25 cells in the CD25+ fraction. Cell viability was greater than 95% in all pre-and post-separation cell samples.
To further test whether all CD25+ T cells had been removed from the CD25-depleted cell fraction, a secondary CD25 selection was performed. Pre- and post-separation cell samples (3 × 107 cells) from donor 1 were enriched for CD25+ cells using a positive magnetic separation system. While 1.4 × 105 CD25+ cells (0.5%) were readily collected from pre-separation samples, few cells (2 × 104; 0.07%) were collected from the CD25-depleted cell fraction, and none expressed CD25 (see Fig. 1B). Attempts to enrich remnant CD25+ T cells from the CD25-depleted cell fraction from donor 2 were unsuccessful, further showing the proficient removal of CD25 cells.
FOXP3 Expression Following Immunomagnetic Separation of CD25+ Cells
To determine whether CD25 depletion had successfully removed Treg cells from the leukapheresis product, we assayed pre- and post-depletion cell products for expression of FOXP3 mRNA, a genetic marker for Treg cells, by TaqMan RT-PCR (Fig. 2A). Eighteen- and 56-fold differences were detected between pre- and post-depletion cell products from donors 1 and 2, with 0.8 and 0.4 copies of FOXP3 per 104 β-actin copies expressed in the post-depletion product compared with 14.1 and 24.4 in the pre-separation phereses, respectively. Compared with the CD25-depleted cell products, the CD25+ cell fraction expressed about 84- and 440-fold higher levels of FOXP3, demonstrating that efficient selection of Treg cells from heterogeneous pheresis samples had occurred. To evaluate the impact of separation on the CD4 cell population, CD4 enrichments were performed on pre-separation cells and CD25-depleted cells and FOXP3 expression was measured (see Fig. 2B). Similar but smaller differences in FOXP3 expression were detected in CD4-enriched pre- and post-separation populations (16- and 42-fold, respectively) as were seen in non-enriched cell samples. Consistent with the slight increase in CD25 detected by flow cytometry, the CD25+ fraction expressed on average only slightly more FOXP3 than purified CD4 cells from the pre-separation pheresis.
FIGURE 2.
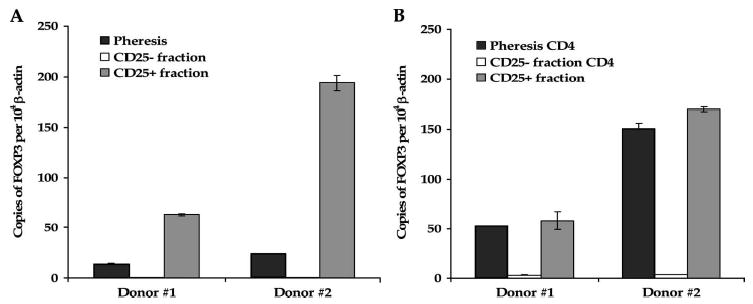
FOXP3 mRNA levels are reduced in CD25-depleted leukapheresis samples. A, Marked differences in FOXP3 expression are noted in post-separation cell fractions. FOXP3 expression was measured in pre- and post-separation leukapheresis products from donors 1 and 2 by TaqMan RT-PCR. B, Similar differences in FOXP3 expression are present in CD4-enriched T-cell population. FOXP3 expression was measured in CD4-enriched pre- separation and CD25-depleted cells, and non-enriched CD25+ fraction cells, which primarily comprised CD4+ T cells (see Fig. 1A). Results represent the relative number of FOX3 copies per 104 copies of β-actin ± SEM.
CD4+ CD25− Cells Do Not Upregulate CD25 or FOXP3 Expression in Short-Term Culture
Since the binding of CD25 microbeads to cells can potentially interfere with CD25 antibody staining, donor 2 cell separation samples were cultured in IL-2-containing medium for 72 hours to facilitate bead detachment and assessed for CD25 and FOXP3 expression. CD25 expression was assessed using the PE conjugated anti-CD25 4A3 clone, which binds CD25 and is detectable in the absence of microbead reagent. After 73 hours in culture, CD25 expression in pre-separation CD4 cells marginally increased from 34% to 40%, with a rise in staining intensity (Fig. 3A). Similarly, CD25 was readily detected on CD4 cells from the CD25+ fraction after culture, while CD25 expression was undetectable on CD25 microbead-bound cells at the initiation of cell culture, confirming that microbead detachment had occurred. In contrast to these results, CD25-depleted cells failed to show any marked increase in CD25 expression after culture in IL-2. FOXP3 expression analysis performed with CD25-depleted cells confirmed the continued absence of Treg cells, with only a slight increase in the number of FOXP3 copies per 104 β-actin after 3 days of culture (3.6 ± 0.2 vs. 4.8 ± 0.2 copies; see Fig. 3B). Following IL-2 culture, FOXP3 expression increased in the pre-separation CD4 cells from 150.8 ± 5.3 to 301.7 ± 12.1 copies per 104 β-actin but slightly decreased in the CD25+ cell fraction from 169.9 ± 2.4 to 125.7 ± 16.3 copies per 104 β-actin, likely as a consequence of CD25 microbead-mediated inhibition of IL-2 binding.
FIGURE 3.
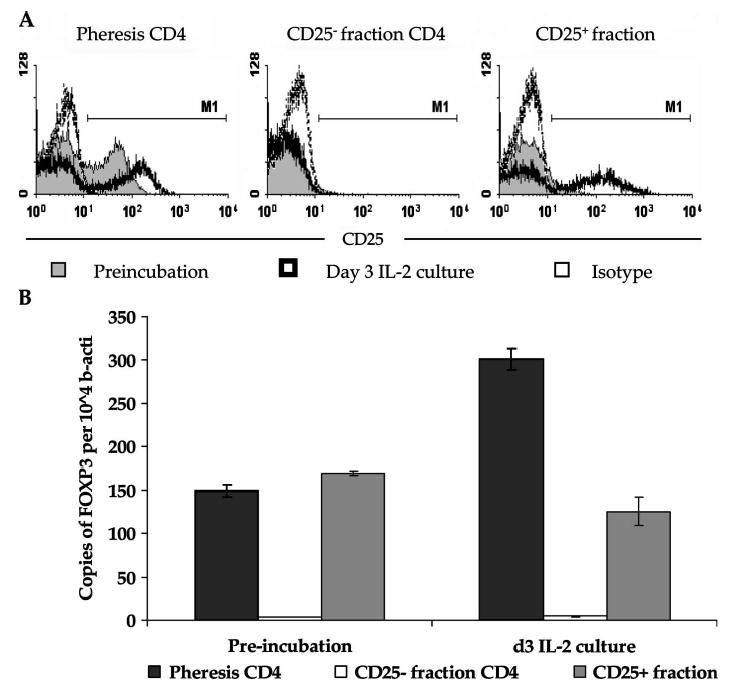
IL-2 incubation results in increased CD25 and FOXP3 expression in pre-separation CD4+ T cells but not CD25-depleted CD4+ T cells. A, CD25 is not re-expressed on CD25-depleted CD4 cells after 3 days in IL-2 culture. CD4-enriched pre-separation and CD25-depleted cells, and non-enriched CD25+ fraction cells were cultured in the presence of 100 CU/mL rhIL-2 for 72 hours and assessed for CD25 using PE-conjugated CD25 4A3 clone antibody or isotype control. Cells were gated through a viable CD4+ gating. B. Foxp3 expression remained low in CD25-depleted CD4+ T cells after IL-2 culture. FOXP3 expression was measured by TaqMan RT-PCR before and after 72 hours of IL-2 incubation. Results represent the relative number of FOX3 copies per 104 copies of β-actin ± SEM.
Suppressive Activity of Enriched CD25+ Cells In Vitro
To determine whether selective depletion of functional Treg cells had occurred, we assessed the suppressive activity of the CD25+ cell fraction. CD25-depleted CD4 T cells were activated with anti-CD3 antibody in the absence or presence of titered numbers of CD25+ cells (Fig. 4). Five days after initiation of culture, robust proliferation was measured in cultures containing CD25-depleted CD4 T cells alone, whereas cultures containing CD25+ cells were hypoproliferative, a characteristic of Treg cells. In co-cultures containing equal numbers of CD25+ and CD25− cells, suppression was first detected 5 days after culture initiation, with nearly 53% suppression detected. This suppressive effect could be titered down with decreasing numbers of CD25+ cells. The microbead-labeled CD25+ cell fraction exhibited suppressive activity against TCR-stimulated autologous CD4+ CD25− T cells fraction despite containing both CD25+ and CD25− cells (see Figs. 1A and 3A). Control cultures containing twice the number of CD25-depleted CD4 cells demonstrated that overcrowding was not the source of suppression (not shown). Incubation in IL-2 reversed the suppressive effect of the CD25+ cell population and restored their ability to proliferate. This demonstrates that successful enrichment of functional Treg cells in the CD25+ cell fraction had occurred.
FIGURE 4.
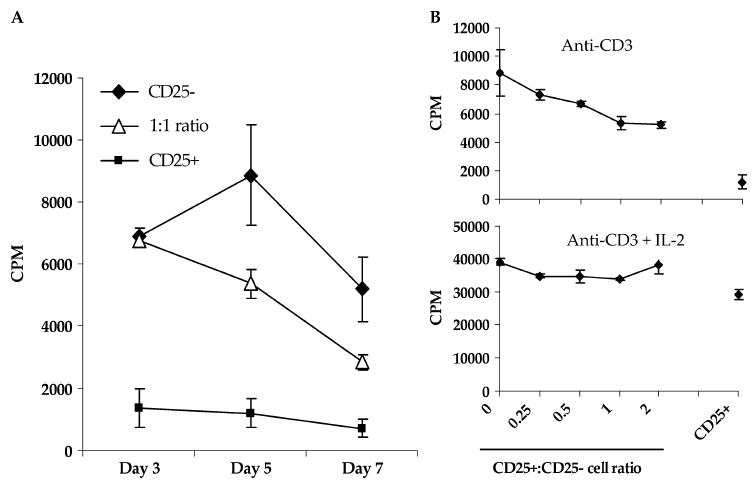
Depleted CD25+ cells are hypoproliferative and exhibit suppressive activity against CD25− CD4+ T cells in vitro. A. CD25+ cell-mediated suppression is measured after 5 days in co-cultures with CD25− CD4+ T cells. Fresh cell cultures were established containing CD25− CD4+ T cells alone or with an equal number of cells from the CD25+ fraction and 1 × 104 autologous irradiated CD3-depleted feeder cells, in triplicate. Secondary magnetic separation was performed on the CD25+ cells to increase the purity of CD25+ cells used in suppression assays. Cells were stimulated with 5 μg/mL soluble OKT3 antibody alone or with 100 CU/mL rhIL-2 for 3, 5, or 7 days and pulsed with 1 μCi [3H]-thymidine per well for the final 18 hours of incubation and harvested for counting. B, CD25+ cell hypoproliferation and mediated suppression is reversed in the presence of IL-2. Cultures of CD25− CD4+ T cells alone or with titered numbers of cells from the CD25+ fraction and CD25+ cells alone were stimulated with 5 μg/mL soluble OKT3 antibody alone or with 100 CU/mL rhIL-2 for 5 days. Incorporation of [3H]-thymidine is expressed in counts per minute (CPM) as the mean of triplicate cultures ± SEM. Percentage suppression was calculated as 1 − ([CD25+ CD25− mixed culture proliferation CD25+ cell proliferation)/CD25− cell proliferation) × 100.
DISCUSSION
CD8+ T cells are known mediators of anti-melanoma responses, but the role of CD4+ T cells in tumor treatment is less well defined.20 CD4+ T cells are capable of providing help to enhance CD8-mediated responses through the production of soluble factors such as IL-2 and ability to activate antigen-presenting cells (APCs).21–23 In the absence of help from CD4+ T cells, CD8+ T cells can be depleted,20 become lethargic,24 or lose the ability to evolve into and remain memory CD8+ T cells upon rechallenge.25–27 In the context of adoptive cell immunotherapy for the treatment of melanoma, self/tumor-reactive CD8+ T cells may undergo a similar fate in the setting of persistent self protein expression. This underlines the potential importance of CD4+ T cell help in facilitating CD8+ T-cell survival, activation, and function in vivo.
In earlier adoptive cell transfer studies, anti-melanoma CD8+ T cell clones were administered to metastatic melanoma patients following lymphodepleting conditioning.28 The preparative lymphoconditioning is thought to improve the host environment for cell transfer by removing Treg cells and cells that compete for APC interaction and homeostatic cytokines such as IL-7 and IL-15.29 No objective clinical responses were observed, and transferred antitumor T-cell clones did not persist in vivo. More recently, tumor infiltrating lymphocytes (TILs), which were heterogenous in CD8/CD4 T-cell composition, were used for adoptive cell transfer to lymphodepleted patients.30 In the context of bulk TILs, transferred T cells persisted and mediated objective clinical responses in nearly 50% of treated patients.30,31 The diversity of this transferred T-cell population may help explain the ability of the bulk TILs to persist and mediate tumor regression in vivo. The relative composition of T-helper and Treg subsets within the CD4+ T-cell component of bulk TIL preparations is under investigation.
Analogous to our mouse studies,9 the ability to enrich for CD25−CD4+ T-helper cells and to administer these enriched cells in conjunction with bulk TILs to lymphoconditioned patients may provide benefit to current adoptive immunotherapeutic approaches. In this study, we evaluated whether CD25+ Treg cells could be efficiently depleted from patient pheresis samples in large scale and under clinical-grade conditions. Following immunomagnetic selection, cells from CD25-depleted fractions expressed little to no FOXP3 mRNA or CD25 on the cell surface. Exposure to IL-2 had little effect on this phenotype, demonstrating that short-term IL-2 culture does not mediate the generation of Treg cells from CD25− CD4+ T cells. CD25+ cells separated from the same leukapheresis product expressed high CD25 and FOXP3, were able to inhibit the proliferation of CD25− CD4+ T cells in standard suppression assays, and were hypoproliferative in the absence of combined TCR stimulation and IL-2. These results demonstrate that selective separation of functional CD25+ Treg cells from large-scale samples can be performed under clinical-grade conditions with sufficient selection, recovery, viability, ability to expand, and function for potential use in adoptive immunotherapy.
Overcoming peripheral self tolerance mechanisms is important in mediating in vivo tumor regression in cancer patients. Immune responses directed against self proteins expressed by both normal tissues and cancers help explain the frequent occurrence of autoimmune vitiligo in melanoma patients responding to IL-2-based therapies.30 Administration of IL-2 alone to metastatic melanoma patients mediates objective tumor regressions in about 15% of treated individuals, although the mechanism of IL-2-mediated tumor regression is unknown.32 Paradoxically, recent studies have shown that one factor that inhibits the potential for immune response is the ability of IL-2 to mediate the in vivo generation of Treg cells that can inhibit immune reactions.33 In vivo generation of Treg cells may thus represent a major obstacle to the success of IL-2-based therapies. Indeed, exogenous IL-2 administration to metastatic melanoma patients results in increased frequencies of functional CD4+ CD25+ Treg cells in the peripheral blood (Ahmadzadeh M, manuscript in preparation). Similarly, in vitro culture of pre-separation CD4+ T cells in IL-2 increased the overall frequency of CD25+ CD4+ T cells, the level at which CD25 was expressed, and the level of FOXP3 expression. This was not true for CD4+ T cells devoid of Treg cells, suggesting that selective expansion of Treg cells had occurred in pre-separation CD4 cultures. Transforming CD4+ CD25− T cells into functional Treg cells in vitro requires strong stimulation such as anti-CD3 and CD28 antibodies in conjunction with TGF-β1 or mitogens, or mitogen-activated APCs.34–36 Since IL-2-mediated Treg cell expansion in vivo may be responsible in part for limiting IL-2 response rates in melanoma patients, the optimal environment for successful IL-2-mediated tumor regressions may be one devoid of Treg cells. To this end, autologous ex vivo CD25-depleted pheresis products for potential adoptive transfer to cancer patients after lymphodepleting chemotherapy may improve the efficacy of IL-2-based therapies. Alternatively, we are evaluating a number of compounds with the potential to target and eliminate the suppressive activity of Treg cells in vivo, including anti-CTLA-4 and toxin conjugated IL-2 cytokine and CD25 antibodies.37–39
In addition to IL-2-mediated responses, a host homeostatic environment devoid of Treg cells can influence the ability to induce CD8- and CD4-mediated antitumor responses.40,41 In the absence of Treg cells, limited tumor outgrowth and augmented reactivity against a surrogate tumor antigen, known to induce both CD4+ and CD8+ T-cell responses, has been shown using a mouse tumor challenge model.41 In another study, the combination of vaccination with CD25+ Treg cell depletion and blockade of CTLA-4, a negative immunoregulatory molecule, could induce a tyrosinase-related protein 2-specific CD8+ T-cell dependent protection from B16 melanoma challenge.40 In our cancer vaccine trials of 440 cancer patients receiving peptide, viral, and dendritic cell vaccines, the overall objective response rate was 2.6%.42 Circulating vaccine-specific T cells could often be detected in vaccinated patients despite this low response rate. For example, in one trial 15 of 18 patients vaccinated with an immunodominant MART-1 peptide showed evidence of immunization, with only one patient experiencing an objective clinical response.43 Similar results have been observed following gp100-specific peptide vaccination.44 The reason for this low response rate remains uncertain; however, the ability to vaccinate patients in a CD25+ Treg-depleted setting, based on either ex vivo or in vivo depletion strategies, deserves evaluation. Infusion of Treg-depleted lymphocytes and subsequent vaccination of lymphodepleted patients may offer added benefit, since mouse studies have shown that tumor-specific T cells preferentially expand in the lymphopenic environment after a melanoma vaccine was given to lymphopenic mice reconstituted with naive T cells from normal mice.45 Furthermore, vaccination of reconstituted lymphopenic hosts could elicit superior antitumor immunity compared with normal hosts, highlighting the potential clinical benefit of performing tumor vaccination during immune reconstitution of the lymphopenic host.
The ability to selectively deplete or enrich CD25+ Treg cells from large-scale patient leukapheresis products has potential clinical implications for the immunotherapeutic treatment of patients with cancer and autoimmunity, respectively. Our aim to improve the efficacy of current immunotherapeutic strategies through the administration of vaccines, cytokines, or tumor-reactive T cells in the absence of Treg cell functional activity relies heavily upon the ability to efficiently deplete Treg cells either in vivo or ex vivo. As the majority of antitumor immune responses are directed against self tissue antigens, loss of regulatory cell function and enhanced access of antitumor effector cells to CD4+ T-cell help may affect the ability to induce clinical tumor regressions in vivo.
References
- 1.Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 2.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 3.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 4.Asano M, Toda M, Sakaguchi N, et al. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med. 1996;184:387–396. doi: 10.1084/jem.184.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McHugh RS, Shevach EM. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. J Immunol. 2002;168:5979–5983. doi: 10.4049/jimmunol.168.12.5979. [DOI] [PubMed] [Google Scholar]
- 6.Golgher D, Jones E, Powrie F, et al. Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur J Immunol. 2002;32:3267–3275. doi: 10.1002/1521-4141(200211)32:11<3267::AID-IMMU3267>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 7.Turk MJ, Guevara-Patino JA, Rizzuto GA, et al. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–782. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antony PA, Piccirillo CA, Akpinarli A, et al. CD8+ T cell immunity against a persisting tumor/self-antigen requires CD4+ T cells and fails in the presence of naturally occurring T regulatory cells. J Immunol. 2005;174:2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang HY, Lee DA, Peng G, et al. Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity. 2004;20:107–118. doi: 10.1016/s1074-7613(03)00359-5. [DOI] [PubMed] [Google Scholar]
- 11.Javia LR, Rosenberg SA. CD4+CD25+ suppressor lymphocytes in the circulation of patients immunized against melanoma antigens. J Immunother. 2003;26:85–93. doi: 10.1097/00002371-200301000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Viguier M, Lemaitre F, Verola O, et al. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol. 2004;173:1444–1453. doi: 10.4049/jimmunol.173.2.1444. [DOI] [PubMed] [Google Scholar]
- 13.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 14.Schoof E, Girstl M, Frobenius W, et al. Decreased gene expression of 11beta-hydroxysteroid dehydrogenase type 2 and 15-hydroxyprostaglandin dehydrogenase in human placenta of patients with preeclampsia. J Clin Endocrinol Metab. 2001;86:1313–1317. doi: 10.1210/jcem.86.3.7311. [DOI] [PubMed] [Google Scholar]
- 15.Lizee G, Aerts JL, Gonzales MI, et al. Real-time quantitative reverse transcriptase-polymerase chain reaction as a method for determining lentiviral vector titers and measuring transgene expression. Hum Gene Ther. 2003;14:497–507. doi: 10.1089/104303403764539387. [DOI] [PubMed] [Google Scholar]
- 16.Heid CA, Stevens J, Livak KJ, et al. Real-time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 17.Ringhoffer M, Wiesneth M, Harsdorf S, et al. CD34 cell selection of peripheral blood progenitor cells using the CliniMACS device for allogeneic transplantation: clinical results in 102 patients. Br J Haematol. 2004;126:527–535. doi: 10.1111/j.1365-2141.2004.05062.x. [DOI] [PubMed] [Google Scholar]
- 18.Schumm M, Lang P, Taylor G, et al. Isolation of highly purified autologous and allogeneic peripheral CD34+ cells using the CliniMACS device. J Hematother. 1999;8:209–218. doi: 10.1089/106161299320488. [DOI] [PubMed] [Google Scholar]
- 19.Knauf W, Fietz T, Schrezenmeier H, et al. CD34 selected alloPBSCT and adoptive immunotherapy. Bone Marrow Transplant. 2000;25(Suppl 2):S2–S5. doi: 10.1038/sj.bmt.1702342. [DOI] [PubMed] [Google Scholar]
- 20.Kurts C, Carbone FR, Barnden M, et al. CD4+ T cell help impairs CD8+ T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J Exp Med. 1997;186:2057–2062. doi: 10.1084/jem.186.12.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bennett SR, Carbone FR, Karamalis F, et al. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 22.Keene JA, Forman J. Helper activity is required for the in vivo generation of cytotoxic T lymphocytes. J Exp Med. 1982;155:768–782. doi: 10.1084/jem.155.3.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahemtulla A, Fung-Leung WP, Schilham MW, et al. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature. 1991;353:180–184. doi: 10.1038/353180a0. [DOI] [PubMed] [Google Scholar]
- 24.Bourgeois C, Veiga-Fernandes H, Joret AM, et al. CD8 lethargy in the absence of CD4 help. Eur J Immunol. 2002;32:2199–2207. doi: 10.1002/1521-4141(200208)32:8<2199::AID-IMMU2199>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 25.Janssen EM, Lemmens EE, Wolfe T, et al. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 26.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dudley ME, Wunderlich JR, Yang JC, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–251. doi: 10.1097/01.CJI.0000016820.36510.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klebanoff CA, Khong HT, Antony PA, et al. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26:111–117. doi: 10.1016/j.it.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenberg SA, Yang JC, Topalian SL, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907–913. [PubMed] [Google Scholar]
- 33.Malek TR. The main function of IL-2 is to promote the development of T regulatory cells. J Leukoc Biol. 2003;74:961–965. doi: 10.1189/jlb.0603272. [DOI] [PubMed] [Google Scholar]
- 34.Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25-naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verhasselt V, Vosters O, Beuneu C, et al. Induction of FOXP3-expressing regulatory CD4pos T cells by human mature autologous dendritic cells. Eur J Immunol. 2004;34:762–772. doi: 10.1002/eji.200324552. [DOI] [PubMed] [Google Scholar]
- 36.Morgan ME, van Bilsen JH, Bakker AM, et al. Expression of FOXP3 mRNA is not confined to CD4+CD25+ T regulatory cells in humans. Hum Immunol. 2005;66:13–20. doi: 10.1016/j.humimm.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 37.Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA. 2003;100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kreitman RJ, Wilson WH, Robbins D, et al. Responses in refractory hairy cell leukemia to a recombinant immunotoxin. Blood. 1999;94:3340–3348. [PubMed] [Google Scholar]
- 39.LeMaistre CF, Saleh MN, Kuzel TM, et al. Phase I trial of a ligand fusion-protein (DAB389IL-2) in lymphomas expressing the receptor for interleukin-2. Blood. 1998;91:399–405. [PubMed] [Google Scholar]
- 40.Sutmuller RP, van Duivenvoorde LM, van Elsas A, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. 2001;194:823–832. doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klein L, Khazaie K, von Boehmer H. In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proc Natl Acad Sci USA. 2003;100:8886–8891. doi: 10.1073/pnas.1533365100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cormier JN, Salgaller ML, Prevette T, et al. Enhancement of cellular immunity in melanoma patients immunized with a peptide from MART-1/Melan A. Cancer J Sci Am. 1997;3:37–44. [PMC free article] [PubMed] [Google Scholar]
- 44.Salgaller ML, Marincola FM, Cormier JN, et al. Immunization against epitopes in the human melanoma antigen gp100 following patient immunization with synthetic peptides. Cancer Res. 1996;56:4749–4757. [PubMed] [Google Scholar]
- 45.Hu HM, Poehlein CH, Urba WJ, et al. Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res. 2002;62:3914–3919. [PubMed] [Google Scholar]