

Abstract
Complexes containing bonds between heteroatoms such as nitrogen and oxygen and “late” transition metals (i.e., those located on the right side of the transition series) have been implicated as reactive intermediates in numerous important catalytic systems. Despite this, our understanding of such M–X linkages still lags behind that of their M–H and M–C analogues. New synthetic strategies have now made possible the isolation and study of a variety of monomeric late-metal alkoxide, aryloxide, and amide complexes, including parent hydroxide and amide species. The heteroatoms in these materials form surprisingly strong bonds to their metal centers, and their bond energies do not necessarily correlate with the energies of the corresponding H–X bonds. The M–X complexes typically exhibit nucleophilic reactivity, in some cases form strong hydrogen bonds to proton donors, and even deprotonate relatively weak acids. These observations, as well as thermodynamic investigations, suggest that late metal–heteroatom bonds are strongly polarized and possess significant ionic character, properties that play an important role in their interactions with organic compounds.
Introduction
Studies of organometallic reaction mechanisms have identified a number of primary transformations such as migratory insertion, β-elimination, oxidative addition, and reductive elimination that are essential to a wide range of metal-mediated processes.1,2 These primary steps act as “building blocks” for more complicated, multistep transformations that result in the formation of organic compounds with new carbon–carbon and carbon–hydrogen bonds (e.g., hydroformylation, hydrogenation, carbon–carbon coupling reactions, etc.). The large majority of intermediates in these reactions have reactive metal–carbon and metal–hydrogen bonds, and consequently most of the understanding that we now have about the mechanisms of organometallic reactions relates to processes involving such bonds.
Organotransition metal complexes also catalyze the formation of bonds between carbon and more electronegative atoms (“heteroatoms”), such as nitrogen, oxygen, phosphorus, and sulfur.3–12 Such processes have long been known in synthetic oxidation chemistry,5,6 they are being increasingly identified in biological systems,8,9 and many new metal-catalyzed processes useful in synthesizing organic compounds with carbon–heteroatom bonds have recently been discovered.3,4,12 Often these reactions have been proposed to proceed through transient intermediates having low-valent late transition metal centers bound to heteroatomic groups, such as unbridged alkoxide (OR) or amide (NR2) ligands. However, until relatively recently, few general methods had been developed for the synthesis of isolable examples of such complexes. As a result, little detailed information has been available on the chemistry of simple monomeric complexes with late metal–heteroatom bonds, or about the mechanisms of their reactions with organic compounds.7,13
It is our view that organometallic chemistry, with the benefit of concepts derived from its classical focus on the study of metal–carbon bonded complexes, can make important contributions to our understanding of transformations that take place at low-valent metal–heteroatom bonds (hereafter referred to as “M–X bonds”), including those that form new bonds between carbon atoms and heteroatoms. To this end, several years ago we set out to develop reliable methods for preparing simple, monomeric metal alkoxides, hydroxides, and amides so that their behavior could be studied systematically. This Account reviews some of the synthetic methodology used by and developed in our group, the novel chemistry exhibited by several of these M–X compounds, and what we have learned so far about the nature of the M–X bond and its relevance to metal-catalyzed carbon–heteroatom bond-forming processes.
Catalytic Reactions Proposed To Involve Reactions at M–O and M–N Bonds.
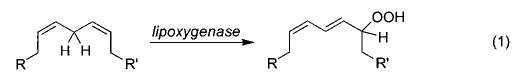
As noted above, complexes with metal–heteroatom single bonds have been implicated in a wide range of biologically and synthetically important catalytic transformations. One important biological case study is the lipoxygenase (a non-heme-iron-containing enzyme)-mediated conversion of 1,4-fatty dienes to alkyl-hydroperoxides (eq 1),14 which is commonly proposed to involve abstraction of an allylic hydrogen atom by an iron hydroxide functionality (eq 2).15,16 Jonas and Stack have

shown that an FeIII(OMe) lipoxygenase model readily oxidizes 1,4-cyclohexadiene to benzene and the corresponding FeII(MeOH) complex,17 implying that the methoxy group can act as an odd-electron fragment by abstracting an H atom from a weak allylic C–H bond of the organic substrate.
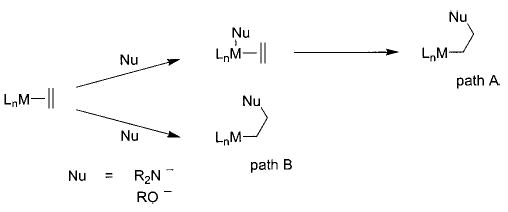
Among the most industrially important synthetic heteroatom-transfer processes are the Wacker process (eq 3) and its mechanistic relative, hydroamination (eq 4).18,19 The key step in these transformations, the net metal-mediated addition of a heteroatom nucleophile to an alkene to form a new carbon–oxygen or –nitrogen bond, has been proposed to take place by either migratory insertion of a coordinated olefin into an M–X bond (Scheme 1, path A) or external nucleophilic attack of water (or amine) at a coordinated olefin (Scheme 1, path B). While there is evidence favoring both of these routes, the mechanism has yet to be definitively established.20,21
Scheme 1.

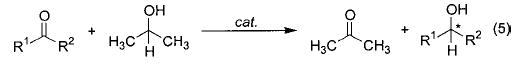
A third important example is transfer hydrogenation of ketones, which is a highly efficient method for enantioselective synthesis of chiral alcohols (eq 5).22–24 Many of the metal catalysts for this transformation have been shown to operate by forming metal alkoxides which subsequently undergo β-hydride elimination. Noyori has also proposed for his catalytic system a novel mechanism involving concerted transfer of two hydrogen atoms from the catalyst complex to the substrate’s carbonyl group (Figure 1).23
FIGURE 1.
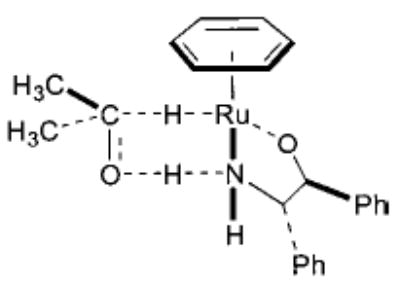
Noyori’s proposed transition state for transfer hydrogenation.
Direct Studies of the Primary Reactions of Metal Alkoxides and Amides.
Reactivity studies provide a good indication of the lability of metal–oxygen and –nitrogen bonds. In the modest number of late transition metal examples that have been examined so far, reactivity at M–O and –N bonds is superficially more analogous to chemistry that might be expected of alkali metal or “free” alkoxides and amides than to the reactivity of charge-neutral organic ethers or amines.7,25 Late-metal alkoxides (and a smaller number of amides) have been observed to attack electrophilic organic compounds such as acid chlorides and also deprotonate a variety of weak acids such as phenylacetylene, alcohols, and amines. Direct observation of migratory insertion reactions with less electrophilic reagents, such as CO and alkenes, appears to be much less common than with metal–carbon bonds.26,27 In an important example, Bryndza and coworkers have observed the migratory insertion reactions of fluorinated alkenes into the M–O bonds of platinum alkoxides.28 However, the lack of reactivity of Bryndza’s complex toward ethylene itself might be taken as evidence of a polar transition state for the fluorinated alkene reaction.
One important issue that has been discussed frequently is the thermochemistry of the metal–heteroatom bond. It has often been asserted that late metal–oxygen and –nitrogen bonds are weaker than their early-metal counterparts, but if we focus on specific homolytic bond dissociation energies (BDEs), rather than average bond energies, only a few studies have been carried out to test this expectation.29,30
More systematic information is available about relative (rather than absolute) bond energies. In a widely cited study by Bryndza, Bercaw, and co-workers, a correlation was proposed between the relative bond strengths of metal–heteroatom bonds (M–X) and their corresponding acids (H–X).30 In these systems, a case was made that a strong H–X bond correlates to a strong M–X bond, and a weak H–X bond correlates to a weak M–X bond (X = OR, NR2, CR3). More specifically, the difference in the BDEs between the [Pt]–OMe and H–OMe bonds is approximately the same as the difference in the BDEs between [Pt]–NMePh and H–NMePh bonds (eq 6).
| (6) |
Finally, there has been substantial discussion of whether the energies of metal–heteroatom bonds are affected by p-π/d-π repulsion between the heteroatom lone pair electrons and electrons in filled d shells on the metal center.31 Caulton has made a strong case that the filled π-symmetry orbitals of the heteroatom of late transition metal M–X complexes can either stabilize the complex if there is an empty d-orbital (such as in 16-electron complexes or intermediates) or undergo a filled/filled repulsion with a filled metal d-orbital, resulting in a higher energy π-orbital (Figure 2). This translates into a relatively reactive heteroatom lone pair, explaining both the nucleophilic and basic reactivity observed at the heteroatom.
FIGURE 2.
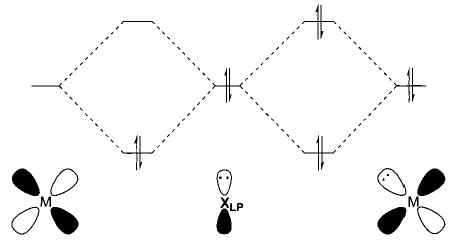
Interaction between a π-symmetry lone pair (LP) orbital on X (=O, N) and the empty and filled d-orbitals of a transition metal.
In our group we have investigated the various properties of late-metal M–X bonds described above. Our approach has focused on the syntheses of complexes containing M–X bonds and the detailed mechanistic investigation of processes involving these complexes. The remainder of this Account summarizes the progress of our recent efforts in these areas.
General Synthetic Routes and Problems
Alkoxides and Amides.
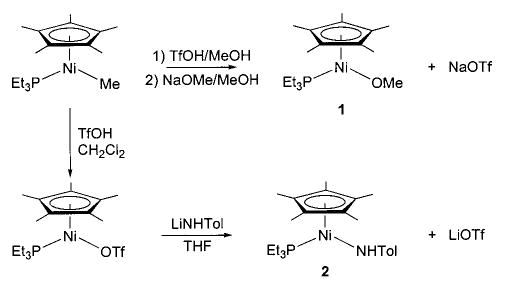
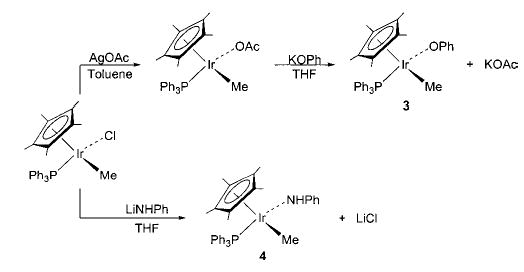
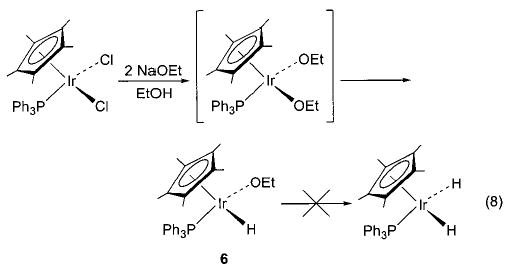
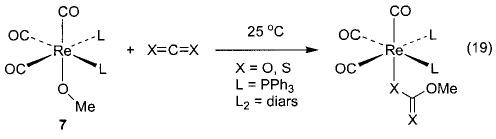
Although we have been able to synthesize several late transition metal alkoxo, aryloxo, and amido complexes, we have found that no single synthetic technique is applicable to all systems. A technique that has proven very convenient in several systems is the metathesis of metal chloride or triflate complexes with alkali metal alkoxides and amides. The syntheses of nickel methoxide 1 and toluidide 2 shown in Scheme 2 use this method, as do the syntheses of the iridium phenoxide and anilide complexes 3 and 4 shown in Scheme 3.32 Similarly, addition of sodium methoxide to fac-(CO)3(PPh3)2Re(OTf) yields the corresponding rhenium methoxide (5, eq 7).33,34 The preparation of Cp*(PPh3)Ir(H)(OEt) (6) proceeds via metathesis from the iridium dichloride (eq 8).35,36 This intriguing process apparently involves one β-hydride elimination by an intermediate ethoxide complex to produce the hydride ligand, but no further elimination by the product to yield the dihydride.
Scheme 2.
Scheme 3.

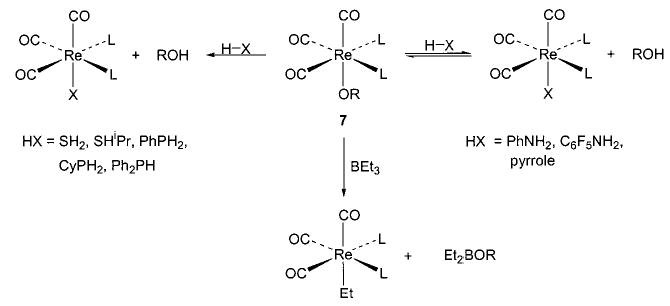
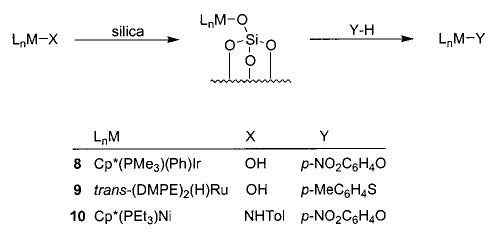
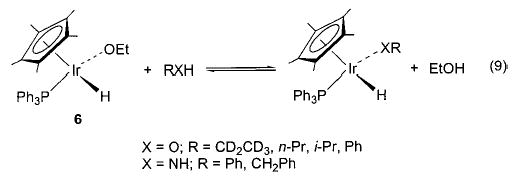
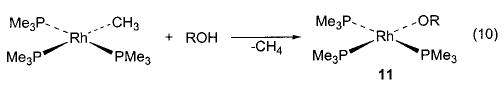
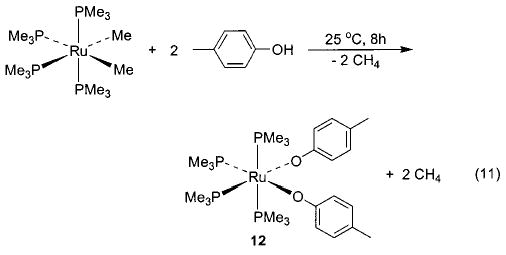
Exchange of one heteroatom group for another has also proven to be a useful synthetic technique in several instances. For example, addition of alcohols and amines to 6 yields the corresponding alkoxides and amides, respectively (eq 9).36 This method is also applicable in the rhenium–alkoxide systems (7) shown in Scheme 4, although in this case it is necessary to use 4 Å molecular sieves (which sequester the methanol that is generated) in order to drive the equilibrium toward the desired products.37 The heteroatom exchange method also allowed access to silica-bound complexes of iridium (8), 38 nickel (9), 29 and ruthenium (10)39 via simple treatment of the respective alkoxides with silica (Scheme 5). In some cases, exchange of a heteroatom for a methyl group (yielding methane) has proven a viable route to either an alkoxide40,41 (11, 12; eqs 10 and 11) or an intermediate triflate complex (Scheme 2).29
Scheme 4.
Scheme 5.
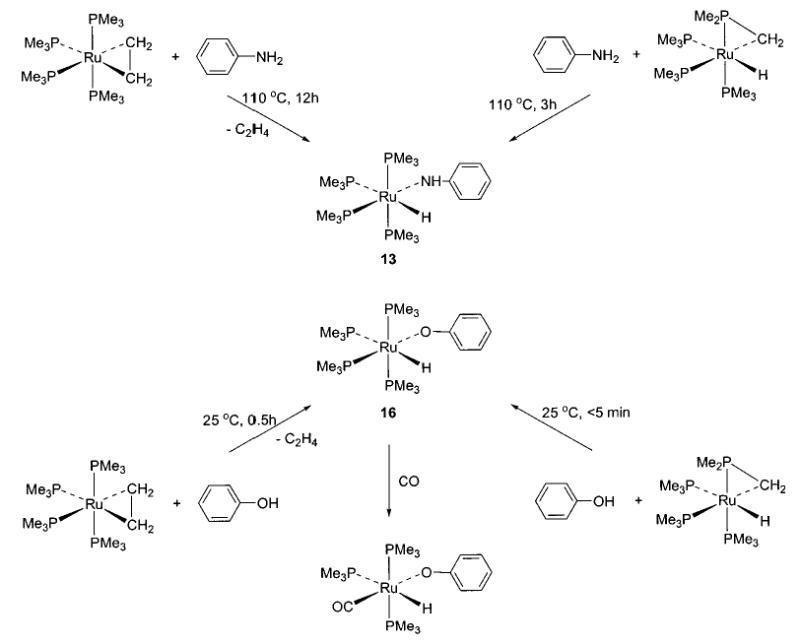
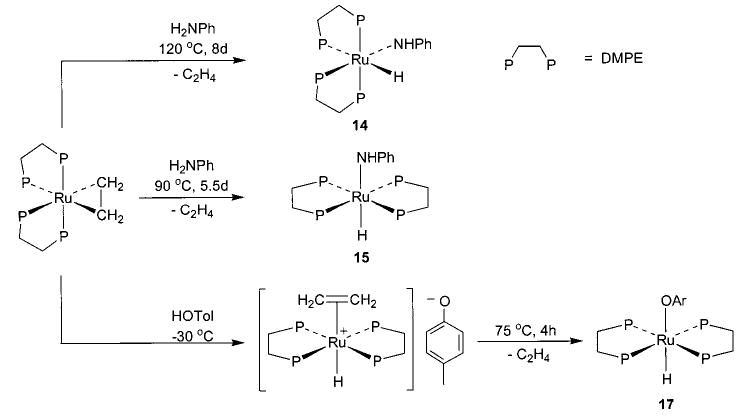
Synthesis of ruthenium tetrakis phosphine amides 13–15 and aryloxide 16 can be achieved using either the appropriate ethylene complex or (PMe3)3Ru(η2-CH2PMe2)-(H), as shown in Schemes 6 and 7.42,43 These processes are thought to proceed via initial protonation of the ruthenium center, as demonstrated by the observation of the [(DMPE)2Ru+(H)(C2H4)][−OTol] ion pair at −30 °C (Scheme 7).43
Scheme 6.
Scheme 7.
Synthesis of Hydroxo and Parent Amido Complexes.
With this range of synthetic routes to alkoxide, aryloxide, and amido complexes available, we were able to devise related methodologies for the synthesis of the much less common parent hydroxo (M–OH) and amido (M–NH2) complexes. In analogy to the oxidative addition of alcohols to (PMe3)4Ru(C2H4) to generate alkoxide complexes, addition of 1 equiv of water resulted in the formation of cis-(PMe3)4Ru(H)(OH) (18) in 80% conversion at room temperature and 30% isolated yield (eq 12).44 Treatment of (DMPE)2Ru(C2H4) (DMPE = Me2PCH2CH2PMe2) with excess water at 80 °C resulted in the formation of a trans-(DMPE)2Ru(H)(OH) dimer bridged by two hydrogen-bonded water molecules (Scheme 8), and this complex was characterized by an X-ray diffraction study. The free hydroxo complex trans-(DMPE)2Ru(H)(OH) (19) was then obtained upon treatment of a solution of the dimer with 3 Å molecular sieves. This material can also be prepared by treating (DMPE)2Ru(H)2 with 1 equiv of N2O (Scheme 9).39
Scheme 8.
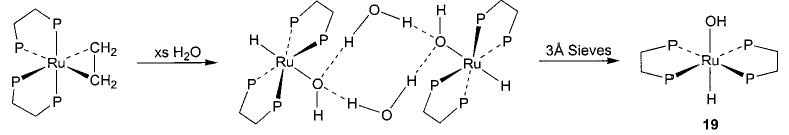
Scheme 9.
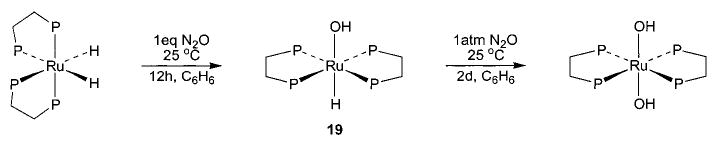
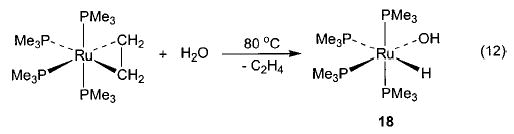
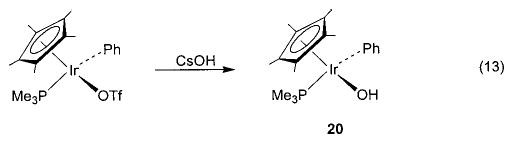
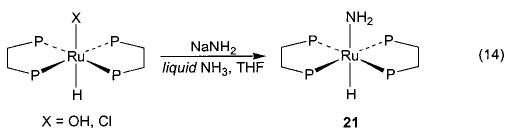
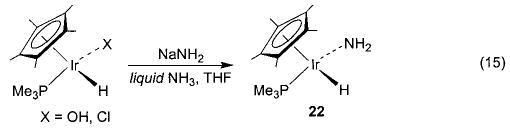
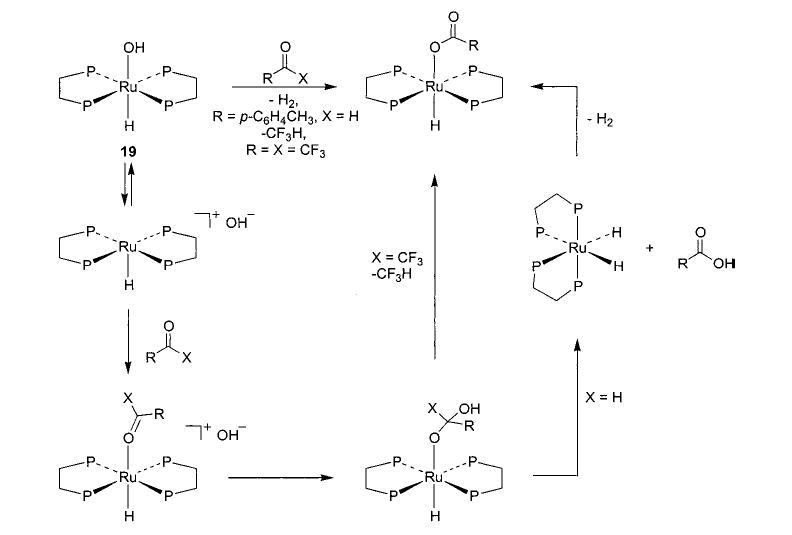
Salt metathesis has also been successfully used to generate late-metal hydroxo and parent amido complexes, although choice of cation and solvent system is crucial to the success of these reactions. Addition of CsOH to a solution of Cp*(PMe3)(Ph)Ir(OTf) in THF resulted in the formation of Cp*(PMe3)(Ph)Ir(OH) (20) in 78% isolated yield (eq 13).45 Synthesis of the corresponding M–NH2 compounds proved to be more difficult. After substantial experimentation, we found that treatment of trans-(DMPE)2Ru(H)(Cl) with NaNH2 yields (DMPE)2Ru(H)-(NH2) (21) in 81% yield (eq 14), but only when a 1:1 mixture of liquid ammonia and THF is used as the solvent system.46 This material is the first structurally characterized monomeric late-metal parent amido complex. Similarly, treatment of Cp*(PMe3)(Ph)Ir(Cl) with NaNH2 under the same solvent conditions afforded Cp*(PMe3)(Ph)-Ir(NH2) (22) with 95% conversion (eq 15).46 This complex is sensitive to concentration and is stable only in solution.
Chemistry of the M–X Bond
Nucleophilic Reactivity with Organic Electrophiles.
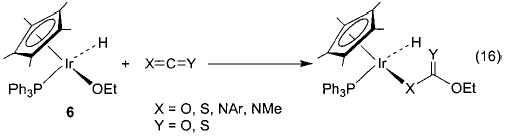
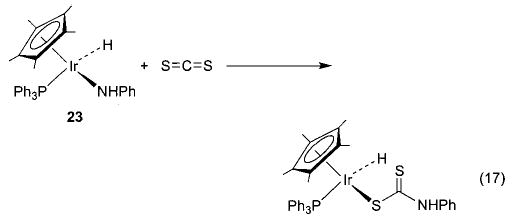
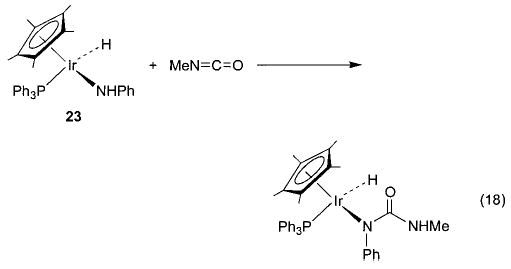
With these new synthetic techniques in hand, we were able to begin to study the nature of late metal–oxygen and -nitrogen single bonds. Heterocumulenes (CO2, CS2, RNCO) have commonly been used to probe the nucleophilicity of metal alkoxide and amide complexes. Iridium ethoxide 6 reacts quite readily with these substrates to generate products of net nucleophilic attack at the electrophilic carbon of the heterocumulene (eq 16).36 For example, addition of CO2 to 6 results in the formation of the metallocarbonate complex Cp*(PPh3)(H)Ir(OC(O)OEt). The corresponding amido complex Cp*(PPh3)(H)Ir(NHPh) (23) undergoes similar nucleophilic insertion chemistry with carbon disulfide to form the metalloxanthate Cp*(PPh3)(H)Ir(SC(S)NHPh) (eq 17). Methyl isocyanate undergoes an overall insertion into the N–H bond to produce Cp*(PPh3)(H)Ir(NPhC(O)NHMe) (eq 18), although this product is likely the result of rearrangement following initial insertion into the M–N bond.
The rhenium series also displays nucleophilic insertion behavior toward heterocumulenes, and the reactivity is dependent on the type of alkoxide complex involved. Addition of CO2 or CS2 to the rhenium methoxide complexes (CO)3L2Re(OMe) (7) results in rapid formation of the metallocarbonate and metalloxanthate complexes, respectively (eq 19).34,47 However, the cresolate complexes of the same series (24) react with CS2 only at elevated temperatures (140 °C; eq 20). No reaction is observed with CO2. This emphasizes the apparent higher stability of aryl-over alkyl-substituted M–OR and M–NR2 complexes.

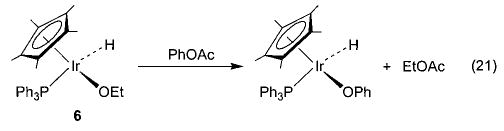
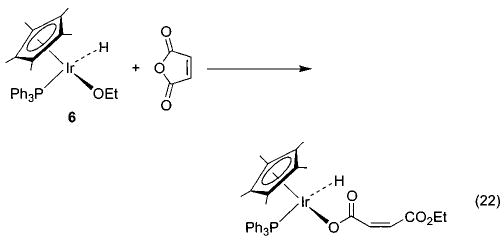
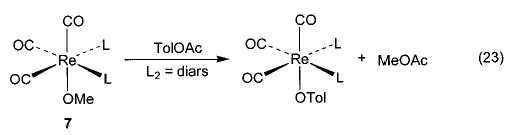
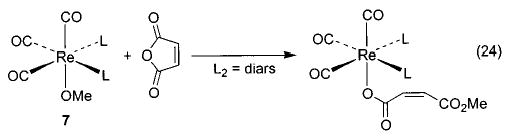
We have observed other examples of apparent nucleophilic reactivity as well. The Ir–OEt complex 6 reacts with phenyl acetate to generate an Ir–OPh complex and ethyl acetate36 (eq 21), and the same complex also induces the nucleophilic ring opening of cyclic anhydrides to generate an iridium carboxylate complex (eq 22). The rhenium methoxide complex 7 displays similar behavior toward these electrophiles (eqs 23 and 24).47 The rhenium complexes also react with acetyl chloride to generate the corresponding rhenium chloride complex.
Hydrogen Bonding.
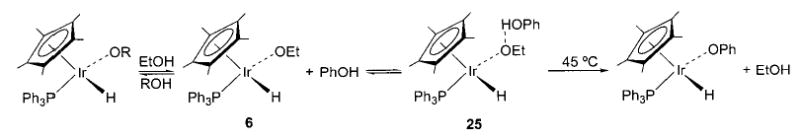
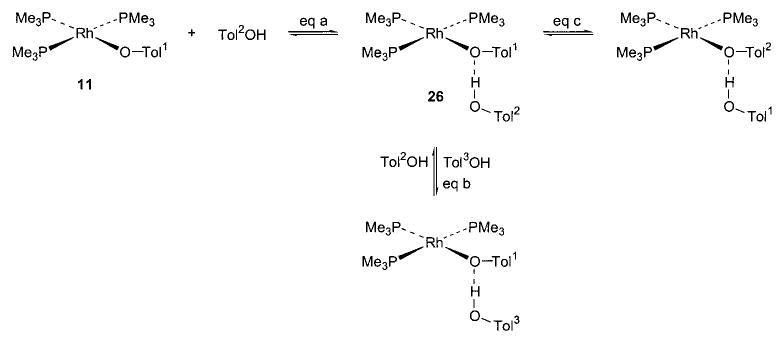
Additional evidence for the considerable polarization of these bonds is provided by several observations of hydrogen bonding to the oxygen atoms of late-metal alkoxides and aryloxides. We first observed hydrogen bonding to the iridium ethoxide complex 6 (Scheme 10),35 and shortly thereafter we noted similar behavior on the part of the rhodium aryloxide complex 11 (Scheme 11, eq a).40 Treatment of either 6 or 11 with phenol produced an equilibrium mixture of the starting material and the hydrogen-bonding adduct 25 or 26, respectively. Both adducts were also stable in the solid state.
Scheme 10.
Scheme 11.
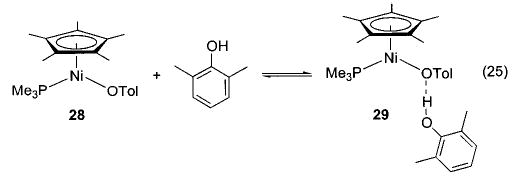
X-ray diffraction studies of the rhenium ethoxide/cresol adduct 27a (Scheme 12)48 and the hydrogen-bonded nickel aryloxide 29 (eq 25)29 proceeded well enough to locate the bridging hydrogen atoms and refine their positions and isotropic thermal parameters. In both structures, the hydrogen atoms are very asymmetrically disposed along nearly linear O–H–O linkages (172° and 166°, respectively). The bond distances in these complexes fall within the range that is typical for hydrogen bonding, and the short O–O and O(ethoxide)–H distances in 27a (2.53 and 1.77 Å, respectively) indicate that this bonding is strong.49–51
Scheme 12.

We have investigated the persistence of these hydrogen bonds in solution using 1H NMR spectroscopy. When rhodium complex 26 is treated with excess p-cresol, the hydrogen-bonded cresol molecule is rapidly exchanged with the free species (Scheme 11, eq b). This process is sufficiently facile that even at −80 °C, the ortho protons of both bound and free cresol appear as a single resonance in the 1H NMR spectrum. The analogous exchange in the rhenium complex 27a is rapid on the NMR time scale at room temperature. Even under vacuum, the hydrogen bonding in the latter case appears to be robust.
The thermodynamics of hydrogen bonding of cresol to rhodium aryloxide 26 were studied both by calorimetry and by 1H NMR using the Scatchard method. The hydrogen bond enthalpy of −11.4 kcal/mol and association constant of 1.56 × 103 thus determined (at 25 °C in benzene) confirm that the hydrogen bond is relatively strong. For comparison, the ΔH value for hydrogen bonding between phenol and anisole is only −3.5 kcal/mol,52 and even between cresol and pyridine it is only −6.88 kcal/mol (both measured in CCl4).53 Larger formation energies for hydrogen bonds have been observed only for bonds to anionic species, and most of these energies have been measured in the gas phase (e.g., −19.3 kcal/mol for HOEt with PhO−).54 The interaction in the 11/26 system appears to fall between these two extremes, suggesting that the Rh–O bond is covalent but highly polarized, and that the metal-bound oxygen is thus unusually basic for a formally neutral atom.
H–X/M–X Exchange
In all of the systems in which hydrogen bonding was observed, as well as the others shown in Schemes 4 and 13–16, we also observed exchange of metal-bound alkoxides and aryloxides with free alcohols and phenols. Both the iridium ethoxide 6 and rhenium alkoxides 7 readily undergo exchange with alcohols at room temperature, but their exchange processes with phenols (in which the hydrogen-bonded species 26 and 27a are observed) occur only at 45 °C.36,40 Exchange takes place between rhodium aryloxide 11 and p-cresol (Scheme 11, eq c) at room temperature. We have also observed exchange in nickel aryloxide 28 (Scheme 13)29 and the iridium hydroxide 20 (Scheme 14).45 After observing the generality of this alkoxide exchange, we were able to exploit it as a synthetic tool (vide supra).
Scheme 13.

Scheme 16.
Scheme 14.
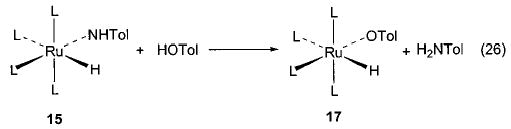
Exchange processes are not limited to cases involving interchange of one oxygen ligand for another. Iridium complex 6 also undergoes exchange with aniline to produce an anilido species,36 and the ruthenium toluido complex 15 exchanges with cresol to generate a ruthenium aryloxide (17, eq 26).42 Both rhenium alkoxides 7 and nickel aryloxide 28 undergo reversible exchange with amines and irreversible exchange with thiols (Schemes 4 and 13).29,37 Finally, the rhenium complexes 7 displayed exchange chemistry with molecules less often involved in such reactions (Scheme 12).37 Exchange with several phosphines yielded phosphido complexes and liberated alcohol, and reactions with alkyl boranes generated rhenium alkyl species by transferring a boryl group (rather than a proton) to the alkoxide. While the last reaction appears to be a substantially different type of transformation than the other exchanges discussed above, it nonetheless involves the cleavage of a polarized bond (Bδ+–Cδ− rather than Hδ+–Xδ−) to place the more electropositive fragment on the heteroatom and the more electronegative moiety on the metal center. It is additionally intriguing due to its relevance to the Suzuki coupling reaction.55
Several mechanisms can be proposed for the more general type of H–X/M–X exchange reaction (Scheme 15). One possibility (Scheme 15, path a) is an ionic dissociation of the alkoxide and subsequent protonation of this species by the alcohol, followed by recombination of the metal center and the newly generated alkoxide. However, both the high rates of these exchanges in nonpolar solvents and the considerably slower exchange rates for phenols relative to alkoxides seem to cast doubt on this possibility. Additionally, a crossover experiment involving two doubly labeled rhenium alkoxides showed that no alkoxide/alkoxide exchange occurred at room temperature when all traces of alcohol were scavenged from the system.48 While the source and role of alcohol in the reaction remain unclear, and further investigation is warranted, these observations nonetheless argue against the involvement of initial ionization in the exchange reactions.
Scheme 15.
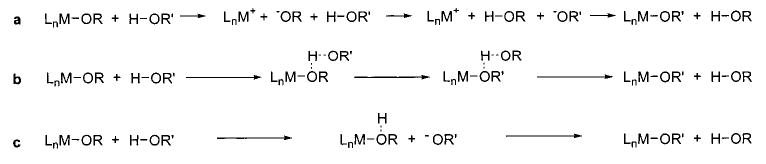
The other mechanistic alternatives (Scheme 15, paths b and c) involve transfer of a proton or hydrogen atom to the bound alkoxide and transfer of the new heteroatom to the metal center. To address these possibilities, we took advantage of the stability of the hydrogen-bonding adducts in the rhenium system48 and studied the kinetics of exchange for the conversion of adducts 27b to exchange products 24b (Scheme 12). The rates of these reactions all displayed clean first-order dependence on [27b] and no dependence on [phenol], suggesting that the hydrogen-bonded species is indeed an intermediate in the exchange process. However, the Hammett plot obtained from these kinetic data was bowl-shaped, with the fastest exchange reactions occurring with both the most electron-donating and the most electron-withdrawing substituents. While such results can be rationalized on the basis of competing BDE and pKa trends, it is difficult to draw any persuasive mechanistic conclusions from these data.
We have observed direct evidence for exchange involving initial proton transfer (Scheme 15, path c) in the case of (DMPE)2Ru(H)(NH2) (21).56 This unusually basic complex deprotonates a wide variety of complexes, and in some cases the resulting coordinated ammonia is replaced by the associated anion. This chemistry is discussed below.
Thermodynamic Investigations.
Throughout this work we have observed qualitative differences in the relative stabilities of various late-metal M–X bonds relative to the corresponding H–X bonds. Some trends are clear: equilibria favor late-metal M–S bonds over M–O bonds, and these are generally favored over M–N bonds. Additionally, M–OAr and M–NHAr are favored over M–OR and M–NHR bonds, respectively. With the intent of quantifying and explaining these differences, exchange reactions (Scheme 13) of the nickel amido complexes 30 were studied in detail, using both calorimetry and the precise measurement of exchange equilibria.29 Utilizing known H–X bond energies, the equilibrium constants obtained were used to construct a scale of relative M–X bond energies.
While a strong correlation between Ni–N and H–N bond energies was observed in the exchange reactions between complexes 30 and 31, the equilibrium constants for the exchange reactions investigated were not equal to 1 as predicted by the 1:1 model of Bercaw and Bryndza.30 Rather, electron-withdrawing anilines favored nickel binding, and electron-donating anilines favored proton binding. While the 1:1 model suggests that the difference between any two H–N bond strengths should be matched by an identical difference between the two corresponding Ni–N bond strengths, we observed a 1.9 kcal/mol Ni–N bond strength difference for every 1 kcal of difference in the H–N bond strengths. The equilibrium constants observed showed a strong correlation with the Hammett parameter σp− (defined by phenol deprotonation), with a ρ value of 3.4. Results deviating from the predictions of the 1:1 model were also observed in exchange reactions involving alkoxide and aryloxide ligands.
These observations are most simply explained by the strong ground-state polarization of late-metal M–X bonds. Because of the substantial ionic component of these bonds, which has been demonstrated repeatedly by their reactivity, stabilization of partial negative charge on the X atom should stabilize the metal complex. In the less ionic H–X bond this stabilization is less marked, so more electronegative atoms bearing delocalizing or electron-withdrawing substituents are favored in binding to the metal center over groups less capable of stabilizing negative charge. We have also analyzed this phenomenon using Drago’s E–C method57,58 and found that this approach complements the qualitative description offered above.29,59 These results reinforce the notion that at least these Ni–X bonds (and possibly M–X bonds involving other late transition metals) include a very large electrostatic component, and suggest that the 1:1 model should be amended to include this consideration.
Unusual Reactivity of Parent M–OH and M–NH2 Complexes
Nucleophilic and Basic Properties.
Some of our most intriguing recent results have come from studies of the novel hydroxo and parent amido complexes prepared in our group. In addition to undergoing the insertion and exchange reactions typical of late-metal alkoxides, these complexes have also displayed exceptional nucleophilicity and basicity. For example, 19 reacts with hexafluoroacetone to release trifluoromethane and generate the fluorinated complex trans-(DMPE)2Ru(H)(OC(O)CF3) (Scheme 16).39 The ruthenium hydroxide will also add to p-tolualdehyde to afford a new ruthenium carboxylate complex and dihydrogen. While this chemistry could be explained by initial M–X heterolysis to give an outer-sphere hydroxide anion, results involving the parent amido complex 21 suggest otherwise (vide infra).
The amido complex 21 undergoes exchange with various weak acids to liberate ammonia. Evidence suggests that ion pairs formed by initial proton transfer are intermediates in this exchange (Scheme 17).56 The ion pair resulting from triphenylmethane deprotonation, [(DMPE)2Ru(H)(NH3)][Ph3C], was spectroscopically observed in equilibrium with the starting materials. When the bulky acid fluorene was used, the exchange process stopped at the ion pair, and the resulting salt 32 was structurally characterized by X-ray diffraction. In cases involving very weak acids, neither the ion pair nor the ammonia-displacement product is observed. However, even with acids as weak as toluene, deuterium-transfer experiments show that deprotonation still takes place. Equilibrium and bracketing studies place the pKa of the protonated ruthenium amide 33 in the vicinity of 31, although this does not consider ion-pairing effects. The pKa of the cationic ruthenium aquo complex [(DMPE)2Ru(H)(OH2)]+ was determined, using the same methodologies, to be approximately 23.
Scheme 17.
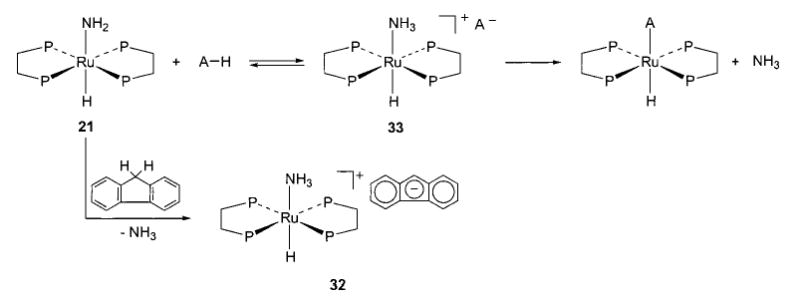
Although rapid H/D exchange occurs between the amido complex and ND3, no 15N is incorporated into 21 upon treatment with 15NH3. This clearly indicates that the Ru–N bond does not dissociate during these proton-transfer reactions, and casts additional doubt on the notion that dissociation of the heteroatom could play a part in other reactions of the amido and hydroxo complexes.
Migratory Insertion with Nonelectrophiles.
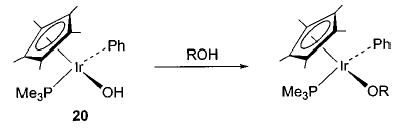
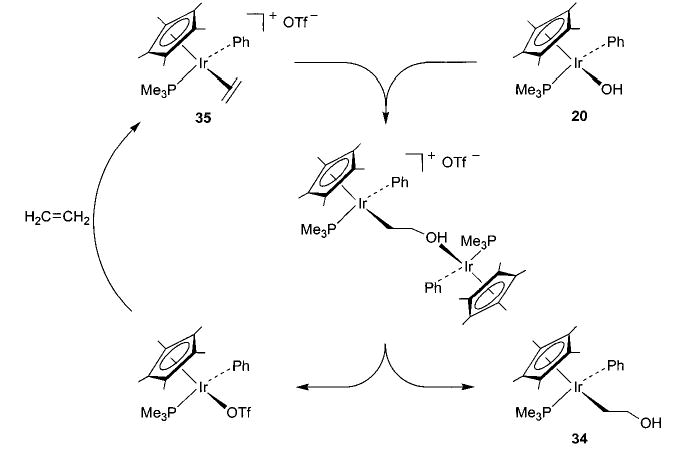
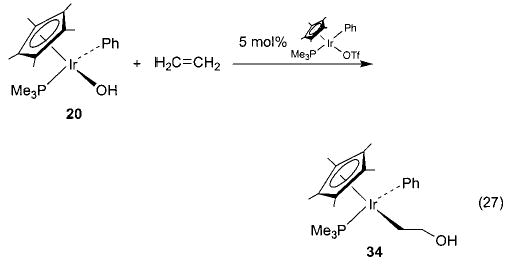
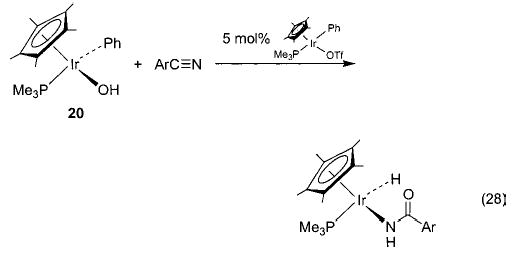
Early studies of the reaction of hydroxoiridium complex 20 with ethylene yielded 34, formed by overall insertion of ethylene into the Ir–OH bond (eq 27).45 Since this reaction is similar to a proposed step in the Wacker process (eq 3), the mechanism was studied in detail. This investigation revealed that different batches of 20 gave widely varying insertion reactivity,60 and repeatedly recrystallized 20 showed no reactivity toward ethylene at all. This result suggested that a catalyst was required for the reaction, and it was found that addition of trace amounts of the triflate complex Cp*(PMe3)(Ph)Ir(OTf) (a precursor to 20) to the purified hydroxide complex 20 renewed its reactivity toward ethylene. Since the triflate ligand can easily dissociate from the iridium metal center, we postulate the binuclear mechanism illustrated in Scheme 18. Here, the cationic center activates ethylene toward nucleophilic attack by the iridium hydroxide complex 20 to generate the hydroxyethyl complex 34 and regenerate the ethylene-coordinated iridium cation 35. A similar process is also observed with arylnitriles.61 The iridium hydroxide complex does not react with benzonitriles, but addition of a catalytic amount of Cp*(PMe3)(Ph)Ir(OTf) to the reaction mixture results in nitrile hydration to form the iridium amide complex Cp*(PMe3)(Ph)Ir(N(H)C(O)Ar) (eq 28).
Scheme 18.
Conclusion and Future Prospects
The number of well-characterized complexes with late transition metal–oxygen and –nitrogen bonds has increased dramatically during the past 20 years. Synthetic methods were developed that initially provided only M–X complexes where the X group was an aryl or electron-deficient moiety, but these have since been extended to give access to much more reactive metal alkoxide and alkylamide complexes. Most recently, even syntheses of monomeric M–OH and M–NH2 complexes have been achieved. The availability of all these materials has allowed our group and others to carefully investigate the chemical transformations of M–X complexes. Studies of these complexes have begun to yield an understanding of the fundamental chemical properties of M–X bonds, as well as the roles they play in a wide range of oxidative and other catalytic processes.
These studies have provided a number of important results, some of which challenge widely accepted ideas about the properties of M–X bonds in low-valent complexes. An important conclusion from our work is that the oft-cited model predicting a general 1:1 correlation between M–X and H–X bond strengths needs to be reevaluated. Additionally, it now seems clear that late metal–nitrogen and –oxygen bonds in simple late-metal alkoxides and amides are quite strongly polarized in the ground state. An expected consequence of this characteristic is that they react most readily with electrophilic organic substrates. This polarization also results in the ability of such complexes to form surprisingly strong hydrogen bonds with organic acceptors and even, in some cases, to display unusually high basicity. The evidence we have obtained indicates that, despite the strong polarization of M–X bonds, the majority of their reactions are initiated not by ionization, but rather by direct interaction of the metal-bound X group with the incoming organic reagent.
We feel certain that these properties are only the first of many that will be revealed as synthetic methods become more general and systematic classes of late transition metal M–X complexes become more readily available. Such progress should lead to increased fundamental understanding of the properties of such complexes and to the application of this understanding to the development of new catalytic C–X bond-forming reactions.
Acknowledgments
We thank our graduate student, postdoctoral fellow, and faculty colleague collaborators, whose names are cited in the references, for their role in generating many of the most significant ideas and results described in this paper. We are also grateful for early funding of this work from the National Institutes of Health, and most recently for support from the National Science Foundation (Grant No. CHE-0094349).
Footnotes
J. Robin Fulton was born in Wichita, Kansas, in 1972. She obtained a B.S. in chemistry and a B.S. in biochemistry in 1995 from the University of Washington, where she worked in the laboratories of James Mayer. She received her Ph.D. from U.C. Berkeley in 2000, and is currently a postdoctoral fellow at the Institute for Arctic and Alpine Research (INSTAAR) at the University of Colorado, Boulder.
Andy Holland was born in Seattle, Washington, in 1975. In 1997 he received his B.S. degree in chemistry from the University of Washington, where he studied with Karen Goldberg. He is currently pursuing his Ph.D. at U.C. Berkeley.
Daniel Fox was born in Poughkeepsie, New York, in 1976. He studied with Robert E. Rosenberg at the State University of New York at Geneseo, where he received his B.S. degree in chemistry in 1998. He is currently pursuing his Ph.D. at U.C. Berkeley.
Robert G. Bergman was born in Chicago, Illinois, in 1942. He received his B.A. at Carleton College and his Ph.D. under the direction of Jerome A. Berson at the University of Wisconsin. After a postdoctoral study with Ronald Breslow at Columbia University, he began his academic career at the California Institute of Technology in 1967. After 11 years at Caltech, he moved his research group to its present location, where he holds appointments at the University of California, Berkeley, and the Lawrence Berkeley National Laboratory. His research interests lie at the interface between organic and inorganic chemistry, with a primary focus on the study of reaction mechanisms. In addition to the topic of the present article, he has made contributions to the areas of diradical and carbocation chemistry, the chemistry of homo- and heterobinuclear complexes, the study of metal–heteroatom multiple bonds, and the use of metal complexes in carbon–hydrogen bond activation reactions.
References
- 1.Spessard, G. O.; Miessler, G. L. Organometallic Chemistry; Prentice Hall: Upper Saddle River, NJ, 1997.
- 2.Collman, J. P.; Hegedus, L. S.; Norton, J. R.; Finke, R. G. Principles and Applications of Organotransition Metal Chemistry; University Science Books: Mill Valley, CA, 1987.
- 3.Hartwig JF. Transition Metal Catalyzed Synthesis of Arylamines and Aryl Ethers from Aryl Halides and Triflates: Scope and Mechanism. Angew Chem Int Ed. 1998;37:2047–2067. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 4.Wagaw S, Rennels RA, Buchwald SL. Palladium-Catalyzed Coupling of Optically Active Amines with Aryl Bromides. J Am Chem Soc. 1997;119:8451–8458. [Google Scholar]
- 5.Simandi, L. I. Catalytic Activation of Dioxygen by Metal Complexes; Kluwer: Dodrecht, 1992.
- 6.Sheldon, R. A.; Kochi, J. K. Metal-Catalyzed Oxidation of Organic Compounds; Academic Press: New York, 1981.
- 7.Bryndza HE, Tam W. Monomeric Metal Hydroxides, Alkoxides, and Amides of the Late Transition Metals: Synthesis, Reactions, and Thermochemistry. Chem Rev. 1988;88:1163–1188. [Google Scholar]
- 8.Bertini, I.; Gray, H. B.; Lippard, S. J.; Valentine, J. S. Bioinorganic Chemistry; University Science Books: Mill Valley, CA, 1994.
- 9.Holm RH, Kennepohl P, Solomon EI. Structural and Functional Aspects of Metal Sites In Biology. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 10.Klinman JP. Mechanisms Whereby Mononuclear Copper Proteins Functionalize Organic Substrates. Chem Rev. 1996;96:2541–2561. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- 11.Roundhill DM. Transition Metal and Enzyme Catalyzed Reactions Involving Reactions with Ammonia and Amines. Chem Rev. 1992;92:1–27. [Google Scholar]
- 12.Ishiyama T, Hartwig JF. A Heck-Type Reaction Involving Carbon–Heteroatom Double Bonds. Rhodium(I) Catalyzed Coupling of Aryl Halides with N-Pyrazyl Aldimines. J Am Chem Soc. 2000;122:12043–12044. [Google Scholar]
- 13.Bergman RG. Synthesis and Reactions of Monomeric Late Transition Metal Alkoxo, Aryloxo and Hydroxo Complexes. Polyhedron. 1995;14:3227. [Google Scholar]
- 14.Samuelsson B, Dahlén S-E, Lindgren JA, Rouzer CA, Serhan CN. Leukotriense and Lipoxins—Structures, Biosynthesis, and Biological Effects . Science. 1987;237:1171–1176. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 15.Nelson MJ, Seitz SP, Cowling RA. Enzyme-Bound Pentadienyl and Peroxyl Radicals in Purple Lipoxygenase. Biochemistry. 1990;29:6897–6903. doi: 10.1021/bi00481a020. [DOI] [PubMed] [Google Scholar]
- 16.Ogo S, Wada S, Watanabe Y, Iwase M, Wada A, Garata M, Jitsukawa K, Masuda H, Einaga H. Synthesis, Structure and Spectroscopic Properties of [Fe(III)(tnpa)(OH)(PhCOO)]ClO4: A Model Complex for an Active Form of Soybean Lipoxygenase-1. Angew Chem, Int Ed. 1998;37:2102–2104. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2102::AID-ANIE2102>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 17.Jonas RT, Stack TDP. C–H Bond Activation by a Ferric Methoxide Complex: A Model for the Rate-Determining Step in the Mechanism of Lipoxygenase. J Am Chem Soc. 1997;119:8566–8567. doi: 10.1021/ja016451g. [DOI] [PubMed] [Google Scholar]
- 18.Tsuji J. Organopalladium Chemistry in the ‘60s and ‘70s. New J Chem. 2000;24:127–135. [Google Scholar]
- 19.Müller TE, Beller M. Metal-Initiated Amination of Alkenes and Alkynes. Chem Rev. 1998;98:675–703. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]
- 20.Bäckvall JE, Akermark B, Ljunggren SO. Stereochemistry and Mechanism for the Palladium(II)-Catalyzed Oxidation of Ethene in Water (the Wacker Process) J Am Chem Soc. 1979;101:2411–2416. [Google Scholar]
- 21.Senn HM, Blochl PE, Togni A. Toward an Alkene Hydroamination Catalyst: Static and Dynamic Ab Initio DFT Studies. J Am Chem Soc. 2000;122:4098–4107. [Google Scholar]
- 22.Alonso DA, Brandt P, Nordin SJM, Andersson PG. Ru-(arene)(amino alcohol)-Catalyzed Transfer Hydrogenation of Ketones: Mechanism and Origin of Enantioselectivity. J Am Chem Soc. 1999;121:9580–9588. [Google Scholar]
- 23.Yamakawa M, Ito H, Noyori R. The Metal–Ligand Bifunctional Catalysis: A Theoretical Study on the Ruthenium(II)-Catalyzed Hydrogen Transfer between Alcohols and Carbonyl Compounds. J Am Chem Soc. 2000;122:1466–1478. [Google Scholar]
- 24.Noyori R, Hashiguchi S. Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Acc Chem Res. 1997;30:97–102. [Google Scholar]
- 25.Lowry, T. H.; Richardson, K. S. Mechanism and Theory in Organic Chemistry, 3rd ed.; Harper & Row: New York, 1987.
- 26.Hauger BE, Huffman JC, Caulton KG. Alkoxide Attack on Coordinated Olefin can be Reversible . Organometallics. 1996;15:1856–1864. [Google Scholar]
- 27.Li JJ, Li W, James AJ, Holbert T, Sharp T, Sharp PR. Phosphine-Based Dinuclear Platinum(II) Diamido, Amido-Hydroxo, Oxo-Amido, Oxo-Imido, Diimido, and Dihydrazido Complexes. Inorg Chem. 1999;38:1563–1572. [Google Scholar]
- 28.Bryndza HE. Mechanism of Olefin Insertion into Metal–Oxygen Bonds. Reaction of [(C6H5)2PCH2CH2P(C6H5)2]Pt(CH3)(OCH3) with Tetrafluoroethylene. Organometallics. 1985;5:406–408. [Google Scholar]
- 29.Holland PL, Andersen RA, Bergman RG, Huang J, Nolan SP. Monomeric Cyclopentadienylnickel Methoxo and Amido Complexes: Synthesis, Characterization, Reactivity, and Use for Exploring the Relationship between H–X and M–X Bond Energies. J Am Chem Soc. 1997;119:12800–12814. [Google Scholar]
- 30.Bryndza HE, Fong LK, Paciello RA, Tam W, Bercaw JE. Relative Metal–Hydrogen, –Oxygen, –Nitrogen, and –Carbon Bond Strengths for Organoruthenium and Organoplatinum Compounds; Equilibrium Studies of Cp*(PMe3)2RuX and (DPPE)MePtX Systems. J Am Chem Soc. 1987;109:1444–1456. [Google Scholar]
- 31.Caulton KG. The Influence of π-Stabilized Unsaturation and Filled/Filled Repulsions in Transition Metal Chemistry. New J Chem. 1994;18:25–41. [Google Scholar]
- 32.Glueck DS, Bergman RG. Mechanism of Ligand Substitution in an Iridium Amide Complex . Organometallics. 1991;10:1479–1486. [Google Scholar]
- 33.While rhenium is not strictly a “late” metal, we have included rhenium(I) alkoxides in this review because their chemistry closely mirrors that of group 8, 9, and 10 alkoxides.
- 34.Simpson RD, Bergman RG. A Dramatic Difference in the Reactivities of Alkoxidorhenium and Aryloxidorhenium Complexes in Insertion Reactions. Angew Chem, Int Ed Engl. 1992;31:220–223. [Google Scholar]
- 35.Newman LJ, Bergman RG. Synthesis, Insertion and Reductive Elimination Reactions of a Hydridoalkoxyirdium Complex. J Am Chem Soc. 1985;107:5314–5315. [Google Scholar]
- 36.Glueck DS, Winslow LJN, Bergman RG. Iridium Alkoxide and Amide Hydride Complexes—Synthesis, Reactivity, and the Mechanism of O–H and N–H Reductive Elimination. Organometallics. 1991;10:1462–1479. [Google Scholar]
- 37.Simpson RD, Bergman RG. Electrophilic Cleavage of Rhenium Oxygen Bonds by Bronsted and Lewis-Acids. Organometallics. 1992;11:3980–3993. [Google Scholar]
- 38.Meyer TY, Woerpel KA, Novak BM, Bergman RG. Silica as a Ligand—Reactivity of the Iridium–Oxygen Bond of Cp*Ir-[Silica](Ph)(PMe3) J Am Chem Soc. 1994;116:10290–10291. [Google Scholar]
- 39.Kaplan AW, Bergman RG. Nitrous Oxide Mediated Synthesis of Monomeric Hydroxoruthenium Complexes. Reactivity of (DMPE)2Ru(H)(OH) and the Synthesis of a Silica-Bound Ruthenium Complex. Organometallics. 1998;17:5072–5085. [Google Scholar]
- 40.Kegley SE, Schaverien CJ, Freudenberger JH, Bergman RG. Rhodium Alkoxide Complexes: Formation of an Unusually Strong Intermolecular Hydrogen Bond in (PMe3)3RhOTol(HOTol) J Am Chem Soc. 1987;109:6563–6565. [Google Scholar]
- 41.Hartwig JF, Andersen RA, Bergman RG. Synthesis of (PMe3)4Ru(Me)(OC(CH2)Me) as an Equilibrium Mixture of Oxygen-Bound and Carbon-Bound Transition-Metal Enolates—Thermal Elimination of Methane to Form an η4-Oxatrimethylenemethane Complex. J Am Chem Soc. 1990;112:5670–5671. [Google Scholar]
- 42.Hartwig JF, Andersen RA, Bergman RG. Synthesis and Chemistry of Ruthenium Hydrido Aryloxides and Arylamides—an Investigation of Structure, N–H and O–H Elimination Processes, Proton-Catalyzed Exchange-Reactions, and Relative Ru-X Bond Strengths . Organometallics. 1991;10:1875–1887. [Google Scholar]
- 43.Burn MJ, Fickes MG, Hollander FJ, Bergman RG. Reactions of (PMe3)4Ru(C2H4) and (DMPE)2Ru(C2H4) with Weak Proton-Donating Electrophiles HX (X = OAr, SAr, NHPh, PHPh)—Synthesis of Complexes with Metal Heteroatom Single Bonds. Organometallics. 1995;14:137–150. [Google Scholar]
- 44.Burn MJ, Fickes MG, Hartwig JF, Hollander FJ, Bergman RG. Synthesis of Monomeric Ruthenium Hydroxide Complexes (PMe3)4Ru(R)(OH) (R = H, Me) and the Isolation of a Unique Example of a Dimeric Ruthenium Hydroxide-Water Complex. J Am Chem Soc. 1993;115:5875–5876. [Google Scholar]
- 45.Woerpel KA, Bergman RG. Synthesis and Reactivity of an Iridium Hydroxo Complex—Insertion of Ethylene into a Metal–Oxygen Bond. J Am Chem Soc. 1993;115:7888–7889. [Google Scholar]
- 46.Kaplan AW, Ritter JCM, Bergman RG. Synthesis and Structural Characterization of Late Transition Metal Parent Amido (LnM–NH2) Complexes: An Acid/Conjugate Base Metathesis Approach. J Am Chem Soc. 1998;120:6828–6829. [Google Scholar]
- 47.Simpson RD, Bergman RG. Comparison of the Reactivity of (CO)3L2ReOR, (CO)3L2ReOAr, and (CO)3L2ReNHAr with CO2 and Other Electrophiles. Organometallics. 1992;11:6–4315. [Google Scholar]
- 48.Simpson RD, Bergman RG. Synthesis, Structure, and Exchange-Reactions of Rhenium Alkoxide and Aryloxide Complexes—Evidence for Both Proton and Hydrogen-Atom Transfer in the Exchange Transition-State. Organometallics. 1993;12:781–796. [Google Scholar]
- 49.Allen FH, Davies JE, Galloy JJ, Johnson O, Kennard O, Macrae CF, Mitchell EM, Mitcehll GF, Smith JM, Wtason DG. The Cambridge Structural Database System. J Chem Inf Comput Sci. 1991;31:187. [Google Scholar]
- 50.Osakada K, Ohshiro K, Yamamoto A. Preparation, Structure, and Formation Mechanism of cis-RuH(OAr)(PMe3)4 (Ar = C6H5, C6H4-p-Me) and cis-RuH(OC6H4-p-CN)(PMe3)4(HOC6H4-p-CN) Organometallics. 1991;10:404–410. [Google Scholar]
- 51.Kim YJ, Osakada K, Takenaka A, Yamamoto A. Alkylnickel and Alklpalladium Alkoxides Associated with Alcohols through Hydrogen Bonding. J Am Chem Soc. 1990;112:1096–1104. [Google Scholar]
- 52.Pimentel, G. C.; McClellan, A. L. The Hydrogen Bond; W. H. Freeman: San Francisco, 1960.
- 53.Spencer JN, Andrefsky JC, Grushow A, Naghadi J, Patti LM, Trader JF. Hydrogen Bond Equilibria of Phenol-Pyridine in Cyclohexane, CCl4, and Benzene Solvents. J Phys Chem. 1987;91:1673–1674. [Google Scholar]
- 54.Meot-Ner M, Sieck LW. The Ionic Hydrogen Bond and Ion Solvation. 5. O–H–O Bonds. Gas-Phase Solvation and Clustering of Alkoxide and Carboxylate Anions. J Am Chem Soc. 1986;108:7525–7529. doi: 10.1021/ja00284a014. [DOI] [PubMed] [Google Scholar]
- 55.Suzuki A. Recent Advances in the Cross-Coupling Reactions of Organoboron Derivatives with Organic Electrophiles. J Organomet Chem. 1999;576:147–168. [Google Scholar]
- 56.Fulton JR, Bouwkamp MW, Bergman RG. Reactivity of a Parent Amidoruthenium Complex: A Transition Metal Amide of Exceptionally High Basicity. J Am Chem Soc. 2000;122:8799–8800. doi: 10.1021/ja001622n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drago RS, Dadmun AP. E- and C-Based Dual Parameter Substituent Constant Correlations. J Am Chem Soc. 1994;116:1792–1799. [Google Scholar]
- 58.Drago RS. Advantages of E and C Based Interpretations of Coordination Chemistry. Polyhedron. 1994;13:2017–2024. [Google Scholar]
- 59.Holland PL, Andersen RA, Bergman RG. An E–C Alternative to p-π/d-π Repulsion in Bonds Between Late Metals and Anionic N or O Ligands. Comments Inorg Chem. 1999;21:115–129. [Google Scholar]
- 60.Ritter JCM, Bergman RG. The Mechanism of Addition of an Ir–OH Bond to Ethylene. Catalytic Tandem Activation by Two [Cp*(Ph)Ir(PMe3)]+ Complex Fragments. J Am Chem Soc. 1997;119:2580–2581. [Google Scholar]
- 61.Tellers DM, Ritter JCM, Bergman RG. Synthesis of Iridium-(III) Carboxamides via the Bimetallic Reaction between Cp*(PMe3)IrPh(OH) and [Cp*(PMe3)Ir(Ph)NCR]+ Inorg Chem. 1999;38:4810–4818. doi: 10.1021/ic9907157. [DOI] [PubMed] [Google Scholar]