

Abstract
In the CNS, fine processes of astrocytes often wrap around dendrites, axons and synapses, which provides an interface where neurons and astrocytes might interact. We have reported previously that selective Ca2+ elevation in astrocytes, by photolysis of caged Ca2+ by o-nitrophenyl-EGTA (NP-EGTA), causes a kainite receptor-dependent increase in the frequency of spontaneous inhibitory post-synaptic potentials (sIPSCs) in neighboring interneurons in hippocampal slices. However, tetrodotoxin (TTX), which blocks action potentials, reduces the frequency of miniature IPSCs (mIPSCs) in interneurons during Ca2+ uncaging by an unknown presynaptic mechanism. In this study we investigate the mechanism underlying the presynaptic inhibition. We show that Ca2+ uncaging in astrocytes is accompanied by a decrease in the amplitude of evoked IPSCs (eIPSCs) in neighboring interneurons. The decreases in eIPSC amplitude and mIPSC frequency are prevented by CPPG, a group II/III metabotropic glutamate receptor (mGluR) antagonist, but not by the AMPA/kainate and NMDA receptor antagonists CNQX/CPP. Application of either the group II mGluR agonist DCG IV or the group III mGluR agonist L-AP4 decreased the amplitude of eIPSCs by a presynaptic mechanism, and both effects are blocked by CPPG. Thus, activation of mGluRs mediates the effects of Ca2+ uncaging on mIPSCs and eIPSCs. Our results indicate that Ca2+-dependent release of glutamate from astrocytes can activate distinct classes of glutamate receptors and differentially modulate inhibitory synaptic transmission in hippocampal interneurons.
Keywords: Astrocytes, metabotropic glutamate receptor, calcium uncaging, interneurons, presynaptic, synaptic transmission
INTRODUCTION
Astrocytes are the most abundant cell type in the CNS. They make extensive close contact with neurons as thin sheets of astrocytic processes intermingle with dendrites, axons and synapses (Ventura and Harris, 1999). This anatomic proximity forms a basis for bidirectional communication between astrocytes and neurons. Although astrocytes are unable to fire action potentials, they display a form of excitability by changing their intracellular Ca2+ concentrations ([Ca2+]i) (Cornell-Bell et al., 1990). Neurotransmitter release can excite astrocytes by increasing their [Ca2+]i (Porter and McCarthy, 1997; Bezzi et al., 1998; Araque et al., 2001; Pasti et al., 2001) Ca2+-dependent and -independent release of neuroactive substances (gliotransmitters) from glial cells, in turn, modulates excitability (Hassinger et al., 1995; Newman and Zahs, 1998; Ransom, 2000; Newman, 2003), [Ca2+]i (Nedergaard, 1994; Parpura et al., 1994; Bezzi et al., 1998; Parri et al., 2001), differentiation (Blondel et al., 2000), synaptic transmission (Pfrieger and Barres, 1997; Araque et al., 1998b; Araque et al., 1998a; Kang et al., 1998; Oliet et al., 2001; Beattie et al., 2002; Zhang et al., 2003; Fiacco and McCarthy, 2004), synapse number (Ullian et al., 2001) and synaptic plasticity (Kang et al., 1998; Yang et al., 2003) of neighboring neurons. Gliotransmitters might also be released at astrocytic endfeet to affect vascular tone and, therefore, blood supply to neurons (Zonta et al., 2003). This feedback loop provides a powerful mechanism for coordinating neuronal activities with energy demand.
To better understand the physiological consequences of astrocyte Ca2+ signaling in situ, we selectively stimulated astrocytes while monitoring stimulation-evoked neuronal responses in hippocampal slices. We have shown previously that selective elevation of [Ca2+]i in astrocytes, induced by Ca2+ uncaging, increases the frequency of action potential-driven sIPSCs recorded from nearby interneurons, an effect that is blocked by kainite-receptor antagonists (Liu et al., 2004). When TTX is included to block action potentials, however, Ca2+ uncaging induces an unexpected decrease in the frequency of mIPSCs, which is not affected by AMPA/kainite- and NMDA-receptor antagonists. The amplitude distribution of mIPSCs remains unchanged during the uncaging, which is consistent with a presynaptic inhibition of GABA release. The present study was undertaken to determine the mechanism underlying the Ca2+-uncaging-induced presynaptic inhibition.
It has been shown that group II/III mGluRs are located at presynaptic terminals of many synapses (Shigemoto et al., 1997), and that agonists of group II/III mGluRs modulate synaptic transmission by a presynaptic mechanism (Gereau and Conn, 1995; Semyanov and Kullmann, 2000). In particular, a group III mGluR agonist induced selective inhibition of eIPSCs in interneurons in stratum radiatum in CA1 region of hippocampus, which is mimicked by high-frequency stimulation of neuronal pathways to trigger glutamate spillover (Semyanov and Kullmann, 2000). If glutamate released from astrocytes activates kainate receptors, it might also activate presynaptic mGluRs and cause presynaptic inhibition of transmitter release. We test this possibility by examining whether a potent, broad-spectrum group II/III mGluR antagonist blocks the Ca2+-uncaging-induced presynaptic inhibition in interneurons.
OBJECTIVE
Using photolysis to uncage calcium in astrocytes associated with interneurons in stratum radiatium of rat hippocampus, together with specific inhibitors of glutamate receptors, we determine if presynaptic inhibition of inhibitory neurons results from activation of group II/III mGluR receptors in response to glutamate released from synaptic astrocytes.
MATERIALS AND METHODS
Preparation of hippocampal slices
Experimental procedures used in this study, including slice preparation, Ca2+ imaging and Ca2+ uncaging, are similar to those described previously (Liu et al., 2004). Briefly, hippocampal slices were prepared from 11–15-day-old Sprague Dawley rats. The whole brains were dissected out after decapitation and immersed in ice-cold solution which contained (mM): 240 sucrose, 2.5 KCl, 0.5 CaCl2, 10 MgSO4, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, saturated with 95% O2:5%CO2. Either transverse or coronal hippocampal slices (300 μm) were cut using a vibratome and transferred to an oxygenated artificial cerebrospinal fluid (ACSF) that contained (mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4 and 10 glucose.
Ca2+ imaging and photolysis of caged compound
After recovering for 30 minutes at room temperature, slices were incubated at 30–32°C for 60–80 minutes in ACSF containing acetoxymethyl (AM) ester of fluo-4 (5 μM) and 0.02–0.04% pluronic F-127, in either the presence or the absence of NP-EGTA AM (10 μM).
Imaging was performed using an Olympus Fluoview (Olympus) laser-scanning photomultiplier tube connected to an Olympus BX60WI upright microscope. For imaging Ca2+ changes in astrocytes, the 488 line of the argon laser was used for excitation and emission was low-pass filtered at 510 nm. Fluorescence intensities were analyzed using Fluoview software. Changes in [Ca2+]i are given as ΔF/F0, where F0 is the baseline fluorescence level, ΔF is determined by the relative change from the base level.
A photolysis system (Prairie Tech) was used to uncage NP-EGTA and release Ca2+. An ultraviolet (UV) laser beam (337 nm) from a nitrogen-pulsed laser (VSL-337ND; Laser Science) was focused through the 40× water-immersion objective to an optical spot of 15 μm diameter. A red spot from a weak 635 nm diode laser, which is visible under infrared differential-interference contrast microscopy (IR-DIC) and under laser-scanning fluorescence microscopy when enabled, was coupled with the UV beam to guide the positioning of the uncaging. To trigger Ca2+ elevation in astrocytes, a train of 12 UV-laser pulses was delivered at 10 second intervals to the targeted astrocytes in stratum radiatum, which often produced reliable uncaging with each pulse, as reflected by changes in Ca2+ fluorescence.
Whole-cell patch-clamp recording
Whole-cell recordings were made from interneurons in stratum radiatum of CA1. Interneurons, pyramidal neurons and astrocytes were identified by their morphology and location under IR-DIC, and by their electrophysiological properties, as described previously (Kang et al., 1998). Neurons with pyramidal-cell morphology in stratum radiatum were excluded for recording of interneurons in case they are misplaced pyramidal cells. The pipette solution used for recording of sIPSCs and eIPSCs contained (mM): 140 CsCl, 2 MgCl2, 10 Hepes, 2 EGTA, 5 sodium phosphocreatine, 4 Mg-ATP, 0.3 GTP and 5 lidocaine N-ethylchloride (QX314). QX314 was omitted when recording mIPSCs. In some recordings, 140 mM CsCl was replace by 10 mM CsCl and 130 mM CsCH3SO3. For intracellular loading of NP-EGTA into pyramidal neurons through whole-cell recording, the pipette solution contained (mM): 10 KCl, 130 KCH3SO3, 2 MgCl2, Hepes 20, 5 sodium phosphocreatine, 4 Mg-ATP, 0.3 GTP, 0.1 Fluo-4 potassium salt and 2 NP-EGTA (preloaded with 1.2 Ca2+). For perforated whole-cell recording, the pipette solution contained (mM): 10 KCl, 130 KCH3SO3, 2 MgCl2, Hepes 20 and 400 μg ml−1 amphotericin B. The pipettes had a resistance of 3–5 MΩ. For recording eIPSCs, a bipolar tungsten stimulation electrode (WPI) was placed in stratum radiatum of the CA1 region, near the recorded interneurons (100–200 μm). Single or paired (separated by 50 ms) pulses of 60 μsecond were delivered at 0.2 Hz with an intensity of 30–100 μA. A Mater-8 stimulator (A.M.P.I) was used to synchronize the electrical stimulation with the uncaging. Membrane currents were filtered at 1–2 KHz, digitized at 5–10 KHz using Axopatch 700A amplifier, Clampex8 software and DigiData 1332A interface (Axon Instruments Inc.). Series resistance (<20 MΩ) was carefully monitored throughout the experiments and experiments were terminated if the series resistance changed >15%. All experiments were done at room temperature (21–23°C).
Data analysis
All data are presented as the mean ± SEM. Miniature IPSCs were analyzed with MiniAnalysis (Synaptosoft). The cumulative probability (Van der Kloot, 1991) of amplitude and interevent intervals was compared using the Kolmogorov-Smirnov test (K-S test). Two cumulative sets of data were considered to be significantly different at P < 0.01. Evoked IPSCs were analyzed with Clampfit 8.0 (Axon Instruments Inc.). For multiple group comparisons, statistical differences were calculated by one-way ANOVA followed by Dunnett's post hoc test. For comparison of means from the same group of cells, Student's paired t-test was used. Results were considered to be significant at P < 0.05.
Chemicals
L(+)-2-amino-4-phosphonobutyric acid (L-AP4), (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG IV), (RS)-α-cyclopropyl-4-phosphonophenylglycine (CPPG), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP) were purchased from Tocris Cookson Inc. Acetoxymethyl (AM) esters and potassium salts of NP-EGTA and fluo-4, BAPTA AM (1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate) and pluronic F-127 were from Molecular Probes Inc. Most other chemicals were from Sigma.
RESULTS
Modulation of eIPSCs by Ca2+ elevation in astrocytes
Hippocampal slices were bulk loaded with calcium-fluorescence-indicator fluo-4 AM and caged Ca2+, NP-EGTA AM. Uncaging NP-EGTA was used to control [Ca2+]i in astrocytes. NP-EGTA releases Ca2+ upon uncaging by lowering its affinity with Ca2+ (Ellis-Davies and Kaplan, 1994). We have showed previously that Ca2+ uncaging in astrocytes induces a decrease in the frequency of mIPSCs in neighboring interneurons and that blocking AMPA/kainate and NMDA glutamate receptors pharmacologically has no effect on this presynaptic inhibition. Here, we test whether eIPSCs are also affected by Ca2+ elevation in astrocytes. Whole-cell recordings were made from interneurons in stratum radiatum in CA1 region. The interneurons were voltage-clamped at −60 mV. AMPA/kainate and NMDA receptor antagonists CNQX (50 μM) and CPP (5 μM) were included to block evoked excitatory postsynaptic currents (EPSCs). Monosynaptic eIPSCs were elicited at 0.2 Hz by focal electrical stimulation. The fine-tipped stimulating electrode was placed within 100 μm of one of the processes of the recorded interneurons. The cell bodies of the astrocytes chosen for uncaging NP-EGTA were within 30 μm of proximal dendrites, which were within 100 μm of the soma of the recorded interneurons (Liu et al., 2004).
After a stable, baseline, whole-cell recording was achieved, an astrocyte near the interneuron process (<30 μm) was targeted and stimulated with a train of 12 UV pulses (0.1 Hz, 2 minutes), the resultant fluorescence change was monitored and eIPSCs were recorded simultaneously. Uncaging NP-EGTA produced a stepwise increase in [Ca2+]i in the astrocyte (Fig. 1A). The astrocyte Ca2+ elevation (mean peak ΔF/F0 = 204 ± 18%, n = 9) was accompanied by a significant reduction in the amplitude of the eIPSCs in 53% (9 of 17) of interneurons (responder, Fig. 1B,C). The mean amplitude of eIPSCs during Ca2+ uncaging was 73 ± 5% of baseline control (n = 9, P < 0.001, paired t-test), and recovered after the uncaging to 97 ± 3% of baseline control (P > 0.3). During the depression the coefficient of variation (CV; SD/mean) of the eIPSCs increased from 0.32 ± 0.03 to 0.38 ± 0.03 (n = 9, P < 0.01), which is consistent with the depression of eIPSCs being mediated by a decrease in the probability of GABA release. The time-course of the decrease in the amplitude of eIPSCs in interneurons followed that of Ca2+ elevation in astrocytes (Fig. 1B). Ca2+ uncaging in astrocytes (peak ΔF/F0 = 194 ± 23%, n = 8) had no significant effect on the amplitude of eIPSCs in 47% (8 of 17) of interneurons (non-responder). The average amplitude of eIPSCs was 96 ± 3% of baseline level (n = 8, P > 0.3). There was no significant changes in CV (control: 0.30 ± 0.03; uncaging: 0.31 ± 0.03; n = 8). The responder and non-responder were classified by testing whether the amplitude of eIPSCs was significantly different during Ca2+ uncaging from the pre-uncaging level (paired t-test). When experiments from both groups were averaged, the depression of eIPSCs during Ca2+ uncaging was still significantly different from the pre-uncaging level (83 ± 4% of control, n = 17, P < 0.01). The peak ΔF/F0 levels between the two groups are not significantly different (P > 0.5). Thus, the difference between responder and non-responder can not be explained by different Ca2+ levels in astrocytes produced by Ca2+ uncaging.
Fig. 1.
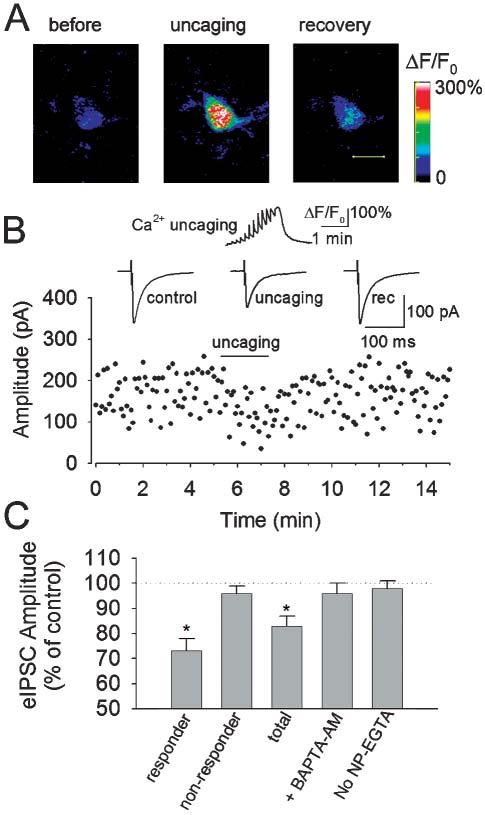
Depression of eIPSCs in interneurons by Ca2+ uncaging in astrocytes. (A) Images from an astrocyte before, during and after stimulation with a train of twelve UV laser pulses (0.1 Hz) to uncage NP-EGTA. Scale bar, 10 μm. (B) Upper: Ca2+ uncaging produces a stepwise increase in ΔF/F0 of the astrocyte in (A) and a reversible decrease in the mean amplitude of eIPSCs (averaged from 24–36 traces) in an interneuron (middle). The amplitudes of eIPSCs over the course of the experiment (lower). (C) Normalized changes in the amplitude of eIPSCs by uncaging. Responder shows the average of nine cells that responded to uncaging by decreasing the amplitude of eIPSCs. Non-Responder shows the average of eight cells that did not respond to uncaging. Total shows the average of all 17 cells. Preloading the slices with BAPTA-AM (10 μM), a calcium chelator, prevents the depression of eIPSCs by uncaging (n = 8). No NP-EGTA indicates slices loaded with fluo-4 alone (n = 9). *P < 0.01 compared with control, BAPTA AM and no NP-EGTA groups by ANOVA with Dunnett's test.
The anatomical proximity of the stimulated astrocytes with the dendrites of the recorded interneurons is required for astrocyte-mediated depression of sIPSCs. In astrocytes that are 60–100 μm from the dendrites of recorded interneurons, Ca2+ uncaging elevated [Ca2+]i in astrocytes (peak ΔF/F0 = 199 ± 24%, n = 7) but did not significantly alter the amplitude of eIPSCs (96 ± 5% of baseline, n = 7, P > 0.4, paired t-test). Therefore, only astrocytes within 30 μm of the proximal dendrites of recorded interneurons were chosen for the following experiments.
UV flashings to astrocytes did not produce appreciable change in Ca2+ fluorescence when hippocampal slices from the same rat brains are loaded with fluo-4 alone (ΔF/F0 = 3 ± 2%; n = 9). This indicates that the astrocyte [Ca2+]i elevation during the uncaging is not due to either photo-damage or unspecific effects produced by the UV laser. Under this condition, UV flashing to astrocytes had no significant effect on the amplitude of eIPSCs recorded from interneurons in these slices (98 ± 3% of control, n = 9, P > 0.3, Fig. 1C). Pre-treatment of the slices with BAPTA-AM (10 μM for 20–30 minutes) abolished the uncaging-induced Ca2+ increase in astrocytes (peak ΔF/F0 = 9±3%, n = 8) and the associated depression of eIPSCs in interneurons (96 ± 4% of preuncaging level, n = 8, P > 0.5). These results indicate that the depression of eIPSCs in interneurons is likely to be caused by Ca2+ elevation in astrocytes.
In agreement with previous studies (Porter and McCarthy, 1996), we found that high frequency stimulation (50–100 Hz, 1 second) caused robust increases in [Ca2+]i in 3–20 astrocytes, but either single or paired-pulse electrical stimulation at low frequency (0.1 Hz) used in this study did not (data not shown). This observation indicates that [Ca2+]i in astrocytes is not affected by the electrical stimulation used to elicit eIPSCs.
The fluo-4 fluorescence was present only in astrocytes, not in neurons, from slices prepared from 11–15-day-old rats. We have showed previously that astrocytes are also loaded preferentially with NP-EGTA in NP-EGTA AM bulk-loaded slices, the neuronal loading of NP-EGTA is insignificant (Liu et al., 2004). First, we made perforated whole-cell recording from pyramidal neurons in the coloaded slices. Pyramidal neurons in CA1 region were voltage-clamped at −60 mV. UV flashing to pyramidal neurons failed to induce Ca2+-activated K+ currents in NP-EGTA AM bulk-loaded slices, but uncaging NP-EGTA loaded into pyramidal neurons through conventional whole-cell recording induced such currents from pyramidal neurons in the same slices (120 ± 19 pA, n = 7, Fig. 2). Second, when fura-2 AM rather than fluo-4 AM was used for coloading with NP-EGTA AM in some slices, UV uncaging at astrocytes caused Ca2+ elevation, but uncaging at either pyramidal neurons or interneurons did not. Unlike fluo-4, fura-2 could still be loaded into some pyramidal neurons and interneurons in the hippocampal slices through bulk loading. These two lines of evidence indicate that NP-EGTA is loaded selectively into astrocytes but not neurons (Liu et al., 2004). The UV-laser beam was focused through a 40× water-immersion object onto astrocytes with a diameter of ∼15 μm. The preferential loading of NP-EGTA into astrocytes and targeted UV stimulation ensure that only astrocytes, not neurons, are stimulated during uncaging NP-EGTA.
Fig. 2.
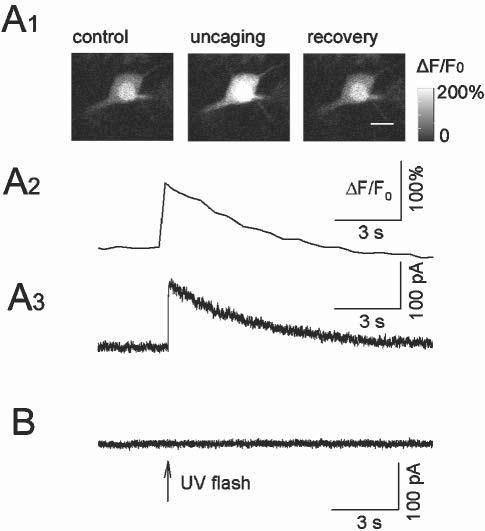
Neuronal loading of NP-EGTA in NP-EGTA-AM-loaded slices is insignificant. (A1) A pyramidal neuron was loaded with potassium salts of NP-EGTA (2 mM, pre-loaded with 2 mM Ca2+) and fluo-4 (0.1 mM) through conventional whole-cell recording. Uncaging NP-EGTA induced Ca2+ elevation as shown by the change in fluo-4 fluorescence. Scale bar, 15 μm. (A2) Time-course of ΔF/F0 of the experiment shown in A1. (A3) Uncaging NP-EGTA induced Ca2+-activated K+ current in the pyramidal neuron. (B) Perforated whole-cell recordings from a pyramidal neuron in NP-EGTA-AM-loaded slices. UV flashing to the neuron did not activate Ca2+-activated K+ currents.
Pharmacological activation of group II/III mGluRs on presynaptic terminals of interneurons
Next we investigated the mechanism that underlie the depression of eIPSCs in interneurons produced by elevation of [Ca2+]i in astrocytes. Ca2+-dependent release of glutamate is reported to account for several neuronal responses (Araque et al., 1998b; Araque et al., 1998a; Bezzi et al., 1998; Kang et al., 1998; Pasti et al., 2001; Fiacco and McCarthy, 2004; Liu et al., 2004). Group II/III mGluRs are expressed on presynaptic terminals and that activation of the receptors reduces transmitter release from many excitatory and inhibitory synapses (Conn and Pin, 1997; Scanziani et al., 1998; Semyanov and Kullmann, 2000). We asked whether the observed depression of eIPSCs is due to activation of presynaptic mGluRs induced by the Ca2+-dependent release of glutamate from astrocytes.
We first established whether presynaptic group II/III mGluRs are present on presynaptic terminals of the inter-neurons using a conventional pharmacological approach. Paired-pulse stimulation with an inter-pulse interval of 50 ms was used to test whether there is an effect at presynaptic or postsynaptic sites. Bath application of 0.5 μM DCG IV, a selective group II mGluR agonist (Hayashi et al., 1993), decreased the mean amplitude of the first eIPSCs to 43 ± 4% of that of baseline (n = 8, P < 0.001, paired t-test). The paired-pulse ratio (PPR), calculated as the ratio of the amplitude of the second eIPSCs to that of the first eIPSCs, increased significantly (P < 0.05). Co-application of 300 μM CPPG, the most potent group II/III mGluR antagonist described (Toms et al., 1996), with the agonist, reversed the agonist-induced inhibition (86 ± 3% of control). CPPG also reversed the DCG IV-induced change in PPR (Fig. 3A,C).
Fig. 3.

Selective group II/III mGluR agonists depress eIPSCs in interneurons. (A) The group II mGluR agonist DCG IV (0.5 μM) decreases the amplitude of eIPSCs (left), accompanied by an increase in the paired-pulse ratio (PPR; right, n=8). Both effects are reversed by CPPG (300 μM), a group II/III mGluR antagonist. *P < 0.05; paired t-test; n = 8. (B) Group III mGluR agonist L-AP4 (50 μM) also depresses eIPSCs and increases the PPR. *P < 0.05, paired t-test; n = 7. CPPG (300 μM) reversed the depression of eIPSCs and change of the PPR. Scale bars,100 pA, 100 ms.. Traces in (A) and (B) are averages of 36 consecutive traces. (C) Normalized values of change of the first IPSC amplitudes by group II/III mGluR agonists and antagonist. **P < 0.001 compared with control, CPPG with agonists, and CPPG alone; ANOVA with Dunnett's test.
Similarly, application of a selective agonist of the group III mGluRs, L-AP4 (50 μM) (Scanziani et al., 1998), reduced the mean amplitude of the first eIPSCs to 44 ± 7% of that of baseline (n = 7, P < 0.001, paired t-test). PPR was increased significantly by L-AP4 (P < 0.05). Co-application of 300 μM CPPG and L-AP4 reversed the agonist-induced inhibition (98 ± 5% of control). CPPG also reversed the L-AP4-induced change in PPR (Fig. 3B,C). Application of CPPG alone did not significantly change the mean amplitude of baseline eIPSCs (103 ± 3% of control, n = 7), which indicates that group II/III mGluRs are not activated tonically under resting conditions (Fig. 3C). Therefore, CPPG was chosen for the following experiment to test whether astrocyte-mediated depression of eIPSCs and mIPSCs is caused by activation of presynaptic mGluRs.
Activation of group II/III mGluRs mediates astrocyte-induced depression of eIPSCs
If astrocytes release glutamate, activate presynaptic group II/III mGluRs and depress GABA release, then CPPG, a potent blocker of group II/III mGluRs, should block the astrocyte-mediated depression of eIPSCs and mIPSCs. We, therefore, examined whether Ca2+ uncaging in astrocytes depresses eIPSCs and mIPSCs in the presence of CPPG. Using the protocol described previously, we uncaged NP-EGTA in astrocytes and recorded eIPSCs in interneurons at a holding potential of −60 mV. As shown in Fig. 4A, uncaging NP-EGTA in the presence of CPPG (300 μM) had no significant effect on the amplitude of eIPSCs in any of the eight interneurons tested (101 ± 3% of pre-uncaging level, P > 0.5, paired t-test). CPPG had no significant effect on the uncaging-induced Ca2+ elevation in astrocytes; the peak ΔF/F0 during uncaging (215 ± 22%, n = 8) in the presence of CPPG was not significantly different from that in the absence of CPPG (P > 0.5).
Fig. 4.
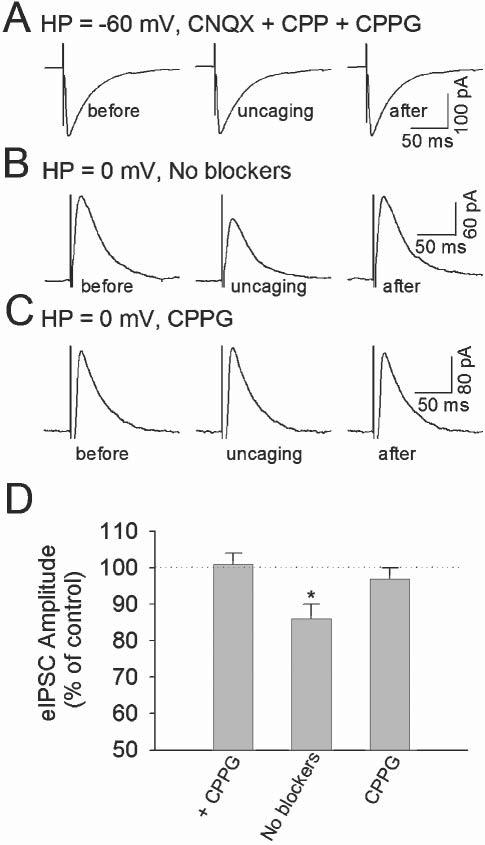
The group II/III mGluR antagonist CPPG blocks the depression of eIPSCs induced by uncaging NP-EGTA in astrocytes. (A) In the presence of AMPA/kainate and NMDA receptor antagonists (50 μM CNQX and 5 μM CPP) and Group II/III mGluR antagonist CPPG (300 μM), Ca2+ uncaging in an astrocyte had no significant effect on eIPSCs in a neighboring interneuron. The holding potential (HP) of the interneuron is −60 mV. Each sample trace of eIPSCs is an average of 24–36 traces. (B) In the absence of glutamate-receptor antagonists, Ca2+ uncaging in an astrocyte reduces the amplitude of eIPSCs in an interneuron. The interneuron was held at 0 mV, which is near the reversal potential of EPSCs. (C) In the presence of CPPG, Ca2+ uncaging has no effect on eIPSCs. (D) Normalized values of the amplitude of eIPSCs during uncaging NP-EGTA. + CPPG, CNQX, CPP and CPPG included in the bathing solution; No blockers, no glutamate-receptor antagonists in the bathing solution; CPPG, CPPG only in the bathing solution. *P < 0.05 compared with control and + CPPG group by ANOVA with Dunnett's test; n = 8–9 for each group.
To separate eIPSCs from evoked EPSCs, AMPA/kainate and NMDA receptor antagonists CNQX (50 μM) and CPP (5 μM) were included in ACSF in the above recordings. These antagonists should block the activation of all ionotropic glutamate receptors including kainate receptors. Activation of either AMPA/NMDA receptors or kainate receptors by glutamate released from astrocytes modulates synaptic transmission (Araque et al., 1998a; Kang et al., 1998; Liu et al., 2004). We tested the effects of Ca2+ uncaging in astrocytes on eIPSCs in the absence of CNQX and CPP in the ACSF. To separate eIPSCs from evoked EPSCs, we clamped interneurons at the reversal potential of EPSCs, 0 mV. The intracellular solution contained 10 mM CsCl and 130 mM CsCH3SO3 to increase the driving force for eIPSCs. Evoked IPSCs appeared outward under this condition. Uncaging NP-EGTA in astrocytes significantly reduced the amplitude of eIPSCs in five out of nine interneurons tested (76 ± 6% of control, n = 5, P < 0.01, paired t-test, Fig. 4B). The mean peak ΔF/F0 during the uncaging in astrocytes was 201 ± 25% (n = 5). The depression of eIPSCs was accompanied by an increase in CV from 0.28 ± 0.02 to 0.35 ± 0.03 (P < 0.01, paired t-test), which is consistent with a presynaptic mechanism (Bekkers and Stevens, 1990; Mitchell and Silver, 2000b). In the remaining four interneurons, Ca2+ uncaging in astrocytes (peak ΔF/F0 = 197 ± 27%, n = 4) had no significant effect on eIPSCs (99 ± 3%, P > 0.5, paired t-test) and did not affect the CV of eIPSCs (baseline 0.30 ± 0.04; uncaging 0.29 ± 0.03). Averaging experiments from the responsive and non-responsive interneurons, the depression of eIPSCs during Ca2+ uncaging is still significantly different from the pre-uncaging level (86 ± 4% of control, n = 9, P < 0.05, Fig. 4B,C). The depression of eIPSCs in the absence of CNQX and CPP is not significantly different from that obtained in the presence of CNQX and CPP (P > 0.2, ANOVA). In the absence of CNQX and CPP, the depression of eIPSCs and increases in the frequency of spontaneous IPSCs occur in the same interneurons (data not shown). The increase in the frequency of sIPSCs is caused by the activation of kainate receptors, as described in our previous study (Liu et al., 2004).
In separate experiments, CPPG (300 μM) was included in ACSF to block group II/III mGluR receptors. Uncaging NP-EGTA in astrocytes caused Ca2+ elevation (ΔF/F0 = 192 ± 20%; n = 9), but had no significant effect on eIPSCs in any of the nine interneurons tested (Fig. 4C,D) The mean amplitude of eIPSCs was 97 ± 3% of that of pre-uncaging level (P > 0.3, Fig. 4D). Thus, exclusion of CNQX and CPP from the ACSF did not unmask any significant modulation of eIPSCs caused by astrocyte activation of AMPA/kainate and NMDA receptors. The depression of eIPSCs in interneurons by astrocytes is likely to be caused by Ca2+-dependent release of glutamate and the subsequent activation of presynaptic group II/III mGluRs, which is blocked by CPPG.
Activation of mGluRs also mediates the presynaptic modulation of mIPSCs
We showed previously that Ca2+ uncaging in astrocytes causes a small, significant decrease in the frequency of mIPSCs in interneurons (Liu et al., 2004). The amplitude distribution of mIPSCs is not affected by the uncaging, which indicates that the depression of mIPSCs has a presynaptic origin. We demonstrated further that pharmacological blocking of AMPA/kainate and NMDA receptors by CNQX and CPP had no effect on the uncaging-induced depression of mIPSCs (Liu et al., 2004). We suspect that depression of mIPSCs shares a similar mechanism with depression of eIPSCs. We, therefore, tested whether CPPG blocks the effect of Ca2+ uncaging on mIPSCs.
We recorded mIPSCs from interneurons in the presence of TTX (0.5 μM) to block action potentials. CNQX (50 μM) and CPP (5 μM) were also included into ACSF to block miniature EPSCs. As reported in our previous study (Liu et al., 2004), Ca2+ uncaging in astrocytes (peak ΔF/F0 = 197 ± 18%; n = 7) produces a significant depression of mIPSCs in neighboring interneurons. The frequency of mIPSCs during the uncaging was 78 ± 3% of baseline frequency (n = 7; P < 0.05, paired t-test; Fig. 5A,C). The amplitude distribution of mIPSCs remained unchanged (Fig. 5A,D), which indicates that a presynaptic mechanism is involved.
Fig. 5.

Activation of presynaptic group II/III mGluRs mediates the Ca2+ uncaging-induced decrease in the frequency of mIPSCs. (A) Ca2+ uncaging in an astrocyte decreases the frequency of mIPSCs in a neighboring interneuron. (B) In the presence of CPPG (300 μM), an antagonist of group II/III mGluRs, uncaging has no significant effect on mIPSCs. (C) Pooled data of normalized frequency of mIPSCs during Ca2+ uncaging. The uncaging produced a significant decrease in the frequency of mIPSCs (uncaging). CPPG prevented the uncaging-induced modulation of mIPSCs (+ CPPG). CPPG perfusion alone had no effect on baseline mIPSCs (CPPG). *P < 0.01 compared with control and + CPPG group by ANOVA with Dunnett's test; n = 7–8 for each group. (D) Cumulative frequency plot of amplitude distribution of the experiment in (A), which shows that uncaging has no significant effect on the amplitude distribution of mIPSCs. This indicates that a presynaptic mechanism is involved in the uncaging-induced depression of mIPSCs.
We tested the effect of CPPG (300 μM) on the uncaging-induced mIPSCs. Bath application of CPPG had no significant effect on baseline frequency of mIPSCs (103 ± 4%, n = 8, P > 0.5; paired-t test, Fig. 5C), but it prevented the uncaging-induced depression of mIPSCs (Fig. 5B). The mean frequency of mIPSCs during uncaging was 98 ± 6% of that of the pre-uncaging level (n = 8; P > 0.5; paired t-test, Fig. 5C). The mean peak δDF/F0 during uncaging was 189 ± 15%, which is not significantly different from that in the absence of CPPG (P > 0.1, ANOVA).
CONCLUSIONS
Presynaptic inhibition of inhibitory interneurons in stratum radiatum in response to astrocytic glutamate release is blocked by inhibitors of group II/III mGluR receptors.
Glutamate that is released from astrocytes can either increase or decrease synaptic transmission in hippocampal neurons by acting on different classes of glutamate receptors.
DISCUSSION
Our results show that elevating [Ca2+]i in astrocytes by uncaging NP-EGTA decreases the amplitude of eIPSCs and the frequency of mIPSCs in neighboring interneurons. The decrease in the amplitude of eIPSCs is accompanied by an increase in the coefficient of variation but the amplitude distribution of mIPSCs remains unchanged. This pattern of changes is typical of a presynaptic effect (Bekkers and Stevens, 1990; Mitchell and Silver, 2000b). Both effects are mediated by presynaptic activation of group II/III mGluRs because they are blocked by CPPG, a potent antagonist of group II/III mGluRs (Toms et al., 1996). Our data are consistent with the idea that Ca2+-dependent glutamate release from astrocytes (Araque et al., 1998b; Araque et al., 1998a; Bezzi et al., 1998; Kang et al., 1998; Pasti et al., 2001) activates presynaptic group II/III glutamate receptors and decreases the release of GABA from presynaptic terminals of interneurons.
Ca2+ elevation in astrocytes mediates the depression of eIPSCs and mIPSCs
The depression of eIPSCs and mIPSCs appears to be caused by elevation of [Ca2+]i in astrocytes. The duration of the synaptic depression follows the time course of the [Ca2+]i elevation in astrocytes, which indicates that the two events are closely linked. The depression is evident only when UV flashing is targeted to astrocytes preloaded with NP-EGTA and fluo-4, but not with fluo-4 alone. As with fluo-4, NP-EGTA seems to load astrocytes preferentially, leaving neurons unloaded, because we detected no Ca2+-activated K currents in these slices during uncaging when perforated whole-cell recordings were used to record pyramidal neurons. Such currents are observed readily when NP-EGTA is loaded directly into the pyramidal neurons through conventional whole-cell recording (Liu et al., 2004). We have also demonstrated previously that UV flashing induces Ca2+ elevation in astrocytes, but not in neurons in NP-EGTA AM-loaded slices, as revealed by changes of fura-2 fluorescence (Liu et al., 2004). The selective loading of NP-EGTA in astrocytes and targeted UV stimulation ensure that Ca2+ elevation is limited to astrocytes. Thus, an increase in [Ca2+]i in neurons is unlikely to contribute to the uncaging-induced depression of eIPSCs and mIPSCs.
Differential neuronal modulation by astrocytes
Astrocytes can release gliotransmitters that affect many neuronal functions (Nedergaard, 1994; Kang et al., 1998; Newman and Zahs, 1998; Blondel et al., 2000; Araque et al., 2001; Bezzi et al., 2001; Ullian et al., 2001; Yang et al., 2003; Fiacco and McCarthy, 2004). The intimate anatomical association (Ventura and Harris, 1999) and diverse functional interaction between astrocytes and neuronal synapses lead to the concept of the ‘tripartite synapse’, in which astrocytes play a crucial role in synaptic transmission (Araque et al., 1999). In recent years, many details of this functional interaction has begun to unfold in intact tissues such as brain slices. Generally, it is now believed that Ca2+-dependent and -independent release of gliotransmitters is essential for astrocyte-mediated neuronal modulation.
Glutamate is a gliotransmitters that has been studied extensively (Mazzanti et al., 2001). It is well established that high-affinity glutamate transporters on astrocytes are responsible for uptake of glutamate (Anderson and Swanson, 2000). Experimental data show that astrocytes can, nevertheless, release glutamate in response to [Ca2+]i elevation (Parpura et al., 1994). Vesicular exocytosis has been proposed as the mechanism of glutamate release (Calegari et al., 1999; Araque et al., 2000; Pasti et al., 2001), but this is controversial (Nedergaard et al., 2002) and other release mechanisms have been proposed (Nedergaard et al., 2002; Ye et al., 2003).
Ca2+-dependent glutamate released from astrocytes produces diverse effects on different synapses and cell types (Araque et al., 1998b; Araque et al., 1998a; Bezzi et al., 1998; Kang et al., 1998; Pasti et al., 2001; Fiacco and McCarthy, 2004; Liu et al., 2004). How can these differences be explained and reconciled? We believe that the nature of astrocyte-mediated neuronal responses is determined initially by the specific target receptors that are activated. For example, astrocyte stimulation potentiates mIPSCs and eIPSCs in pyramidal neurons by activating presynaptic AMPA/NMDA receptors (Kang et al., 1998), but it depresses mIPSCs and eIPSCs in interneurons by activating presynaptic group II/III mGluRs, as demonstrated in this study. The expression of presynaptic mGluRs (group II/III mGluRs) is often target-specific, thus, mGluRs are present if the postsynaptic neuron is an interneuron but not if it is a pyramidal neuron (Shigemoto et al., 1996; Scanziani et al., 1998). Indeed, agonists of group II and III mGluRs depress eIPSCs in interneurons but not in pyramidal neurons (Gereau and Conn, 1995; Scanziani et al., 1998; Semyanov and Kullmann, 2000). This might explain why mGluR-mediated depression of eIPSCs and mIPSCs is observed in interneurons in this study, but not in pyramidal neurons in a previous study (Kang et al., 1998).
Glutamate released from astrocytes can modulate excitatory and inhibitory synapses in the same type of neurons through different mechanisms. Astrocyte stimulation potentiates IPSCs and EPSCs in hippocampal pyramidal neurons (Kang et al., 1998; Fiacco and McCarthy, 2004). However, the potentiation of EPSCs is mediated by activation of group I mGluRs (Fiacco and McCarthy, 2004) whereas the potentiation of IPSCs is mediated by activation of AMPA/NMDA receptors (Kang et al., 1998). We have shown that astrocyte Ca2+ elevation increases the frequency of sIPSCs by activating either axonal or somatodendritic kainate receptors in interneurons (Liu et al., 2004), whereas it decreased the amplitude of eIPSCs and the frequency of mIPSCs by activating presynaptic group II/III mGluRs. Thus, astrocyte activation can differentially modulate inhibitory synapses in the same interneurons by activating distinct classes of glutamate receptors. Astrocytes and other glial cells might also use different gliotransmitters to affect the same types of neurons. For example, stimulation of glial cells in retinal slices either increases or decreases action-potential firing in the same type of ganglion neurons (Newman and Zahs, 1998). It has been reported recently that astrocyte-mediated ATP release and its conversion into adenosine accounts for the neuronal inhibition (Newman, 2003). The mechanism of glial-induced increases in action-potential firing is not clear, but it is likely to be caused by activation of other gliotransmitters. Thus, gliotransmitters released from glial cells can cause bidirectional modulation of neuronal activity and synaptic transmission depending on the target receptors that are activated.
The spatial distribution of glutamate receptors is crucial for their activation by glutamate released from astrocytes. High-affinity glutamate transporters on astrocyte membranes limit glutamate diffusion (Anderson and Swanson, 2000). Anatomical segregation between glutamate release sites and glutamate transporters must exist so that glutamate that is released can reach target receptors. The anatomical proximity between astrocyte processes and neuronal glutamate receptors is required for astrocyte-mediated neuronal modulation. It is conceivable that the astrocyte processes contact different target glutamate receptors at different synapses. This might explain why glutamate released from astrocytes produces such diverse effects on different synapses and cell types. Electron microscopy indicates that astrocyte processes surround the axon-spine interface but are not in direct contact with the postsynaptic sites (Ventura and Harris, 1999). This indicates that gliotransmitters released from astrocyte processes are likely to activate extra-synaptic receptors rather than postsynaptic receptors (Araque et al., 1998a). Thus, astrocytes might modulate rather than participate in synaptic transmission.
The sensitivity of target glutamate receptors also shapes the responses to stimulating astrocytes. Different glutamate receptors have different affinity with glutamate. High affinity glutamate receptors are more likely to be activated by glutamate. Generally mGluRs have a high affinity for glutamate, for example, the affinity for glutamate for group II mGluRs is 4–12 μM (Pin and Duvoisin, 1995). Kainate receptors can also be activated by low micromolar concentration of glutamate (Rodriguez-Moreno et al., 2000). The close apposition of presynaptic terminals with astrocytic processes (Ventura and Harris, 1999) and the high affinity for glutamate may allow glutamate to reach a sufficient concentration to activate mGluRs and kainate receptors.
Functional implication
By selectively stimulating astrocytes while monitoring neuronal responses, we have found that Ca2+-dependent release of glutamate from astrocytes can activete distinct classes of glutamate receptors on interneurons. Activation of kainate receptors caused an increase in the frequency of sIPSCs (Liu et al., 2004), while activation of group II/III mGluRs led to a decrease in the amplitude of eIPSCs and the frequency of mIPSCs. Astrocyte-mediated modulation of synaptic transmissions through activation of mGluRs and AMPA/NMDA receptors has also been reported in hippocampal slices and hippocampal cell culture (Araque et al., 1998b; Araque et al., 1998a; Kang et al., 1998; Fiacco and McCarthy, 2004). These findings suggest that astrocyte activation could bidirectionally modulate synaptic transmission through activation of different glutamate receptors. The ‘net’ effect of stimulating astrocytes on hippocampal circuit function is hard to predict, nevertheless, astrocytes provide a non-synaptic source of glutamate that could fine-tune the strength of excitatory and inhibitory synapses. This is particularly meaningful for inhibitory synapses. Although mGluRs and kainate receptors are expressed on presynaptic terminals of inhibitory synapses, how glutamate released from excitatory synapses reaches the glutamate receptors on the inhibitory terminals is unclear. Recent experimental evidence showed that under the condition of high frequency of synaptic activity, inhibition of glutamate uptake, or close apposition of excitatory and inhibitory terminals in certain brain structure, glutamate could ‘spillover’ and activate mGluRs on inhibitory terminal to modulate inhibitory synaptic transmission (Mitchell and Silver, 2000a; Semyanov and Kullmann, 2000). The physiological significance for this heterosynaptic inhibition is not clear. Synapses are often wrapped by astrocyte processes (Ventura and Harris, 1999), Ca2+-dependent release of glutamate from astrocytes could readily access glutamate receptors on inhibitory terminals. As such, astrocyte-derived glutamate should represent a common mechanism by which inhibitory synaptic transmission can be regulated.
ACKNOWLEDGEMENT
This work was supported by National Institutes of Health Grants NS38073 and NS41031 to M.N.
REFERENCES
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG. Dynamic signaling between astrocytes and neurons. Annual Review of Physiology. 2001;63:795–813. doi: 10.1146/annurev.physiol.63.1.795. [DOI] [PubMed] [Google Scholar]
- Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. Journal of Neuroscience. 1998a;18:6822–6829. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. European Journal of Neuroscience. 1998b;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends in Neurosciences. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Araque A, Li N, Doyle RT, Haydon PG. SNARE protein-dependent glutamate release from astrocytes. Journal of Neuroscience. 2000;20:666–673. doi: 10.1523/JNEUROSCI.20-02-00666.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, et al. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nature Neuroscience. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Blondel O, Collin C, McCarran WJ, Zhu S, Zamostiano R, Gozes I, et al. A glia-derived signal regulating neuronal differentiation. Journal of Neuroscience. 2000;20:8012–8020. doi: 10.1523/JNEUROSCI.20-21-08012.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calegari F, Coco S, Taverna E, Bassetti M, Verderio C, Corradi N, et al. A regulated secretory pathway in cultured hippocampal astrocytes. Journal of Biological Chemistry. 1999;274:22539–22547. doi: 10.1074/jbc.274.32.22539. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annual Review of Pharmacology and Toxicology. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- Ellis-Davies GC, Kaplan JH. Nitrophenyl-EGTA, a photolabile chelator that selectively binds Ca2+ with high affinity and releases it rapidly upon photolysis; Proceedings of the National Academy of Sciences of the U.S.A; 1994. pp. 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. Journal of Neuroscience. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gereau RW, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. Journal of Neuroscience. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassinger TD, Atkinson PB, Strecker GJ, Whalen LR, Dudek FE, Kossel AH, et al. Evidence for glutamate-mediated activation of hippocampal neurons by glial calcium waves. Journal of Neurobiology. 1995;28:159–170. doi: 10.1002/neu.480280204. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Momiyama A, Takahashi T, Ohishi H, Ogawa-Meguro R, Shigemoto R, et al. Role of a metabotropic glutamate receptor in synaptic modulation in the accessory olfactory bulb. Nature. 1993;366:687–690. doi: 10.1038/366687a0. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nature Neuroscience. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Liu QS, Xu Q, Arcuino G, Kang J, Nedergaard M. Astrocyte-mediated activation of neuronal kainate receptors; Proceedings of the National Academy of Sciences of the U.S.A; 2004. pp. 3172–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzanti M, Sul JY, Haydon PG. Glutamate on demand: astrocytes as a ready source. Neuroscientist. 2001;7:396–405. doi: 10.1177/107385840100700509. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Glutamate spillover suppresses inhibition by activating presynaptic mGluRs. Nature. 2000a;404:498–502. doi: 10.1038/35006649. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. GABA spillover from single inhibitory axons suppresses low-frequency excitatory transmission at the cerebellar glomerulus. Journal of Neuroscience. 2000b;20:8651–8658. doi: 10.1523/JNEUROSCI.20-23-08651.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Takano T, Hansenm AJ. Beyond the role of glutamate as a neurotransmitter. Nature Reviews Neuroscience. 2002;3:748–755. doi: 10.1038/nrn916. [DOI] [PubMed] [Google Scholar]
- Newman EA. Glial cell inhibition of neurons by release of ATP. Journal of Neuroscience. 2003;23:1659–1666. doi: 10.1523/JNEUROSCI.23-05-01659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. Journal of Neuroscience. 1998;18:4022–4028. doi: 10.1523/JNEUROSCI.18-11-04022.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–926. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nature Neuroscience. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- Pasti L, Zonta M, Pozzan T, Vicini S, Carmignoto G. Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. Journal of Neuroscience. 2001;21:477–484. doi: 10.1523/JNEUROSCI.21-02-00477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. Journal of Neuroscience. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Astrocytic neurotransmitter receptors in situ and in vivo. Progress in Neurobiology. 1997;51:439–455. doi: 10.1016/s0301-0082(96)00068-8. [DOI] [PubMed] [Google Scholar]
- Ransom BR. Glial modulation of neural excitability mediated by extracellular pH: a hypothesis revisited. Progress in Brain Research. 2000;125:217–228. doi: 10.1016/S0079-6123(00)25012-7. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Lopez-Garcia JC, Lerma J. Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons; Proceedings of the National Academy of Sciences of the U.S.A; 2000. pp. 1293–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Gahwiler BH, Charpak S. Target cell-specific modulation of transmitter release at terminals from a single axon; Proceedings of the National Academy of Sciences of the U.S.A; 1998. pp. 12004–12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semyanov A, Kullmann DM. Modulation of GABAergic signaling among interneurons by metabotropic glutamate receptors. Neuron. 2000;25:663–672. doi: 10.1016/s0896-6273(00)81068-5. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kulik A, Roberts JD, Ohishi H, Nusser Z, Kaneko T, et al. Target-cell-specific concentration of a metabotropic glutamate receptor in the presynaptic active zone. Nature. 1996;381:523–525. doi: 10.1038/381523a0. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, et al. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. Journal of Neuroscience. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toms NJ, Jane DE, Kemp MC, Bedingfield JS, Roberts PJ. The effects of (RS)-alpha-cyclopropyl-4-phosphonophenylglycine ((RS)-CPPG), a potent and selective metabotropic glutamate receptor antagonist. British Journal of Pharmacology. 1996;119:851–854. doi: 10.1111/j.1476-5381.1996.tb15750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Van der Kloot W. The regulation of quantal size. Progress in Neurobiology. 1991;36:93–130. doi: 10.1016/0301-0082(91)90019-w. [DOI] [PubMed] [Google Scholar]
- Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. Journal of Neuroscience. 1999;19:6897–6906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, et al. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine; Proceedings of the National Academy of Sciences of the U.S.A; 2003. pp. 15194–15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. Journal of Neuroscience. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, et al. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nature Neuroscience. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]