

Abstract
A parallel synthetic strategy to the 9-aminoacridine scaffold of the classical anti-malarial drug quinacrine (2) is presented. The method features a new route to 9-chloroacridines that utilizes triflates of salicylic acid derivatives, which are commercially available in a variety of substitution patterns. The route allows ready variation of the two diversity elements present in this class of molecules: the tricyclic aromatic heterocyclic core, and the disubstituted diamine sidechain. In this study, a library of 175 compounds was designed, although only 93 of the final products had purities acceptable for screening. Impurity was generally due to incomplete removal of 9-acridones (18), a degradation product of the 9-chloroacridine synthetic intermediates. The library was screened against two strains of Plasmodium falciparum, including a model of the drug-resistant parasite, and six novel compounds were found to have IC50 values in the low nanomolar range.
1. Introduction
Malaria is a devastating infectious disease caused by the protozoan parasite Plasmodium.1 Four species of Plasmodium are known to infect humans: P. vivax, P. malariae, P. ovale, and P. falciparum, the latter being the most deadly. While the controversial use of polychlorinated biphenyl pesticides (e.g. DDT), has generally led to the eradication of malaria in North America and most European countries,2 the disease is still widespread in Africa, Central and South America, and Southeast Asia. Malaria continues to affect 200-500 million people, and causes 1-2 million fatalities annually.3 As a result, malaria has produced devastating social and economic burdens on the countries most afflicted by it.1
The development of anti-malarial compounds continues to be a major focus of many laboratories,4 and as such, a variety of compounds have been explored in the prophylaxis and treatment of this disease (Figure 1). The efficacy of quinine (1), a derivative of cinchona-bark, as an anti-malarial compound was recognized as early as the 1600s. The earliest synthetic anti-malarial agent was quinacrine (2). Quinacrine was supplemented by chloroquine (3),5 along with various other 4- and 8-substituted quinolines, including mefloquine (4).6 Chloroquine was found to be highly effective against all four human-infecting species of Plasmodium, and its inexpensive production led it to quickly become a standard anti-malarial treatment.5 Unfortunately, over several decades this resulted in the development of chloroquine-resistant strains of Plasmodium, particularly of P. falciparum.7,8 Certain substituted quinolines, including mefloquine (4) have been efficacious against chloroquine-resistant P. falciparum (CRPF), but this drug is also associated with a variety of neuropsychiatric side-effects.9 Another promising class of compounds with activity against CRPF are the artemisinins.10-12 The continuing exploration of compounds active against CRPF is an active pursuit of our laboratory.13,14
Figure 1.
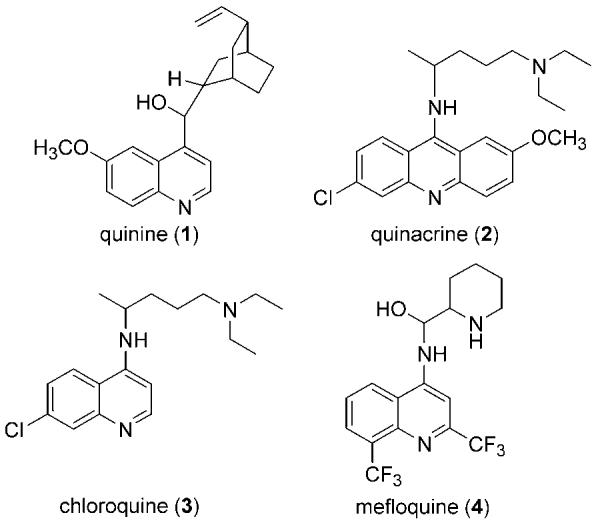
Structures of common quinoline and acridine anti-malarial compounds.
In the context of exploring the 9-aminoacridine scaffold of quinacrine (3) to target the propagation of prions,15-17 we became interested in re-evaluating this class of molecules for anti-malarial activity, particularly against CRPF. Herein we report a parallel synthetic strategy to generate libraries of 9-aminoacridines based on the general structure of quinacrine, along with in vitro cell-based screening results against drug-sensitive and drug-resistant strains of P. falciparum. In addition to having anti-prion and anti-malarial properties, 9-aminoacridines are interesting for other types of biological activity. For example, 9-aminoacridines are known DNA intercalators,18 inhibitors of mammalian topoisomerase I19 and acetylcholinesterase,20 and these compounds are also active against African trypanosomes.21-23 Cancer chemotherapeutics based on the 9-aminoacridine scaffold have been developed24 (e.g. Ledakrin and Amascrine). Additionally, 9-aminoacridines have been explored as photoaffinity labels25 and as fluorescent probes used to detect cancer cells.26
Our general synthetic strategy to 9-aminoacridines is summarized in Figure 2. Derivatives of substituted salicylic acid chemset 5 were coupled to aniline chemset 6 to generate 9-chloroacridine chemset 7, as was previously introduced.27 The use of salicylic acid precursors, activated as triflates, is novel and deviates from an earlier approach to 9-chloroacridines based on the Ulmann coupling of anilines with 2-bromoaryl carboxylic acids,28-30 of which much fewer are commercially available. To approach the amine side-chain, 1,3-diaminopropane (8) was dually functionalized to generate a library of requisite diamine building block chemset 9. Finally, parallel coupling of 9-chloroacridines with the diamines was done to generate the desired 9-aminoacridine chemset 10. A notable feature of this strategy is that it allows quick access to variations of the tricyclic acridine heterocycle as well as the amine sidechain, which will allow probing of the cooperativity of these two structural features.
Figure 2.

A parallel synthetic strategy to generate libraries of 9-aminoacridines.
2. Results
Chemistry
The synthesis of the 9-chloroacridine heterocycles is illustrated in Scheme 1. The salicylic acid chemset (5) was methylated and then activated for crosscoupling as the corresponding aryl triflate chemset (11), generally in near quantitative yield giving compounds of high purity. These species were coupled to substituted aniline chemset 6 to generate diarylamine chemset 12, using conditions developed by Buchwald et al.31, which proceeded smoothly. Next, hydrolysis of the methyl ester groups was performed using non-aqueous conditions designed for parallel synthesis,27 followed by Friedel-Crafts cyclization with POCl3 to generate the 9-chloroacridine chemset (7). The transformation of 11 to 7 was accomplished in a parallel 3-step, one pot procedure, without intermediate workup, using a 12-place reaction carrousel (Radleys Discovery Technologies). The 9-chloroacridine products (7) were isolated in acceptable yield and >95% purity after automated flash column purification using a CombiFlash system (Isco).
Scheme 1.

Synthesis of 9-chloroacridines. Reagents and conditions: (a) MeI (1.05 eq.), Cs2CO3 (0.5 eq.) DMF (90-100% yield). (b) Tf2O (1.1 eq.), Et3N (2.0 eq.), CH2Cl2, −78 °C to rt (95-100% yield). (c) substituted anilines 6 (1.05 eq.), Pd(OAc)2 (0.05 eq.), BINAP (0.08 eq.), Cs2CO3 (1.4 eq.), toluene. (d) Ba(OH)2-8H2O (2.5 eq.), CH3OH, 80 °C overnight; (e) POCl3 (neat) 120 °C, 1h (5-50% yield).
The synthesis of the diamine building blocks is illustrated in Scheme 2. 1,3-diaminopropane (8) was monosulfonylated under dilute conditions to attach the thiolate-labile 2-nitrobenzenesulfonyl (nosyl = Ns) protecting group32,33 to generate 13 in high yield and purity. Amidation with carboxylic anhydride reagents (14) followed by reduction with borane-dimethylsulfide complex34 generated the corresponding secondary amine chemset 15 in good yield and excellent purity. An alternate acylation reaction using acid chloride reagents formed a byproduct with an additional acyl group, apparently attached to the sulfonamide nitrogen atom. Lastly, reductive amination with aldehyde chemset 16 and sodium triacetoxyborohydride35 followed by deprotection of nosyl groups by treatment with cesium benzenethiolate32,33 afforded the free amine chemset 9. Steps a-c were carried out in large scale (>60 g initially) using traditional glassware, while steps d and e (1.5 g/each) were done in parallel using the Radleys carrousel apparatus. The final two heterogeneous reactions proceeded slowly in the 12-place Radleys carrousel, presumably due to poor mixing; magnetic stirring using the 6-place carrousel alleviated this problem. The reductive amination reaction (step d) could alternatively be performed using the smaller reaction vessels of the 12-place carrousel, but agitated in a sonicating wash bath rather than by magnetic stirring. The products of the last two steps were isolated in high purity, either by aqueous workup, or in parallel using solid-phase extraction (SPE) with Dowex 50WX2-400 ion-exchange resin. Chromatographic purification was not required in any of the steps of the sequence.
Scheme 2.

Synthesis of diamines. Reagents and conditions: (a) 2-nitrobenzenesulfonyl chloride (0.1 eq.), CH2Cl2, °C to rt, overnight (70 g scale, 85% yield). (b) carboxylic anhydrides (14) (1.2 eq.), pyridine (1.1 eq.), THF (90-100% yields). (c) BH3·dimethylsulfide complex (4.0 eq.), THF, 60 °C, 30 min. (67-77% yields) (d) aldehydes 16 (1.5 eq.), sodium triacetoxyborohydride (1.5 eq.), THF, sonicated, 35 °C, 1h. (e) benzenethiol (5 eq.), Cs2CO3 (2.5 eq.), degassed CH3CN under argon.
The coupling of 9-chloroacridines with diamines to generate the desired 9-aminoacridine products is illustrated in Scheme 3. 9-chloroacridine chemset 7 was converted to the reactive intermediate 9-phenoxyacridine chemset 17 by treatment with phenol in DMSO. Subsequent reaction with the diamine chemset 9 afforded the desired 9-aminoacridine products 10. Direct treatment of 9-chloroacridines with diamines, i.e. omitting the activation step, afforded little or no product. Furthermore, in some of the coupling reactions, the formation of 9-acridone byproducts (chemset 18) was observed, presumably due to hydrolysis of 9-chloroacridines.36 This side reaction could be partially suppressed by the addition of 3Å molecular sieves during the coupling, although decomposition also occurred during ambient storage of the precursor chloroacridines.
Scheme 3.
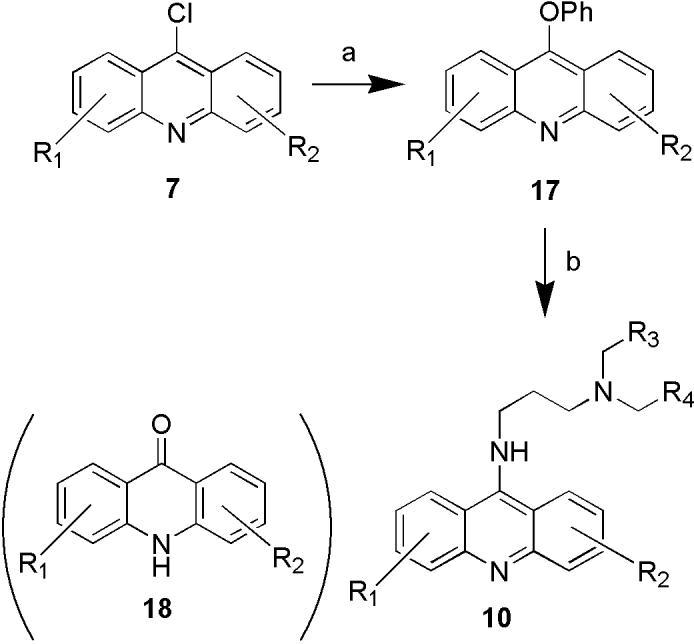
Coupling of 9-chloroacridines with diamines. Reagents and conditions: (a) phenol (15 eq.), Cs2CO3 (1 eq.), 3Å molecular sieves, DMSO, 100 °C, 2 h. (b) diamines (9) (4 eq.), DMSO, 100 °C, 4h; then methyl isocyanate polystyrene resin (NovaBiochem, 8 eq.), overnight, rt.
The coupling reactions are carried out in 48-position parallel synthesis blocks (Bohdan), and agitated with the Bohdan MiniBlock High Capacity Shaking and Washing Station (600 RPM, 100 °C). After coupling was achieved, the remaining diamine was then scavenged by treatment with electrophilic methyl isocyanate resin. Subsequent workup was performed by SPE with Dowex 50WX2-400 ion-exchange resin. A three-step parallel SPE protocol was devised to purify the 9-aminoacridine products: first, the crude product was loaded on the column and washed with TFA (5% in MeOH) to elute DMSO and phenol; next, the resin was washed with pyridine (5% in MeOH) to elute unreacted 9-phenoxyacridines (17) and 9-acridones (18); finally, the desired 9-aminoacridine product chemset (10) was eluted using Et3N (5% in MeOH).
Synthesis of 9-aminoacridine and diamine components
A selection of 15 diversely substituted 9-chloroacridine components and 16 diamine components was defined. The precursor components of the 9-chloroacridine library are defined in Figure 3. Various substituent patterns were chosen to test the scope of the chemistry, specifically electronic effects in the Buchwald coupling and cyclization steps. The synthesis results of the 9-chloroacridine component are outlined in Table 1. The purified yields after the three-step sequence ranged from moderate to fairly low. A degree of degradation was observed in nearly all of the 9-chloroacridines synthesized, namely hydrolysis to form 9-acridones (18).
Figure 3.

Composition of 9-chloroacridine (7) chemset.
Table 1.
9-chloroacridine (7) chemset synthesis results.
| Entry | Salicylic Acid | Aniline | Product | Yielda |
|---|---|---|---|---|
| 1 | 5{1} | 6{1} | 7{1,1} | 48% |
| 2 | 5{1} | 6{2} | 7{1,2} | 26% |
| 3 | 5{1} | 6{3} | 7{1,3} | 21% |
| 4 | 5{1} | 6{4} | 7{1,4} | 44% |
| 5 | 5{2} | 6{1} | 7{2,1} | 42% |
| 6 | 5{2} | 6{2} | 7{2,2} | 37% |
| 7 | 5{2} | 6{3} | 7{2,3} | 6% |
| 8 | 5{2} | 6{4} | 7{2,4} | 31% |
| 9 | 5{3} | 6{1} | 7{3,1} | 25% |
| 10 | 5{3} | 6{2} | 7{3,2} | 30% |
| 11 | 5{3} | 6{3} | 7{3,3} | 31% |
| 12 | 5{4} | 6{1} | 7{4,1} | 18% |
| 13 | 5{4} | 6{2} | 7{4,2} | 40% |
| 14 | 5{4} | 6{4} | 7{4,4} | 23% |
| 15 | 5{1} | 6{5} | 7{1,5} | 49% |
Purified yield of the final three-step one-pot sequence.
The design of the disubstituted diamine chemset (9) is outlined in Figure 4. All amines in this library were based on a 1,3-diaminopropane linker. Two carboxylic anhydrides (acetic and propionic) were used to vary R3, while the majority of variation was achieved with a selection of aldehydes to vary R4. The results of the synthesis of the diamines are outlined in Table 2. Of the products, the volatile low molecular weight dialkyl-substituted diamines (entries 1-2, 9-10) were isolated as trifluoroacetate salts. Overall, yields were moderate to very good in the reductive amination and nosyl deprotection reactions. Each of the diamine compounds was produced in very high purity, as assessed by NMR.
Figure 4.

Composition of disubstituted diamine (9) chemset.
Table 2.
Disubstituted diamine chemset (9) synthesis results.
| Entry | Anhydride | Aldehyde | Product | Yield: Reductive Amination | Yield: Nosyl Deprotection |
|---|---|---|---|---|---|
| 1 | 14{1} | 16{1} | 9{1,1} · 2TFA | 46% | 81% |
| 2 | 14{1} | 16{2} | 9{1,2} · 2TFA | 82% | 85% |
| 3 | 14{1} | 16{3} | 9{1,3} | 84% | 91% |
| 4 | 14{1} | 16{4} | 9{1,4} | 71% | 87% |
| 5 | 14{1} | 16{5} | 9{1,5} | 70% | 91% |
| 6 | 14{1} | 16{6} | 9{1,6} | 87% | 93% |
| 7 | 14{1} | 16{7} | 9{1,7} | 71% | 33% |
| 8 | 14{1} | 16{8} | 9{1,8} | 58% | 94% |
| 9 | 14{2} | 16{1} | 9{2,1} · 2TFA | 75% | 89% |
| 10 | 14{2} | 16{2} | 9{2,2} · 2TFA | 83% | 92% |
| 11 | 14{2} | 16{3} | 9{2,3} | 73% | 100% |
| 12 | 14{2} | 16{4} | 9{2,4} | 79% | 97% |
| 13 | 14{2} | 16{5} | 9{2,5} | 69% | 74% |
| 14 | 14{2} | 16{6} | 9{2,6} | 66% | 84% |
| 15 | 14{2} | 16{7} | 9{2,7} | 63% | 73% |
| 16 | 14{2} | 16{8} | 9{2,8} | 42% | 79% |
Proof-of-concept library
The coupling of 9-chloroacridines with disubstituted diamines to generate a proof-of-concept library of 9-aminoacridines (10) is outlined in Table 3. In the first set of entries (1-15) the synthesized 9-chloroacridines were coupled with commercially available sidechain 9{1,9}, while in the second set of entries (16-31), the commercially available 9-chloroacridine heterocycle 7{4,6} of quinacrine was coupled with the set of synthesized diamines. Yields and purities of the coupled products were highly variable. Notable degradation of the synthesized 9-chloroacridine starting materials to 9-acridones (18) accounts for the overall lower yields in the first set of entries. Impurity was generally accounted for by the presence of 9-acridones not effectively separated during the SPE purification. We suspect that the second set of entries was less prone to impurity problems due to the shelf stability of the commercially available quinacrine 9-chloroacridine 7{4,6} starting material. Products where final purity was less than 50% by RP-HPLC analysis were indicated as failed entries, and constituted 20% of this library.
Table 3.
Proof library: Coupling reactions to generate a library of 9-aminoacridines (10)
| Entry | 9-chloroacridine | Diamine component | Product | Yield (purity)a |
|---|---|---|---|---|
| 1 | 7{1,1} | 9{1,9} | 10{1,1,1,9} · 2HCl | 52% (100%) |
| 2 | 7{1,2} | 9{1,9} | 10{1,2,1,9} · 2HCl | 16% (83%) |
| 3 | 7{1,3} | 9{1,9} | 10{1,3,1,9} · 2HCl | Failed |
| 4 | 7{1,4} | 9{1,9} | 10{1,4,1,9} · 2HCl | 19% (100%) |
| 5 | 7{2,1} | 9{1,9} | 10{2,1,1,9} · 2HCl | 22% (95%) |
| 6 | 7{2,2} | 9{1,9} | 10{2,2,1,9} · 2HCl | 4% (81%) |
| 7 | 7{2,3} | 9{1,9} | 10{2,3,1,9} · 2HCl | Failed |
| 8 | 7{2,4} | 9{1,9} | 10{2,4,1,9} · 2HCl | 21% (92%) |
| 9 | 7{3,1} | 9{1,9} | 10 {3,1,1,9} · 2HCl | 8% (56%) |
| 10 | 7{3,2} | 9{1,9} | 10{3,2,1,9} · 2HCl | 15% (50%) |
| 11 | 7{3,3} | 9{1,9} | 10{3,3,1,9} · 2HCl | Failed |
| 12 | 7{4,1} | 9{1,9} | 10{4,1,1,9} · 2HCl | 22% (81%) |
| 13 | 7{4,2} | 9{1,9} | 10{4,2,1,9} · 2HCl | 22% (73%) |
| 14 | 7{4,4} | 9{1,9} | 10{4,4,1,9} · 2HCl | Failed |
| 15 |
7{1,5} |
9{1,9} |
10{1,5,1,9} · 2HCl |
10% (73%) |
| 16 | 7{4,6} | 9{1,1} · 2TFA | 10{4,6,1,1} · 2HCl | 32% (81%) |
| 17 | 7{4,6} | 9{1,2} · 2TFA | 10{4,6,1,2} · 2HCl | 18% (100%) |
| 18 | 7{4,6} | 9{1,3} | 10{4,6,1,3} · 2HCl | 48% (100%) |
| 19 | 7{4,6} | 9{1,4} | 10{4,6,1,4} · 2HCl | 34% (100%) |
| 20 | 7{4,6} | 9{1,5} | 10{4,6,1,5} · 2HCl | 52% (100%) |
| 21 | 7{4,6} | 9{1,6} | 10{4,6,1,6} · 2HCl | 32% (95%) |
| 22 | 7{4,6} | 9{1,7} | 10{4,6,1,7} · 2HCl | Failed |
| 23 | 7{4,6} | 9{1,8} | 10{4,6,1,8} · 2HCl | 19% (68%) |
| 24 | 7{4,6} | 9{2,1} · 2TFA | 10{4,6,2,1} · 2HCl | 34% (100%) |
| 25 | 7{4,6} | 9{2,2} · 2TFA | 10{4,6,2,2} · 2HCl | 60% (100%) |
| 26 | 7{4,6} | 9{2,3} | 10{4,6,2,3} · 2HCl | 69% (100%) |
| 27 | 7{4,6} | 9{2,4} | 10{4,6,2,4} · 2HCl | 62% (100%) |
| 28 | 7{4,6} | 9{2,5} | 10{4,6,2,5} · 2HCl | 73% (100%) |
| 29 | 7{4,6} | 9{2,6} | 10{4,6,2,6} · 2HCl | 47% (90%) |
| 30 | 7{4,6} | 9{2,7} | 10{4,6,2,7} · 2HCl | Failed |
| 31 | 7{4,6} | 9{2,8} | 10{4,6,2,8} · 2HCl | 51% (87%) |
Purity determined by RP-HPLC-MS integration detected at 254 nm.
Cross-coupled library
Having established our parallel synthesis methodology in a proof-of-concept library, we then prepared a cross-coupled library of synthesized 9-chloroacridines with synthesized diamines. The proof-of-concept library showed some classes of 9-chloroacridines achieving better yields and purities than others during the coupling reaction, with the some being particularly more prone hydrolysis to the 9-acridone, possibly due to electron-withdrawing substitutuents increasing electrophilicity at C9. Thus, we designed a library utilizing a subset of the 9-chloroacridines (7), specifically compounds 7{1,1}, 7{1,2}, 7{1,4}, 7{2,1}, 7{2,2}, 7{2,4}, 7{4,1}, 7{4,2}, and 7{1,5}, that appeared be the most promising and likely to generate compounds acceptable for screening. These species were cross-coupled with the entire series of synthesized amines (9), to generate a library of 144 compounds (Table S1, Supporting information). Using the same criteria established for the proof library, 53% of the compounds were marked as failed entries, due to final purities being less than 50% after the initial SPE purification. While the number of failed couplings is significant, it is notable that the relative success of certain 9-chloroacridines in the proof-of-concept library translated quite well to success in the cross-coupled library. Specifically, certain 9-chloroacridines (7) that worked relatively well in the proof library achieved a reasonable degree of yield and purity in the cross-coupled library: 7{1,1}, 7{2,1}, 7{2,4}, 7{4,1}, and 7{4,2}.
Screening and biological evaluation
The compounds of the proof-of-concept and cross-coupled libraries were screened for in vitro antimalarial activity against P. falciparum cultured in human erythrocytes. Growth inhibition of the parasite was measured using a fluorescent-active cell sorting (FACS) assay.13,14 Two strains of the parasite were examined: 3D7 and W2, which are established models of chloroquine-sensitive and chloroquine, mefloquine-resistant P. falciparum, respectively. Initially, activity was screened at a fixed concentration of 30 nM (Figure 3), with data represented as parasite growth relative to untreated controls. Chloroquine (3) serves as a positive control for the 3D7 strain and a negative control for the W2 strain. Quinacrine (2) also serves as a positive control for 3D7, while this compound also has partial activity against the W2 strain. In this assay, several compounds of the proof-of-concept library were found to be highly active against both strains of the parasite, with notable decrease in parasitic growth. The 9-aminoacridines in the proof-of-concept library that appeared the most interesting, particularly in inhibiting the growth of the chloroquine-resistant W2 strain, were 10{1,4,1,9}, 10{4,6,1,2}, 10{4,6,1,4}, 10{4,6,2,1}, 10{4,6,2,2}, and 10{4,6,2,4}. Of the products derived from novel 9-chloroacridines, only 10{1,4,1,9}, derived from 7{1,4}, appeared to have significant activity at 30 nM. Notably, use of the commercially available acridine heterocycle scaffold 7{4,6} with variation of the amine (Table 3, entries 16-31) appears to be a promising strategy for optimizing 9-aminoacridines for antimalarial activity. Unfortunately, fewer compounds in the cross-coupled library appear as interesting, as many entries in this library failed in synthesis and thus were not screened. Several compounds were active against the 3D7 strain, but only one, 10{1,1,1,8} was found to have promising activity against the chloroquine resistant W2 strain.
Figure 3.
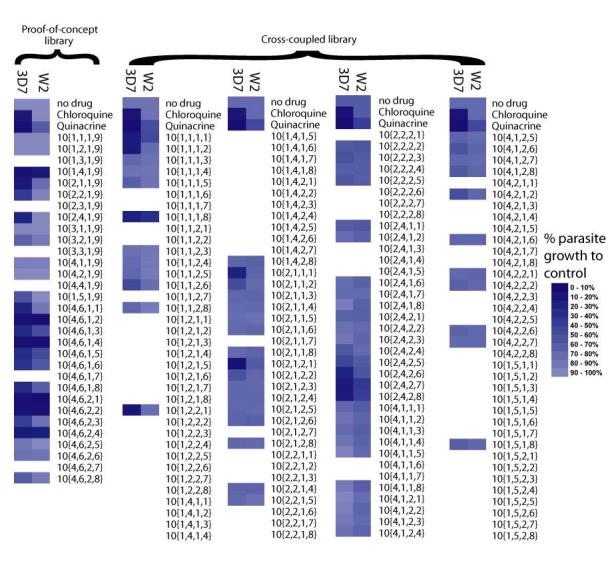
Relative activity of the library defined in Table 3 against P. falciparum strains 3D7 and W2 cultured in human erythrocytes (measured at fixed concentration: 30 nM). Data is represented in blue scale, as percent parasite growth relative to un-treated controls, with darker squares indicating a higher activity against the parasites. Omitted entries are due to failure during synthesis.
Structures and 50% effective inhibitory concentration (EC50) values of the most promising 9-aminoacridines 10{1,4,1,9}, 10{4,6,1,2}, 10{4,6,1,4}, 10{4,6,2,1}, 10{4,6,2,2}, and 10{4,6,2,4} from the initial screen are presented in Figure 5. Of this set, compounds 10{1,4,1,9} and 10{4,6,2,1} appear to be the most potent, having sub-nanomolar EC50 values. The slightly less potent compounds can be grouped by their amine substitution patterns: 10{4,6,1,2} and 10{4,6,2,2} (both containing an isobutyl substituent), as well as 10{4,6,1,4} and 10{4,6,2,4} (both containing a furylmethyl substituent). The tolerance for fairly different styles of amine substituents, e.g. branched alkyl groups and a furyl heterocyclic group, is an interesting feature of the pharmacophore. EC50 values were also determined for quinacrine (2) and chloroquine (3). Notably all of the synthesized compounds had improved potencies, when compared to 2 and 3, against both strains of P. falciparum, and none of the compounds have previously been described in the literature.
Figure 5.

Structures and EC50 values of the most active compounds against P. falciparum strains 3D7 and W2 in cultured human erythrocytes, as well as for quinacrine (2) and chloroquine (3). Activity at a series of 12 concentrations was determined as percent parasite growth relative to un-treated controls, EC50 values were determined with SigmaPlot.
3. Conclusion
A parallel synthetic strategy to generate 9-aminoacridines is described. The method features a new route to 9-chloroacridines that utilizes triflates of salicylic acid derivatives, which are commercially available in fairly diverse substitution patterns. Additionally, a procedure for the synthesis of disubstituted diaminoalkanes was presented. Two libraries were designed totaling 175 compounds, although only 93 of the final products had purities suitable for in vitro screening. Low purity was generally attributed to contamination by 9-acridones (18), a hydrolytic degradation product of the 9-chloroacridine synthetic intermediates. The compounds were screened initially at fixed concentration (30 nM), with the most active hits re-analyzed to determine dose-response. Six previously undescribed compounds were found to have nanomolar EC50 values, and were particularly interesting due to improved potency against the W2 parasite strain, relative to quinacrine (2) and chloroquine (3). Of this set, most were derived from the commercially available acridine heterocycle 7{4,6} with novel substituents on the amine side chain, suggesting that further modification of this component may be a viable strategy for future optimizations.
4. Experimental
General
NMR spectra were recorded on a Varian Model AS 400-MHz machine. The following abbreviations are used to describe peak splitting when appropriate: s=singlet, d=doublet, t=triplet, q=quartet, bs=broad singlet, mult=multiplet. Reactions were carried out under an atmosphere of argon. Reagents were of commercial quality unless otherwise indicated. Analytical thin layer chromatography (TLC) was carried out using plastic plates coated with silica gel 60 F254 (Whatman Part #4420 222). Developed plates were visualized using short wave UV light (254 nm), and were typically stained in an iodine/silica chamber and/or using a staining dip (e.g. p-anisaldehyde or ceric ammonium molybdate) followed by heating with a heat gun.
Measurement of in vitro antimalarial activity
Growth inhibition of P. falciparum cultured in human erythrocytes was measured with flow cytometry.37 Synchronized cultures of ring-stage parasites (0.8% parasitemia, 0.5% hematocrit) were grown in 96-well tissue culture plates (Falcon) in the presence of compounds for screening, at fixed concentration of 30 nM or serially diluted for dose-response. Cultures were grown in atmospherically regulated (6% CO2, 5% O2) incubators (Sanyo) at 37 °C for 72 h. Following drug incubation, cultures were fixed with 1% paraformaldehyde for 1 hr at RT and subsequently stained with 50 nM YOYO-1 (Molecular Probes) in 1X PBS for 18 hr at RT in the dark. Fluorescence of each sample was then obtained on a Becton-Dickenson LSR2 flow cytometer. YOYO-1 is a DNA intercalator that allows for distinction between parasitized and unparasitized red blood cells (RBCs), as RBCs lack DNA. Percent of parasitized RBCs was determined from fluorescent signal due to YOYO-1, and growth inhibition values were then calculated as the fraction of parasitized RBCs relative to cultures without drug. All screening was done in triplicate. Compounds were screened initially at a fixed concentration (30 nM). The most promising compounds were then evaluated for dose-response at 12 concentrations: 2000, 1000, 500, 250, 125, 62.5, 31.3, 15.6, 15.6, 7.8, 3.9, 2.0, and 0.9 nM. Fifty percent effective inhibitory concentrations (EC50 values) were determined using SigmaPlot.
General procedure for the synthesis of salicyl triflate chemset (11)
A solution of substituted salicylic acid 5 (11.7 mmol) in DMF (100 ml) was treated with Cs2CO3 (0.5 eq) and iodomethane (1.05 eq.). The solution was stirred for 1.5 h, taken up in Et2O:EtOAc (2:1) (100 ml), washed with H2O (50 ml), NaHCO3 (10% aq., 50 ml), and NaCl (satd. aq.) (25 ml). The organic product layer was dried over MgSO4, and concentrated in vacuo to yield methyl esters in excellent purity (80-100% yield).
The methyl esters (5.5 mmol) were dissolved in CH2Cl2 (50 ml), cooled to −78 °C and stirred under argon. Treatment with Et3N (2 eq.) was followed by the dropwise addition of trifluoromethanesulfonic anhydride (1.1 eq.). The mixture was stirred at −78 °C for 30 min, and then warmed to RT, whereupon TLC indicated quantitative conversion. The solution was taken up in Et2O (100 ml) and washed with HCl (1M aq.) (25 ml). The aqueous layer was back-extracted with additional Et2O (25 ml), and the combined organic product layer was washed with NaCl (satd. aq.) (25 ml), dried over MgSO4, and concentrated in vacuo to yield salicyl triflate chemset (11) in excellent purity (95-100% yield).
General procedure for the parallel synthesis of the diarylamine chemset (12)
Following the general procedure of Buchwald,31 each of the vessels of a Radleys 12-place carrousel were loaded with a solution of salicyl triflate chemset 11 (0.728 mmol) in toluene (7 ml). The vessels were purged under argon, and each was then treated with anilines (6) (1.2 eq.), Cs2CO3 (1.4 eq.), rac-2,2'-bis(diphenylphosphino)-1,1'-binahthalene (BINAP) (0.08 eq.), followed by Pd(OAc)2 (0.05 eq.). The carrousel was heated to 120 °C for 16 h, after which TLC of reaction mixtures generally indicated complete conversion to the diarylamine products (12). The products were concentrated in vacuo using a Genevac HT-4 parallel evaporation system, and then used in the directly in the next step without purification or workup.
General procedure for the parallel synthesis of the 9-chloroacridine chemset (7)
The crude diarylamines (12) (ca. 0.728 mmol), loaded into the vessels of a Radleys 12-place carrousel, were re-dissolved in methanol (7 ml), and treated with barium hydroxide octahydrate (1.5 eq.). The carrousel was heated to 90 °C overnight, after which TLC showed complete ester hydrolysis. The crude carboxylic acid species were concentrated in vacuo using a Genevac Parallel Evaporation System, and used directly in the next step without purification or workup. To affect cyclization, the species were treated with POCl3 (7 ml), and heated to 120 °C under air for 2 h. Parallel removal of POCl3 was accomplished by heating the Radleys carrousel to 160 °C, while water (60 °C) was pumped through the Radleys upper water jacket using a Julabo MB-5 heated water circulator, and while house vacuum was applied to the carrousel (with an in-line dry ice/ethanol vacuum trap). After the removal of POCl3, the crude 9-chloroacridines were azeotropically dried with EtOAc and then dry-loaded onto silica for flash column chromatography. Automated flash column purification using the CombiFlash system by Isco was done to yield the 9-chloroacridine products (7) in high purity determined by homogenous 1H-NMR spectra. A general solvent gradient for this class of compounds was devised: 5:95 → 55:45 ethyl acetate:hexanes (2% Et3N) over 25 min.
N-(3-Aminopropyl)-2-nitrobenzenesulfonamide (13)
1,3-diaminopropane (236 ml, 2807 mmol) was cooled to 0 °C under argon, and then a solution of 2-nitrobenzenesulfonyl chloride (62.2 g, 280.7 mmol) in CH2Cl2 (2400 ml) was slowly added by pressure-equalizing dropping funnel, over a period of ca. 2 h. The reaction mixture was stirred overnight, and then split into two equal portions for workup. Each portion was cooled to °C, followed by dropping-funnel addition of H2O (300 ml) and HCl (conc. aq.) (150 ml) (Caution: both additions are exothermic!). The CH2Cl2 layers were extracted and disposed. The aqueous layers were then cooled to 0 °C, treated directly with solid NaOH until the mixtures were visibly heterogeneous, and then extracted with CH2Cl2 (6 x 500 ml). The combined organic layers were dried over Na2SO4, and concentrated in vacuo to yield the title compound (13) (60.27 g, 83% yield), which can also be purchased commercially (TCI America).
General procedure for amidation/reduction to generate the secondary amine chemset (15)
A solution of N-(3-aminopropyl)-2-nitrobenzenesulfonamide (13) (31.0 g, 120 mmol) in THF (400 ml) was stirred at 25 °C under argon in an ambient water bath, and treated with pyridine (11 ml, 131 mmol, 1.1 eq.) followed by carboxylic anhydride reagents (14) (1.2 eq.). The solution was stirred at 25 °C for 1.5 h, after which RP-HPLC analysis indicated quantitative conversion to the amide. The reaction mixture was quenched with ammonium hydroxide (30% aq.) (70 ml, 598 mmol, 5 eq.), stirred an additional 15 min, and then concentrated in vacuo. The residue was treated with ammonium chloride (satd. aq.):NaCl (satd aq.) (2:1, 300 ml), and acidified with HCl (1M aq.) to pH 1, and then the product was extracted with Et2O:EtOAc (1:1) (2×200 ml). The organic layer was washed with HCl (1M aq):NaCl (satd aq.) (1:1, 100 ml), dried over Na2SO4, and concentrated in vacuo to yield the crude amide intermediate, used in the next step without further purification.
The subsequent borane-methylsulfide reduction step required an apparatus to remove of dimethylsulfide by distillation:34 the reaction was carried out in a 2000 ml single-neck round bottom flask attached to a 2-way Claisen adaptor. The adaptor was equipped with a rubber septum for reagent addition, and a vacuum distillation head with a trap for distilled dimethylsulfide. The system was charged with a solution of crude amide (ca. 120 mmol) in anhydrous THF (400 ml), stirred at 25 °C under argon, and treated dropwise with borane-dimethyl sulfide complex (46 ml, 478 mmol, 4 eq.). The solution was heated in an oil bath at 60 °C for 30 min, with concomitant distillation of dimethyl sulfide from the reaction mixture. RP-HPLC analysis showed the reaction to be complete. The distillation head was replaced with a condenser, and H2O was slowly and carefully added over 20 min., until major bubbling ceased. Next, HCl (1M aq., 100 ml) was added and the solution was heated to 60 °C for 45 min. The solution was cooled to °C, treated with Et2O (500 ml), and extracted with HCl (1M aq., 2×300 ml). The combined aqueous layers were washed with Et2O (200 ml), cooled to 0 °C, treated directly with solid NaOH until the mixture was visibly heterogeneous, and then extracted with EtOAc (3×200 ml), dried over Na2SO4, and concentrated in vacuo to yield the desired secondary amines (15) in high purity (50-71% yield).
General procedure for parallel reductive amination and nosyl deprotection (9)
Secondary amines (14) generated in the previous step were loaded into the vessels of a Radleys 12-place reaction carrousel as 0.3M stock solutions in THF (12 ml each, 3.5 mmol), followed by aldehydes (16) (1.5 eq.), and sodium triacetoxyborohydride (1.5 eq.). The vessels were purged under argon, and then agitated by placement in a Fisher Scientific FS20 sonicating wash bash for 30-60 min., after which RP-HPLC showed the reductive amination reaction to be quantitative. Parallel solvent removal in vacuo was accomplished using a GeneVac HT-4 instrument. Parallel workup by SPE: initially columns (Teledyne Isco Inc., # 69-3873-146) were loaded with Dowex 50WX2-400 ion-exchange resin (∼15 g), and the resin was then pre-conditioned with MeOH (1% CF3CO2H) (50 ml). The concentrated reaction mixtures were redissolved in MeOH (5% CF3CO2H) (50 ml) and loaded onto the columns. The resin was washed with MeOH, and then the products eluted with MeOH (10% Et3N) (50 ml). Finally, the products of reductive amination were obtained after parallel solvent removal in vacuo using the GeneVac HT-4 instrument. Alternative aqueous workup: The crude reaction mixture in THF was carefully quenched with HCl (1M aq.) (25 ml), treated with Et2O (150 ml), and the aqueous layer extracted followed by an additional aqueous extraction with HCl (1M aq.) (25 ml). The aqueous layers were combined and washed with Et2O (2 × 50 ml), basified with NaOH (1M aq.) until visibly heterogeneous, and the products extracted with Et2O (3 × 30 ml). The organic layer was dried over Na2SO4, and concentrated in vacuo to yield the products.
The crude tertiary amine products (ca. 3.5 mmol) were dissolved in CH3CN (35 ml) and loaded into the vessels of a Radleys 6-place reaction carrousel. The vessels were purged of air by applying three iterations of house vacuum followed by positive pressure of argon. Each vessel was then treated with Cs2CO3 (2.5 eq.) followed by benzenethiol (5.0 eq.), and then the vessels were stirred vigorously under argon for 1 h at rt., after which RP-HPLC showed near quantitative (>80%) conversion. More benzenethiol (5.0 eq.) was added, and the vessels stirred for 1 h, where upon RP-HPLC analysis showed the reactions were complete. An SPE workup identical to that described above for reductive amination was performed. Alternative aqueous workup: The crude reaction mixture in CH3CN was taken up in EtOAc (150 ml), and extracted with HCl (1M aq.) (2 × 50 ml). The aqueous layer was washed with EtOAc (50 ml), basified with NaOH (1M aq.) to pH 14, and extracted with EtOAc (3 × 30 ml). The combined organic layer was dried over Na2SO4, and for volatile alkyl-substituted diamine products, CF3CO2H (2 eq.) was added. Finally, the products were obtained after concentration in vacuo.
General procedure for the parallel synthesis of the 9-aminoacridine chemset (10)
DMSO stock solutions of 9-chloroacridines (7), diamines (9), and phenol were prepared at 0.077 M, 0.60 M, and 2.9 M, respectfully. The 2 ml polypropylene vessels of a 48-position Bohdan MiniBlock were loaded with 9-chloroacridines (7) (250 μl DMSO stock, 19.3 μmol), phenol (100 μl DMSO stock, 288 μmol, 15 eq.), Cs2CO3 (5 mg, 19.3 μmol, 1 eq.), and 3Å molecular sieves (ca. 0.010 g). The latter two items were conveniently added in parallel via the Bohdan Resin Dispenser accessory. The block was simultaneously heated to 100 °C and agitated at 600 RPM using the Bohdan MiniBlock High Capacity Shaking and Washing Station for 2 h. RP-LCMS usually showed quantitative conversion to the 9-phenoxyacridine chemset (17). Next, diamines (9) were added (125 μL DMSO stock, 4 eq.) and the blocks heated again to 100 °C at 600 RPM for 4 h. RP-LCMS analysis showed the presence of 9-aminoacridine product (10), as well as varying amounts of 9-acridone (17). Unreacted primary amine was scavenged by treatment with methyl isocyanate polystyrene resin (NovaBiochem, 1.40 mmol/g loading, 8 eq.) followed by agitation at 600 RPM overnight at ambient temperature. Parallel workup by SPE: pre-packed SPE columns (Bohdan SCX #13511218) were conditioned with H2O (1% CF3CO2H). The reaction mixtures (in DMSO) were treated with H2O (1% CF3CO2H) (1 ml) and loaded onto the columns. The resin was sequentially washed with H2O (1% CF3CO2H) (2 ml), MeOH (2 ml), and MeOH (1% pyridine) (8 ml); followed by product elution with EtOAc (10% Et3N) (3 ml). The desired 9-aminoacridine chemset (10) was finally obtained after parallel solvent removal in vacuo using the GeneVac HT-4 instrument.
Supplementary Material
5. References
- 1.Sachs J, Malaney P. Nature. 2002;415:680–5. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- 2.Kitron U, Spielman A. Rev. Infect. Dis. 1989;11:391–406. doi: 10.1093/clinids/11.3.391. [DOI] [PubMed] [Google Scholar]
- 3.Gilles HM. Management of Severe Malaria: A Practical Handbook. 2nd ed. World Health Organization; Geneva: 2000. [Google Scholar]
- 4.Ridley RG. Nature. 2002;415:686–93. doi: 10.1038/415686a. [DOI] [PubMed] [Google Scholar]
- 5.Foley M, Tilley L. Pharmacol. Ther. 1998;79:55–87. doi: 10.1016/s0163-7258(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 6.Palmer KJ, Holliday SM, Brogden RN. Drugs. 1993;45:430–75. doi: 10.2165/00003495-199345030-00009. [DOI] [PubMed] [Google Scholar]
- 7.Slater AF. Pharmacol. Ther. 1993;57:203–35. doi: 10.1016/0163-7258(93)90056-j. [DOI] [PubMed] [Google Scholar]
- 8.Ward SA. Trends. Pharmacol. Sci. 1988;9:241–6. doi: 10.1016/0165-6147(88)90153-8. [DOI] [PubMed] [Google Scholar]
- 9.Van Riemsdijk MMS,MC, Pepplinkhuizen L, Stricker BH. J. Clin. Psychiatry. 2005;66:199–204. doi: 10.4088/jcp.v66n0207. [DOI] [PubMed] [Google Scholar]
- 10.Frederich M, Dogne JM, Angenot L, De Mol P. Curr. Med. Chem. 2002;9:1435–56. doi: 10.2174/0929867023369691. [DOI] [PubMed] [Google Scholar]
- 11.O'Neill PM, Posner GH. J. Med. Chem. 2004;47:2945–64. doi: 10.1021/jm030571c. [DOI] [PubMed] [Google Scholar]
- 12.Tang Y, Dong Y, Vennerstrom JL. Med. Res. Rev. 2004;24:425–48. doi: 10.1002/med.10066. [DOI] [PubMed] [Google Scholar]
- 13.Madrid PB, Sherrill J, Liou AP, Weisman JL, Derisi JL, Guy RK. Bioorg. Med. Chem. Lett. 2005;15:1015–8. doi: 10.1016/j.bmcl.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 14.Madrid PB, Wilson NT, DeRisi JL, Guy RK. J. Comb. Chem. 2004;6:437–42. doi: 10.1021/cc0340473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korth C, May BC, Cohen FE, Prusiner SB. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9836–41. doi: 10.1073/pnas.161274798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.May BC, Fafarman AT, Hong SB, Rogers M, Deady LW, Prusiner SB, Cohen FE. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3416–21. doi: 10.1073/pnas.2627988100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vogtherr M, Grimme S, Elshorst B, Jacobs DM, Fiebig K, Griesinger C, Zahn R. J. Med. Chem. 2003;46:3563–4. doi: 10.1021/jm034093h. [DOI] [PubMed] [Google Scholar]
- 18.Jaycox GD, Gribble GW, Hacker MP. J. Heterocyclic. Chem. 1987;24:1405–8. [Google Scholar]
- 19.Ciesielska E, Pastwa E, Szmigiero L. Acta. Biochim. Pol. 1997;44:775–80. [PubMed] [Google Scholar]
- 20.Walker TM, Starr B, Dewhurst BB, Atterwill C. Human. Exp. Toxicol. 1995;14:469–74. doi: 10.1177/096032719501400601. [DOI] [PubMed] [Google Scholar]
- 21.Bonse S, Santelli-Rouvier C, Barbe J, Krauth-Siegel RL. J. Med. Chem. 1999;42:5448–5454. doi: 10.1021/jm990386s. [DOI] [PubMed] [Google Scholar]
- 22.Inhoff O, Richards Jonathan M, Briet Jan W, Lowe G, Krauth-Siegel RL. J. Med. Chem. 2002;45:4524–30. doi: 10.1021/jm020885k. [DOI] [PubMed] [Google Scholar]
- 23.Obexer W, Schmid C, Barbe J, Galy JP, Brun R. Tropical Med. Parasitol. 1995;46:49–53. [PubMed] [Google Scholar]
- 24.Denny WA. Curr. Med. Chem. 2002;9:1655–65. doi: 10.2174/0929867023369277. [DOI] [PubMed] [Google Scholar]
- 25.Kopacz SJ, Mueller DM, Lee CP. Biochim. Biophys. Acta. 1985;807:177–88. doi: 10.1016/0005-2728(85)90121-5. [DOI] [PubMed] [Google Scholar]
- 26.Schwarz G, Wittekind D. Anal. Qual. Cytol. 1982;4:44–54. [PubMed] [Google Scholar]
- 27.Anderson MO, Moser J, Sherrill J, Guy RK. Synlett. 2004:2391–2393. [Google Scholar]
- 28.Bui Vinh L, Boux LJ, Cheung HTA, Holder GM. J. Heterocyclic. Chem. 1983;20:281–4. [Google Scholar]
- 29.Csuk R, Barthel A, Raschke C. Tetrahedron. 2004;60:5737–5750. [Google Scholar]
- 30.Ledochowski A, Glowacki A, Wysocka-Skrzela B, Jazdzewski S. Pol. J. Chem. 1981;55:1721–4. [Google Scholar]
- 31.Ahman J, Buchwald SL. Tetrahedron Lett. 1997;38:6363–6366. [Google Scholar]
- 32.Fukuyama T, Jow C-K, Cheung M. Tetrahedron Lett. 1995;36:6373–4. [Google Scholar]
- 33.Nihei K-I, Kato MJ, Yamane T, Palma MS, Konno K. Synlett. 2001:1167–1169. [Google Scholar]
- 34.Brown HC, Choi YM, Narasimhan S. J. Org. Chem. 1982;47:3153–63. [Google Scholar]
- 35.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. J. Org. Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 36.Skonieczny S. Heterocycles. 1980;14:985–1032. [Google Scholar]
- 37.Barkan D, Ginsburg H, Golenser J. Int. J. Parasitol. 2000;30:649–53. doi: 10.1016/s0020-7519(00)00035-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.