

Abstract
Using model acidic glycans, we demonstrate the benefits of permethylation for matrix-assisted laser desorption/ionization time-of-flight/time-of-flight (MALDI/TOF-TOF) tandem mass spectrometry. With both the linear and branched structures, extensive cross-ring fragmentation product ions were generated, yielding valuable information on sugar linkages. Elimination of the negative charges commonly associated with sialylated structures through permethylation allowed their structural analysis in the positive ion mode. Extensive A- and X-type ions were observed for the linear structures, and slightly weaker signals for the branched sialylated structures. The diagnostic cross-ring fragments, permitting a distinction between α2-3 and α2-6 linkages of the sialic acid residues, were seen in abundance. Importantly, the cross-ring fragmentation with the branched structures provides adequate information to assign sialic acid residues, with a specific linkage, to a particular antenna.
While mass spectrometry (MS) using single mass analyzers can now readily assist the acquisition of carbohydrate compositional data in terms of isobaric monosaccharides,1 a complete structural analysis of glycans requires determination of branching, linkage position and a monomer anomericity. With the relative inefficiency of most post-source decay (PSD) ion fragmentation processes, these structural attributes necessitate the use of tandem mass spectrometry (MS/MS) techniques, although structural studies in less complex cases can also be aided by the use of exoglycosidase mixtures in concert with MS measurement.2
Not surprisingly, the fragmentation mechanisms for glycans have been studied extensively for a number of years, utilizing different combinations of ionization techniques and mass analyzers: fast atom bombardment (FAB)-MS,3-8 infrared-laser desorption MS,9,10 matrix-assisted laser desorption/ionization (MALDI)/magnetic sector,11-13 electrospray ionization (ESI)-MS,14,15 MALDI time-of-flight/MS,16-18 ESI ion-trap MS,19,20 MALDI Fourier-transform ion cyclotron resonance MS,21 and, very recently, ESI22,23 and MALDI/quadrupole time-of-flight MS.24,25 However, it is still difficult to deduce some structural details on a glycan structure from MS data alone. The considerations of combining an effective ionization technique and energetic collision-induced dissociation (CID), together with our current efforts26 to combine microcolumn separations of glycans with MALDI-MS, has prompted us to investigate the merits of MALDI time-of-flight/time-of-flight (TOF/TOF) instrumentation in the elucidation of detailed oligosaccharide structures without the use of exoglycosidases.
The fragmentation of glycans observed in MALDI-MS is similar to that seen earlier in FAB-MS and ESI-MS, showing dependence on several factors such as the type of ion formed, its charge state, the energy deposited into an ion, and the time available for fragmentation. These situations have been discussed extensively in several recent reviews.1,27,28 In general, glycans undergo two types of cleavages: glycosidic cleavages that result from breaking the bond linking two sugar residues; and cross-ring cleavages that involve rupturing any two bonds on the same sugar residue. The former provide information pertaining mainly to the sequence and branching, while the latter may reveal additionally some details on a linkage.
Recently, we have exploited the potential of MALDI-TOF/TOF to investigate linear oligosaccharides and branched oligosaccharides including high-mannose-type N-glycans derived from ribonuclease B.29 Extensive cross-ring fragmentation ions, which are very informative on the linkages of the monosaccharide residues forming these molecules, were readily observed in the MALDI-TOF/TOF-MS/MS spectra of oligosaccharides. These ions, in some cases, were more abundant than the commonly observed Y and B ions. The A-type ions observed for the simple oligosaccharides allowed the distinction between α1-4- and α1-6-linked isobaric structures. This distinction was not based merely on the differences in the type of ions formed, but also on their abundances. For example, the α1-4- and α1-6-linked isobaric structures both produce ions resulting from the loss of ca. 120 Da, but with different abundances, as they correspond to two different ions (i.e., 0,4A and 2,4A ions), which require different energies of formation. Highly abundant A- and X-type ions were also observed for the high-mannose N-glycans, allowing the determination of linkages. In addition, the high spectral resolution furnished by MALDI-TOF/TOF also allowed determination of certain ions that had previously been overlooked by MALDI-TOF or MALDI magnetic sector instruments as a result of their low resolution. Moreover, the fact that MS/MS spectra for the precursor ions and all product ions are acquired under the same experimental conditions permitted an accurate and simultaneous determination of the molar ratios of isomeric glycans in any mixture through MALDI-TOF/TOF-MS/MS. Quite recently, this technique has also been demonstrated to be highly effective for the characterization of neutral carbohydrates.30-32
In this study, we wish to describe the utility of MALDI-TOF/TOF-MS/MS for the analysis of sialylated oligosaccharides. Analyses of the sialylated structures have been made possible through permethylation which effectively stabilizes the sialic acid moieties of oligosaccharides. This derivatization eliminates the negative charge associated with such sialylated structures, thus permitting their analysis in the positive-ion mode. The studies presented here show the characterization of cross-ring cleavages of sialylated oligosaccharides as well as the glycosidic cleavages. Additionally, we discuss the formation of fragments pertaining only to the α2-6-linked sialic acid moiety, which permits the differentiation between the isomeric sialylated oligosaccharide structures.
EXPERIMENTAL
Materials
3′-Sialyllactose, 6′-sialyllactose of human origin, as well as the N-acetylneuraminyl-lacto-N-neo-tetraose a (LST a), N-acetylneuraminyl-lacto-N-neo-tetraose c (LST c), and N-acetylneuraminyl-lacto-N-neo-hexaose (all from human origin), were purchased from Sigma (St. Louis, MO, USA). The MALDI matrix, 2,5-dihydroxybenzoic acid (DHB), and sodium hydroxide beads were purchased from Aldrich (Milwaukee, WI, USA). Chloroform and iodomethane were from EM Science (Gibbstown, NJ, USA), while dimethyl sulfoxide (DMSO) was from Sigma.
Permethylation
All glycans were permethylated adopting the method of Ciucanu and coworkers.33,34 Briefly, iodomethane, a trace of water, and sodium hydroxide powder were suspended in DMSO and mixed at room temperature for 10 min at room temperature. Typically, a 1–10 μg sample aliquot was suspended in 30 μL of DMSO, to which 3.6 mg of sodium hydroxide powder, 0.3 μL of water and 5.6 μL of iodomethane were added. The permethylated glycans and oligosaccharides were subsequently extracted from the mixture with chloroform and washed repeatedly with cold water prior to evaporation. Ice-cold water was added to the derivatization mixture and placed in an ice bath prior to the addition of chloroform. The mixture was then vortexed for several minutes. The aqueous layer was then discarded and the chloroform layer washed repeatedly with water. The pH of the aqueous layer was continuously monitored with pH indicators. Five times washing with water was sufficient to eliminate residual sodium hydroxide, side products and excess iodomethane.
MALDI spot preparation
The dried permethylated sample was resuspended in 50:50 methanol/water solution containing 1 mM sodium acetate. The sample was spotted directly on the MALDI plate and mixed with the DHB matrix. The DHB matrix was prepared by suspending 10 mg in 1 mL of water to produce 10 mg/mL matrix concentration. Sample and matrix were mixed on the MALDI plate at a ratio of 1:1. The sampled spots were dried under vacuum.
Instrumentation
An 4700 proteomics analyzer from Applied Biosystems (Framingham, MA, USA) was utilized in this study. This TOF/TOF instrument is equipped with an Nd:YAG laser with 355-nm wavelength. The instrument uses a two-stage mirror for a better focusing across the broad energy range of product ions in the MS/MS mode, without sacrificing performance in the MS-only mode. In both MS and MS/MS mode, the instrument laser operates at 200 Hz repetition rate. The MALDI-MS/MS spectra were acquired in the positive-ion mode at 1 kV accelerating voltage difference between sources 1 and 2. The transmission of precursor ions into the collision cell is optimized by the use of a decelerating lens. The precursor mass window was set to ±10 m/z around the precursor mass, thus eliminating the inclusion of underpermethylated products, oxidation products, and potassium adducts. Argon was used as a collision gas, and the collision cell pressure was set to 6.5 × 10−6 or 1 × 10−7 Torr for high- and low-pressure experiments, respectively. The acquired spectra were the average of 1000 laser shots. MS and MS/MS data were further processed using DataExplorer 4.0 (Applied Biosystems).
RESULTS AND DISCUSSION
As mentioned above, the permethylation procedure developed by Ciucanu and coworkers33,34 was utilized in this study to permit efficient permethylation of glycans. Permethylation of samples was employed to eliminate the negative charges associated with the sialylated structures, thus permitting their MS/MS analysis in positive-ion mode. The efficient permethylation of glycan structures not only allowed the analysis of sialylated structures in positive-ion mode, which is on average 1–2 orders of magnitude more sensitive than the negative-ion mode, not to mention the increase of sensitivity associated with permethylation,33,34 but also increased the mass of the analyzed structures. This increase in mass permitted detection of human milk oligosaccharides and O-glycans with native molecular weights less than 800 Da. The matrix cluster region of the spectrum is observed at m/z values lower than 800. All permethylated precursor ions and their product ions were predominantly detected as sodium adducts as a result of the addition of a small amount of sodium acetate to the matrix (Fig. 1(b)).35
Figure 1.
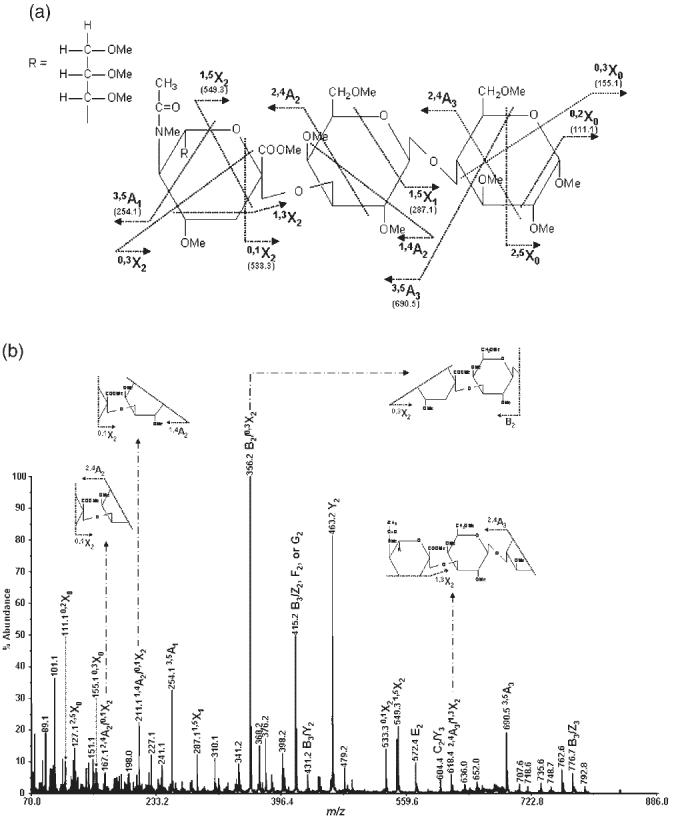
MS/MS spectrum of permethylated 3′-sialyllactose isolated from human milk (precursor ion: m/z 895.7): (a) structure and assignment of cross-ring fragments observed and (b) the spectrum with insets illustrating internal fragments.
The Domon and Costello nomenclature of carbohydrate fragmentation was employed in this study,36 with addition of the new nomenclature for the mechanisms of fragmentation described by Spina et al.30 and discussed by Stephens and coworkers.31 According to this nomenclature, the ions retaining the charge at the reducing terminus are designated as X for cross-ring cleavages, and Y and Z for glycosidic bond cleavages. Those retaining a charge at the nonreducing terminus are designated as A for cross-ring cleavages, and B and C for glycosidic bond cleavages. Ions are designated by a subscript number that follows the letter showing the fragment type. Sugar rings are numbered from the nonreducing end for A, B, and C ions and from the reducing end for the others. Greek letters are used to distinguish fragments from branched-chain glycans, with the letter α representing the largest branch. In the case of ring cleavages, superscript numbers are given to show the cleaved bonds. In addition, ions produced as a result of more than one cleavage are designated with a slash between sites of cleavages (e.g., B5/0,1A5). Other fragments which involve six-atom rearrangement are labeled E, F and G.30,31 E and F fragments entail the elimination of the substituent in position 2 of the 3-linked monosaccharide or the terminal monosaccharides, respectively. On the other hand, G fragments entail the elimination of 3-linked and 4-linked monosaccharides.
MS/MS of permethylated sialylated human milk oligosaccharides
A challenging aspect of the structural determination of sialylated glycans has been the inability to conclusively determine the antenna to which the sialic acid residues are attached, as well as the inability to distinguish the presence of structural isomers with sialylation on different antennae using MS either alone, or in combination with enzymatic sequencing. The lability of sialic acid residues is the most serious obstacle that must be overcome prior to any successful use of MS/MS analysis in the characterization of sialylated structures. Generally, MS/MS of sialylated glycans on a MALDI-TOF/TOF instrument yielded only the fragments that retain the sialic acid on the nonreducing end of the structure. No other fragments could be observed, as expected, since the sialic acid residue is the site of a charge on the precursor ion or any resulting fragment. This is attributed to the ease with which sialic acid residues, possessing the negative charge needed for adsorption/ionization and subsequent detection, are cleaved. Accordingly, losing these moieties totally eliminates their gas-phase presence and their subsequent detection. A suitable alternative appears to be a derivatization approach, which renders the otherwise negatively charged structures amenable to positive-ion MALDI-TOF/TOF-MS analysis.
In this study, we exploit the potential of permethylation for the characterization of sialylated glycan structures by MALDI-TOF/TOF-MS/MS. In such analyses, all polar moieties of sialylated oligosaccharides are effectively derivatized. This derivatization primarily eliminates the negative charge associated with such sialylated structures, thus permitting their analysis in the positive-ion mode. The permethylated, sialylated glycans all demonstrated characteristic cross-ring cleavages, in addition to the glycosidic bond cleavages. Importantly, this technique allows the distinction between α2-3- and α2-6-linked sialic acid residues, as discussed below.
The MS/MS spectrum of permethylated 3′-sialyllactose (for structure, see Fig. 1(a)) is illustrated in Fig. 1(b), while the corresponding spectrum of permethylated 6′-sialyllactose (for structure, see Fig. 2(a)) is shown in Fig. 2(b). The abundance of cross-ring fragmentation is apparent in both spectra. Cross-ring fragments attributed to α2-3 linkage, such as 1,4A 2 in 1,4A2/0,1X2 and 2,4A3 in 2,4A3/0,3X2, are distinguishable in the MS/MS spectrum of permethylated 3′-sialyllactose (Fig. 1(b)). These fragments are not distinguishable from the isobaric 3,5A2/2,5X2 and 2,4A2/1,3X2 ions which are present in the MS/MS spectrum of permethylated 6′-sialyllactose (Fig. 2(b)). However, ions at m/z 458.2, 486.3 and 588.4, corresponding to 0,4A2, 3,5A2 and 2,4A3/2,4X1 fragments, respectively, are only observable in the MS/MS spectrum of 6′-sialyllactose and are attributed to the α2-6 linkage of the sialic acid residue in this structure. Accordingly, the presence of these cross-ring fragments easily permits the distinction between the 3′- and 6′-sialyllactose isobaric structures which differ only in the sialic acid linkages. These characteristic fragmentations were observed previously by FAB-MS.6-8
Figure 2.
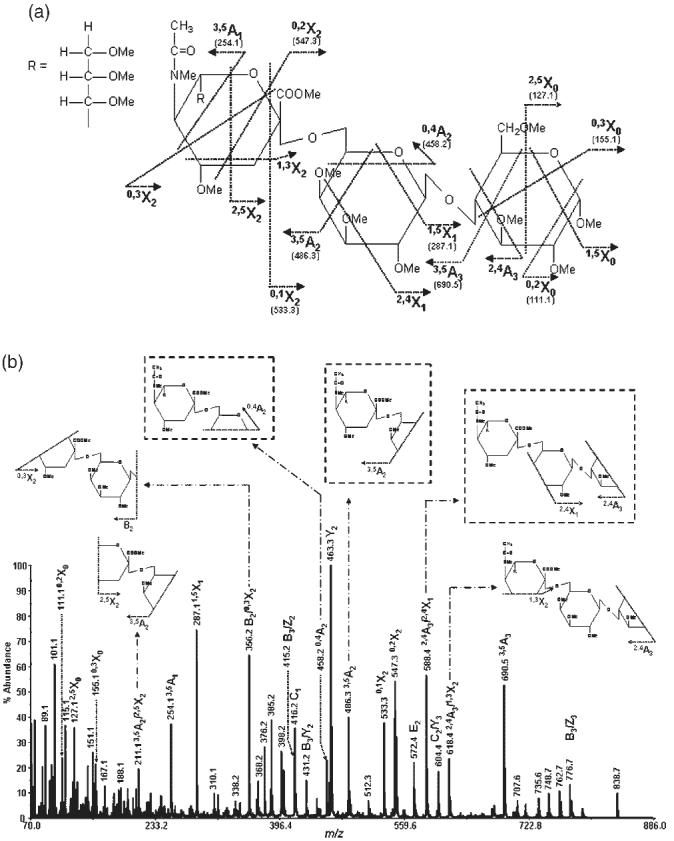
MS/MS spectrum of permethylated 6′-sialyllactose isolated from human milk (precursor ion: m/z 895.7): (a) structure and assignment of cross-ring fragments observed and (b) the spectrum with insets illustrating internal and characteristic fragments.
Moreover, an ion at m/z 416.2 is only observable in the MS/MS spectrum of the 6′-sialyllactose structure (Fig. 3). This ion corresponds to the C1 fragment which does not form in the case of the 3′-sialyllactose structure. Although an ion at m/z 416 is observed in the MS/MS spectrum of the 3′-sialyllactose structure (Fig. 3(a)), it corresponds to the C13 isotope of the ion at m/z 415 (corresponding to B3/Z2, F2 or G2 fragment), as deduced from the natural isotopic abundance. On the other hand, the higher abundance of the ion observed at m/z 416 in the MS/MS spectrum of the 6′-sialyllactose structure (Fig. 3(b)) indicates the presence of an ion other than the C13 isotope of the ion at m/z 415. This suggests that the difference in the stability of Z ions produced for the 3′ and 6′ structures accounts for the formation of the stable C1 ion in the case of the 6′ structure, but not in the 3′ structure. In other words, the Z ion produced in the case of the 3′ structure is expected to be very stable and exclusively retain the sodium ion, consequently eliminating the detection of the C1 ion formed in this case. On the other hand, it seems that the Z ion formed in the case of the 6′ structure is as stable as the C1 fragments which would result in an equal possibility of either structures retaining the sodium ion; consequently, both structures are being observed. This fragment was not observed in the FAB-MS studies6-8 due to lack of sufficient mass resolution.
Figure 3.

Zoomed MS/MS spectra of permethylated 3′-sialyllactose (A) and 6′-sialyllactose (B) to illustrate the C1 fragment.
This ability to distinguish between isobaric structures was not only limited to these structures, but was also observed in the case of many other structures including LST a (for structure, see Fig. 4(a)) and LST c (for structure, see Fig. 5(a)), two pentasaccharides differing in the linkage of the terminal sialic acid and the internal linkage between GlcNAc and Gal residues. The MALDI-TOF/TOF-MS/MS spectra of these structures are depicted in Figs. 4(b) and 5(b), respectively. Once again, the abundance of cross-ring fragmentation is noticeable in both MS/MS spectra. The diagnostic α2-6 linkage ions, namely 0,4A2, 3,5A2, and 2,4A3/2,4X3, are observed in the MS/MS spectrum of LST c (Fig. 5(b)), but not in that of LST a (Fig. 4(b)). Accordingly, it can be concluded from the MS/MS spectra that the LST c structure possesses a terminal sialic acid residue that is α2-6-linked, while the LST a structure has its terminal sialic acid α2-3-linked. Moreover, C1 ions (m/z 416.1) are detected in the MS/MS spectrum of the LST c structure, but not with LST a.
Figure 4.

MS/MS spectrum of permethylated N-acetylneuraminyl-lacto-N-neo-tetraose isolated from human milk (precursor ion: m/z 1287.6): (a) structure and observed cross-ring fragments and (b) the spectrum with insets illustrating internal fragments.
Figure 5.
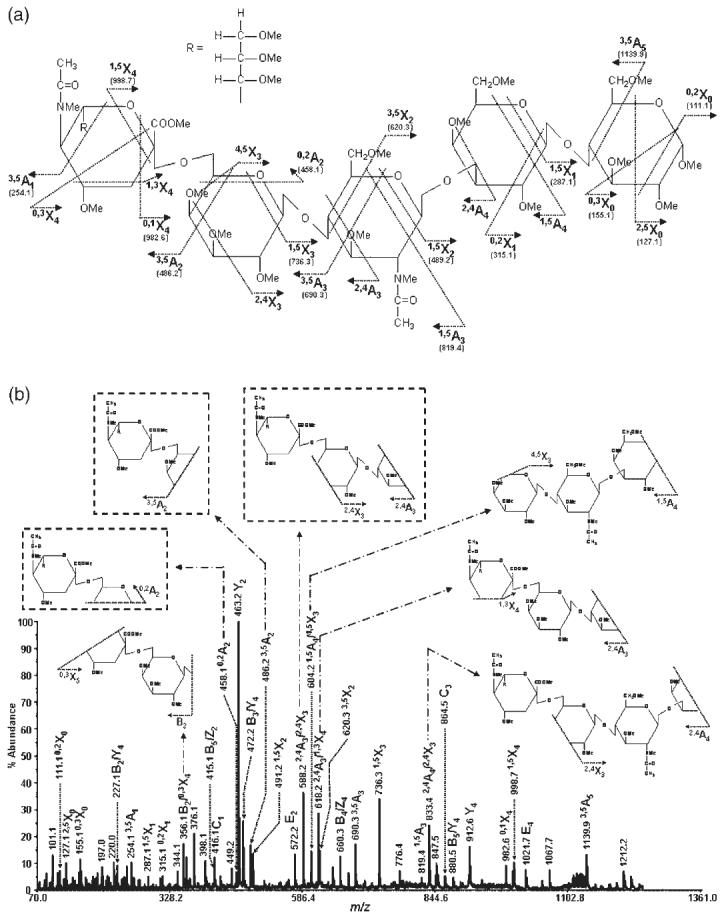
MS/MS spectrum of permethylated N-acetylneuraminyl-lacto-N-neo-tetraose c isolated from human milk (precursor ion: m/z 1287.6): (a) structure and observed cross-ring fragments and (b) the spectrum with insets illustrating internal and characteristic fragments.
Thus far, we have demonstrated the ability to assign sialic acid linkages by MALDI-TOF/TOF-MS/MS. The next example will illustrate the ability of this technique to assign sialic acid residues to a particular antenna. Nuclear magnetic resonance (NMR) spectrometry is currently the only analytical technique known to be capable of assigning the antenna to which sialic acid residues are attached. MS, in conjunction with enzymatic digestion, is not sufficiently specific to determine the antenna to which sialic acid residues are attached. N-Acetylneuraminyl-lacto-N-neohexaose from human milk (for structure, see Fig. 6(a)) is a branched sialylated oligosaccharide which has both a 6-linked antenna and a 3-linked antenna. The sialic acid residue in this structure is present on the 3-linked antenna. The MS/MS spectrum of N-acetylneuraminyl-lacto-N-neohexaose is shown in Fig. 6(b). The most informative fragments in the spectra are 1,5X2β, 3,5X1, 0,4A4, and 2,4A4/2,4X3α. These fragments conclusively allowed the assignment of the sialic acid residue to the 3-linked antenna. Furthermore, the presence of the diagnostic fragments associated with α2-6 linkage, as discussed above, as well as the presence of the C1 fragment (m/z 416.1), suggests an attachment of the terminal sialic acid residue to be through a α2-6 linkage. Accordingly, MALDI-TOF/TOF-MS/MS allows the determination of linkages and branching of glycan structures through the ability to produce cross-ring fragments that conclusively permit the assignment of such structures.
Figure 6.
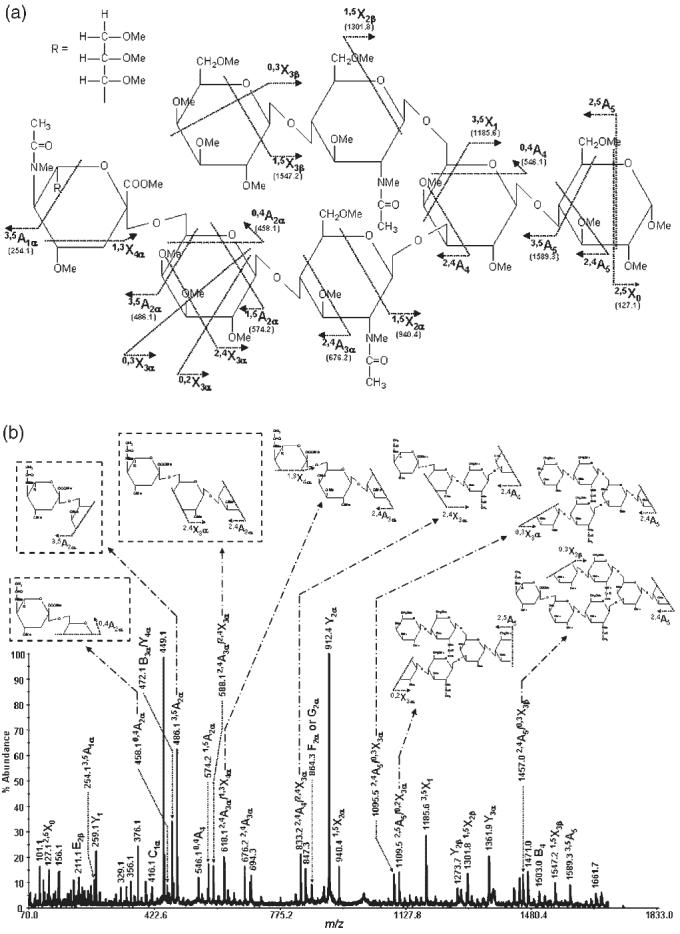
MS/MS spectrum of permethylated N-acetylneuraminyl-lacto-N-neo-hexaose isolated from human milk (precursor ion: m/z 1736.8): (a) structure and observed cross-ring fragments and (b) the spectrum with insets illustrating internal and characteristic fragments.
CONCLUSIONS
While branching, different linkages and other forms of sugar isomerism are often important in the biological functions of glycoconjugates, they present a formidable challenge in structural studies. The previous investigations involving MALDI-TOF/TOF-MS in oligosaccharide analysis have demonstrated the merits of high-energy CID in fragmenting reproducibly neutral oligosaccharide molecules into diagnostically useful ionic entities. The permethylation of oligosaccharides combined with this form of MS/MS analysis has now extended our structural capabilities to biologically important sialylated structures. Most notably, characteristic A- and X-type ions, generated in our studies from sialylated oligosaccharides, allow the distinction of their linkages. This information is usually not available without the use of tedious and indirect procedures. Additionally, the demonstrated ability to detect diagnostically important ions for sialic acid residues allowed the definitive determination of the attachment antenna which has traditionally been achieved primarily through NMR measurements.
The stabilization effect of permethylation on glycan molecules has the positive consequences of enhanced measurement sensitivity and the capability of addressing complex mixtures of oligosaccharides (neutral and acidic) in the positive-ion MS mode. Conveniently, the oligosaccharides are sufficiently hydrophobic for a prior separation by reversed-phase capillary liquid chromatography (LC)37,38 and a microdeposition on MALDI plate26 for MS/MS analysis. Combined with suitable microscale cleavages,39-41 capillary LC/MALDI-TOF/TOF-MS appears potentially to provide a powerful analytical platform for exploratory studies in functional glycomics.
Acknowledgements
This work was supported by Grant No. GM24349 from the National Institute of General Medical Sciences, U.S. Department of Health and Human Services and a center grant from the Indiana 21st Century Research and Technology Fund. In addition, the mass spectrometer used in this study was facilitated through the Indiana Genomics Initiative (INGEN), which is funded in part by the Lilly Endowment, Inc.
Footnotes
Contract/grant sponsor: National Institute of General Medical Sciences, U.S. Department of Health and Human Services; contract/grant number: GM24349.
REFERENCES
- 1.Mechref Y, Novotny MV. Chem. Rev. 2002;102:321. doi: 10.1021/cr0103017. [DOI] [PubMed] [Google Scholar]
- 2.Mechref Y, Novotny MV. Anal. Chem. 1998;70:455. doi: 10.1021/ac970947s. [DOI] [PubMed] [Google Scholar]
- 3.Domon B, Müller DR, Richter WJ. Biomed. Environ. Mass Spectrom. 1990;19:390. doi: 10.1002/bms.1200150805. [DOI] [PubMed] [Google Scholar]
- 4.Domon B, Müller DR, Richter WJ. Int. J. Mass Spectrom. Ion Processes. 1990;100:301. [Google Scholar]
- 5.Garozzo D, Giuffrida M, Impallomeni G, Ballistreri A, Montaudo G. Anal. Chem. 1990;62:279. [Google Scholar]
- 6.Boulenguer P, Leroy Y, Alonso JM, Montreuil J, Ricart G, Colbert C, Duquet D, Dewaele C, Fournet B. Anal. Biochem. 1988;168:164. doi: 10.1016/0003-2697(88)90024-3. [DOI] [PubMed] [Google Scholar]
- 7.Lemoine J, Strecker G, Leroy Y, Fournet B, Ricart G. Carbohydr. Res. 1991;221:209. doi: 10.1016/0008-6215(91)80057-t. [DOI] [PubMed] [Google Scholar]
- 8.Lemoine J, Fournet B, Despeyroux D, Jenning KR, Rosenberg R, de Hoffmann E. J. Am. Soc. Mass Spectrom. 1993;4:197. doi: 10.1016/1044-0305(93)85081-8. [DOI] [PubMed] [Google Scholar]
- 9.Spengler B, Dolce JW, Cotter RJ. Anal. Chem. 1990;62:1731. doi: 10.1021/ac00207a004. [DOI] [PubMed] [Google Scholar]
- 10.Carroll JA, Ngoka L, Mccullough S, Gard E, Jones AD, Lebrilla CB. Anal. Chem. 1991;63:2526. [Google Scholar]
- 11.Carroll JA, Ngoka L, Beggs CG, Lebrilla CB. Anal. Chem. 1993;65:1582. doi: 10.1021/ac00059a017. [DOI] [PubMed] [Google Scholar]
- 12.Harvey DJ, Rudd PM, Bateman RH, Bordoli RS, Howes K, Hoyes JB, Vickers RG. Org. Mass Spectrom. 1994;29 [Google Scholar]
- 13.Harvey DJ, Naven TJP, Kuster B, Bateman RH, Green MR, Critchley G. Rapid Commun. Mass Spectrom. 1995;9:1556. doi: 10.1002/rcm.1290091517. [DOI] [PubMed] [Google Scholar]
- 14.Viseux N, deHoffmann E, Domon B. Anal. Chem. 1997;69:3193. doi: 10.1021/ac961285u. [DOI] [PubMed] [Google Scholar]
- 15.Weiskopf AS, Vouros P, Harvey DJ. Rapid Commun. Mass Spectrom. 1997;11:1493. doi: 10.1002/(SICI)1097-0231(199709)11:14<1493::AID-RCM40>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 16.Naven TJP, Harvey DJ, Brown J, Critchley G. Rapid Commun. Mass Spectrom. 1997;11:1681. doi: 10.1002/(SICI)1097-0231(19971015)11:15<1681::AID-RCM34>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 17.Harvey DJ. Mass Spectrom. Rev. 1999;18:349. doi: 10.1002/(SICI)1098-2787(1999)18:6<349::AID-MAS1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 18.Mechref Y, Novotny MV. Carbohydr. Res. 1999;313:145. doi: 10.1016/s0008-6215(98)00264-x. [DOI] [PubMed] [Google Scholar]
- 19.Weiskopf AS, Vouros P, Harvey DJ. Anal. Chem. 1998;70:4441. doi: 10.1021/ac980289r. [DOI] [PubMed] [Google Scholar]
- 20.Sheeley DM, Reinhold VN. Anal. Chem. 1998;70:3053. doi: 10.1021/ac9713058. [DOI] [PubMed] [Google Scholar]
- 21.Solouki T, Reinhold BB, Costello CE, O'Malley M, Guan SH, Marshall AG. Anal. Chem. 1998;70:857. doi: 10.1021/ac970562+. [DOI] [PubMed] [Google Scholar]
- 22.Harvey DJ. J. Am. Soc. Mass Spectrom. 2000;11:900. doi: 10.1016/S1044-0305(00)00156-2. [DOI] [PubMed] [Google Scholar]
- 23.Harvey DJ. J. Mass Spectrom. 2000;35:1178. doi: 10.1002/1096-9888(200010)35:10<1178::AID-JMS46>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 24.Harvey DJ, Bateman RH, Bordoli RS, Tyldesley R. Rapid Commun. Mass Spectrom. 2000;14:2135. doi: 10.1002/1097-0231(20001130)14:22<2135::AID-RCM143>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 25.Hunnam V, Harvey DJ, Priestman DA, Bateman RH, Bordoli RS, Tyldesley R. J. Am. Soc. Mass Spectrom. 2001;12:1220. doi: 10.1016/S1044-0305(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 26.Tegeler TJ, Mechref Y, Boraas K, Reilly JP, Novotny MV. Anal. Chem. 2004;76:6698. doi: 10.1021/ac049341b. [DOI] [PubMed] [Google Scholar]
- 27.Harvey DJ, Hunter AP, Bateman RH, Brown J, Critchley G. Int. J. Mass Spectrom. 1999;188:131. [Google Scholar]
- 28.Reinhold VN, Reinhold BB, Costello CE. Anal. Chem. 1995;67:1772. doi: 10.1021/ac00107a005. [DOI] [PubMed] [Google Scholar]
- 29.Mechref Y, Novotny MV, Krishnan C. Anal. Chem. 2003;75:4895. doi: 10.1021/ac0341968. [DOI] [PubMed] [Google Scholar]
- 30.Spina ESL, Romeo D, Impallomeni G, Garozzo D, Waidelich D, Glueckmann M. Rapid Commun. Mass Spectrom. 2004;18:392. doi: 10.1002/rcm.1350. [DOI] [PubMed] [Google Scholar]
- 31.Stephens E, Maslen SL, Gree LG, Williams DH. Anal. Chem. 2004;76:2343. doi: 10.1021/ac030333p. [DOI] [PubMed] [Google Scholar]
- 32.Morelle W, Slomianny M-C, Diemer H, Schaeffer C, van Dorsselaer A, Michalski J-C. Rapid Commun. Mass Spectrom. 2004;18:2637. doi: 10.1002/rcm.1668. [DOI] [PubMed] [Google Scholar]
- 33.Ciucanu I, Kerek F. Carbohydr. Res. 1984;131:209. [Google Scholar]
- 34.Ciucanu I, Costello CE. J. Am. Chem. Soc. 2003;125:16213. doi: 10.1021/ja035660t. [DOI] [PubMed] [Google Scholar]
- 35.Chen P, Baker AG, Novotny MV. Anal. Biochem. 1997;244:144. doi: 10.1006/abio.1996.9874. [DOI] [PubMed] [Google Scholar]
- 36.Domon B, Costello CE. Glycoconjugate J. 1988;5:397. [Google Scholar]
- 37.Delaney J, Vouros P. Rapid Commun. Mass Spectrom. 2001;15:325. doi: 10.1002/rcm.230. [DOI] [PubMed] [Google Scholar]
- 38.Kang P, Mechref Y, Klouckova I, Novotny MV. Rapid Commun. Mass Spectrom. 2005;19:3421. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang Y, Konse T, Mechref Y, Novotny MV. Rapid Commun. Mass Spectrom. 2002;16:1199. doi: 10.1002/rcm.701. [DOI] [PubMed] [Google Scholar]
- 40.Huang Y, Mechref Y, Novotny MV. Anal. Chem. 2001;73:6063. doi: 10.1021/ac015534c. [DOI] [PubMed] [Google Scholar]
- 41.Palm AK, Novotny MV. Rapid Commun. Mass Spectrom. 2005;19:1730. doi: 10.1002/rcm.1979. [DOI] [PubMed] [Google Scholar]