


Abstract
Human ribonuclease-1 (hRNase-1) is an extracellular enzyme found in exocrine pancreas, blood, milk, saliva, urine and seminal plasma, which has been implicated in digestion of dietary RNA and in antiviral host defense. The enzyme is characterized by a high catalytic activity toward both single-stranded and double-stranded RNA. In this study, we explored the possibility that hRNase-1 may also be provided with a ribonuclease H activity, i.e. be able to digest the RNA component of RNA:DNA hybrids. For this purpose, we developed an accurate and sensitive real-time RNase H assay based on a fluorogenic substrate made of a 12 nt 5′-fluorescein-labeled RNA hybridized to a complementary 3′-quencher-modified DNA. Under physiological-like conditions, hRNase-1 was found to cleave the RNA:DNA hybrid very efficiently, as expressed by a kcat/Km of 330 000 M−1 s−1, a value that is over 180-fold higher than that obtained with the homologous bovine RNase A and only 8-fold lower than that measured with Escherichia coli RNase H. The kinetic characterization of hRNase-1 showed that its hybridase activity is maximal at neutral pH, increases with lowering ionic strength and is fully inhibited by the cytosolic RNase inhibitor. Overall, the reported data widen our knowledge of the enzymatic properties of hRNase-1 and provide new elements for the comprehension of its biological function.
INTRODUCTION
Human ribonuclease-1 (hRNase-1), also known as human pancreatic ribonuclease, is a basic protein of 128 amino acid residues (1,2). It belongs to the superfamily of extracellular ribonucleases of tetrapods (3) that counts eight members in humans (4). The enzyme represents the human counterpart of the well-known bovine pancreatic RNase A (5,6) with which it shares 70% sequence identity and the ability to cleave RNA specifically on the 3′ side of pyrimidine bases (7). Despite the high degree of sequence identity, hRNase-1 differentiates from the bovine enzyme for a very high activity toward double-stranded RNA (dsRNA) (8). Under physiological-like conditions, the kcat/Km of hRNase-1 for cleavage of the homopolymeric substrate poly(rA):poly(rU) is 148 500 M−1 s−1, a value that is ∼500-fold higher than that measured with RNase A (9). The activity of the human enzyme toward single-stranded RNA (ssRNA) is instead few-fold lower than that of RNase A. Few other members of the ribonuclease superfamily are able to digest dsRNA but less efficiently than hRNase-1. Among them, whale pancreas RNase whose activity towards poly(rA):poly(rU) is ∼10-fold lower than that of hRNase-1 (8). Mutational studies have shown that the potent activity of hRNase-1 on dsRNA is related to the presence of noncatalytic basic residues which cooperatively contribute to the binding and destabilization of the double-helical RNA molecule (9).
Human RNase-1 is expressed at high levels in exocrine pancreas and in endothelial cells (10,11), from which it is believed to be secreted in duodenum and in blood plasma, respectively. Plasma concentration of hRNase-1 has been estimated to be ∼0.4 mg/l (11). The enzyme is also expressed in testis, ovary, brain, mammary gland and other tissues (10,12), and is found in other body fluids such as milk, urine, saliva and seminal plasma (13–15). The fraction of hRNase-1 that is produced by the pancreas is likely responsible for the digestion of dietary RNA (16) whereas the hRNase-1 found in blood plasma and in the other body fluids plays a role yet to be assessed.
Many viruses have dsRNA genomes or ssRNA genomes that replicates through dsRNA intermediates. These include flavivirus, picornavirus and influenza virus whose replication is assisted by an RNA-dependent RNA polymerase. Mammals have evolved a variety of defense mechanisms in response to exposition to dsRNA. These include the interferon system and the intracellular degradation of dsRNA operated by Dicer (17). Since hRNase-1 is very effective in digesting dsRNA and since is present in blood and other body fluids, it has been proposed that the enzyme plays an antiviral role (9,11). In this regard, other human extracellular ribonucleases have been found recently to play not-digestive roles and to be involved in host defense. Human RNase-3 (also named eosinophil cationic protein) (18) has helminthotoxic and anti-bacterial activity (19,20) and hRNase-7 from human skin has a very potent antimicrobial activity against various pathogenic microorganisms, including Staphylococcus aureus, Pseudomonas aeruginosa, Propionibacterium acnes and Candida albicans (21). It should also be noted that hRNase-1 has been shown to suppress the replication of HIV in cultured lymphocytes (22).
A wider characterization of the enzymatic properties of hRNase-1 may provide new elements for the comprehension of the biological function of the enzyme. Given the peculiar ability of hRNase-1 to bind and cleave dsRNA, we considered another double-helical substrate, an RNA:DNA hybrid and tested the enzyme for ribonuclease H activity (23,24), i.e. for the ability to digest the RNA component of RNA:DNA hybrids.
MATERIALS AND METHODS
Materials and general procedures
Water, buffers and salt solutions were made RNase-free by filtration through a Sep-Pak C18 column (Waters) activated with 5 ml of methanol. Acetylated BSA, RNase A and human plasma were obtained from Sigma. RNase A was quantified spectrophotometrically using a ɛ278 value of 9800 M−1 cm−1 (25). Human RNase-1 was obtained from a recombinant expression system in Escherichia coli (26) and stored in 0.1 M Tris–HCl (pH 8.0), containing 0.3 M NaCl, as it is poorly soluble at low ionic strength. The stock concentration of hRNase-1 was determined by amino acid analysis. Cytosolic RNase inhibitor was obtained from Promega. E.coli RNase H was from USB. Its stock concentration was 5 U/µl, which is equivalent to 12 ng/µl (27). Fluorescence measurements were carried out on a Perkin-Elmer LS50B Luminescence Spectrometer equipped with semimicro (1.5 ml) glass cuvettes. Ion-pair reversed-phase high pressure liquid chromatography (HPLC) of oligonucleotides was performed using a Symmetry® C18 column (Waters) thermostated at 65°C and a 13 min linear gradient of acetonitrile concentration from 10 to 40% in 0.1 M triethylammonium acetate (pH 7.0), at a flow rate of 0.9 ml/min. The absorbance was monitored at 260 nm.
Preparation of the substrate
The RNA strand, fluorescein-5′-rA-rA-rC-rA-rG-rG-rA-rG-rG-rA-rG-rG-3′ and the DNA strand, 5′-dC-dC-dT-dC-dC-dT-dC-dC-dT-dG-dT-dT-3′-dabcyl, were custom synthesized on a 1 µmol scale by MWG Biotech. Briefly, the RNA oligo was prepared via the 5′-silyl-2′-acetoxy ethyl orthoester chemistry, purified by polyacrylamide gel electrophoresis, and provided in the stable 2′-orthoester protected form. It was then deprotected by mild acidic treatment, according to the manufacturer's instructions, and quantified spectrophotometrically using a ɛ260 value of 154 700 M−1 cm−1. The DNA oligo was synthesized by standard phosphoramidite chemistry, purified by reversed-phase HPLC and quantifed using a ɛ260 value of 102 700 M−1 cm−1. The two oligonucleotides were then mixed at 10 µM concentration each in 50 mM Tris–HCl (pH 7.4), containing 50 mM KCl, heated at 80°C for 5 min, and cooled at room temperature over a 2 h period to allow hybrid formation. The resulting substrate solution was stored in aliquots at −20°C.
Enzyme kinetics
Hybridase assays were performed at 37°C in 1 ml of the specified buffer containing 10–400 nM RNA:DNA hybrid. Initial rates of substrate cleavage were determined by measuring the rate of change in fluorescence intensity at 520 nm upon excitation at 490 nm. Initial rates in units of fluorescence intensity per second were converted to moles per liter per second by dividing by the maximum change in fluorescence intensity and multiplying by the initial concentration of the substrate. The maximum change in fluorescence intensity for each substrate concentration and pH was obtained by incubation with a large excess of the enzyme under testing. Initial rates were determined by considering only those portions of the reaction progress curves that yielded <7% of substrate cleavage. Values of kcat/Km were calculated from the equation kcat/Km = V0/[E][S] by measuring the initial rates of reaction at a substrate concentration, 10 nM, that is well below Km of all the enzymes tested. This was verified by running control assays at 3- to 4-fold higher substrate concentrations that yielded proportionally increased V0 values. Ribonucleolytic assays toward the isolated RNA strand of the hybrid were performed in 100 µl of 40 mM Tris–HCl (pH 7.4), containing 60 mM NaCl, 60 mM KCl, 20 µg/ml BSA, 1 µM fluorescein-5′-rA-rA-rC-rA-rG-rG-rA-rG-rG-rA-rG-rG-3′ and 0.3 nM hRNase-1. After 20–40 min of incubation at 37°C, substrate and products were quantified by ion-pair reversed-phase HPLC, performed as described in Materials and Methods except that the acetonitrile gradient ranged from 10 to 15% in 5 min. Values of kcat/Km were then calculated from the formula kcat/Km = ln ([S0]/[S])/t[E], were [S0] and [S] are substrate concentrations at the initial time and time t, respectively. The reported value of kcat/Km of hRNase-1 for cleavage of the RNA strand is the mean ± SEM of six measurements. Control assays at 3 µM substrate concentration yielded values of kcat/Km undistinguishable from those obtained at 1 µM.
RESULTS AND DISCUSSION
Design and preparation of a fluorogenic RNA:DNA hybrid
Traditional methods for assaying ribonuclease H activity are largely based on radiolabeled substrates and are discontinuous (28,29). Assay systems for real-time detection of RNase H activity have been developed only recently. In 2002, Rizzo et al. (27) reported an assay method for E.coli RNase H based on the fluorescence quenching mechanism of molecular beacons. The substrate was a single-stranded hairpin RNA:DNA chimeric oligonucleotide comprising homopolymeric 5′-fluorescein-oligo(rA) duplexed with oligo(dT)-3′-dabcyl. The fluorophore of the substrate was held in close proximity to the quencher (dabcyl) by the RNA:DNA stem–loop structure. When the RNA sequence of the RNA:DNA hybrid stem was cleaved, the fluorophore was separated from the quencher producing an increase in fluorescence. This assay permits accurate measurement of RNase H activity but cannot be easily implemented since the substrate is not commercially obtainable. It should also be noted that the RNA component of this substrate, made of poly(rA), doesn't meet the substrate specificity of hRNase-1 that cleaves at pyrimidine sites. In 2003, Parniak et al. (30) reported a similar fluorometric assay based on commercially obtainable oligonucleotides. The substrate was a 18 nt 3′-fluorescein-labeled RNA annealed to a complementary 18 nt 5′-dabcyl-modified DNA. The RNA sequence, containing eight pyrimidines, and the overall structure of this substrate had been optimized for testing the HIV reverse transcriptase-associated RNase H (30). This feature makes the assay poorly suitable to kinetic studies with hRNase-1 because this enzyme, if endowed with RNase H activity, will likely cleave the substrate at the several pyrimidine sites making the measurement of true kcat or kcat/Km values very difficult.
In order to develop a real-time RNase H assay suitable to test hRNase-1, we designed a new fluorogenic substrate based on commercially obtainable oligonucleotides and provided with a single cleavable bond (Figure 1). The double-helical hybrid is made of a 12 nt 5′-fuorescein-labeled RNA hybridized to a complementary 12 nt 3′-dabcyl-modified DNA. The melting temperature (Tm) of the hybrid was predicted to be 52°C, a value high enough to ensure a good stability of the secondary structure at the assay temperature (37°C). This is important to minimize the baseline fluorescence signal by ensuring that the fluorophore and the quencher stay in close proximity. The RNA strand of the hybrid contains only one pyrimidine, three bases apart from the 5′ end, where the cleavage by hRNase-1 is expected to occur (Figure 1). Upon cleavage, the resulting fluorescein-labeled trinucleotide is expected to dissociate readily from the complementary DNA, producing a large increase in fluorescence emission at 520 nm upon excitation at 490 nm. On the basis of these theoretical considerations, the two oligonucleotides were custom synthesized, purified and annealed to form the double-helical substrate.
Figure 1.

Structure of the fluorogenic RNA:DNA hybrid and principle of the ribonuclease H assay. In the intact substrate the fluorophore (fluorescein) and the quencher (dabcyl) are in close proximity and fluorescence is hence quenched. Upon cleavage of the RNA strand, the fluorophore is liberated from the hybrid, resulting in a large increase in fluorescence.
Evaluation of the substrate with E.coli RNase H and specificity tests
As a first step in the evaluation of the newly prepared substrate, an aliquot of it was excited at 490 nm and an emission spectrum was recorded. As shown in Figure 2, emission at 520 nm is very weak, suggesting that the dabcyl efficiently quenches the emission of the fluorescein label. The substrate was then treated with an excess of E.coli RNase H (31), used as a positive control of the enzymatic reaction, and a new spectrum was recorded (Figure 2). As shown, the complete degradation of the substrate produced a large fluorescence enhancement, with an emission signal at 520 nm that is 19-fold higher than that of the intact substrate. This result indicates that the substrate is suitable to test the enzymatic activity of RNases H. Kinetic measurement were then performed at lower concentrations of E.coli RNase H, within the pM range and fluorescence was measured continuously soon after the addition of the enzyme. As shown in Figure 3, the resulting fluorescence increases are time-dependent and RNase H concentration-dependent. Each reaction progress trace shows a good initial linearity and enzyme concentrations as little as 28 pM can be easily assayed. This enzyme concentration is at least 75-fold lower that those tested in the previously reported molecular beacon assay for E.coli RNase H (27). Overall, the kinetic data were used to measure an apparent kcat/Km value of 2.54 × 106 M−1 s−1 (Table 1).
Figure 2.

Fluorescence emission spectra of the substrate and its cleavage products. Spectra of the substrate before (dashed line) and after (solid line) treatment with E.coli RNase H were recorded at 37°C with excitation at 490 nm. Digestion had been performed in 40 mM Tris–HCl (pH 7.4), containing 60 mM NaCl, 60 mM KCl, 2 mM MgCl2, 4 mM DTT, 20 µg/ml BSA, 10 nM substrate and 1 nM E.coli RNase H, at 37°C for 1 h.
Figure 3.

Cleavage of the substrate by E.coli RNase H. Assays were performed at 37°C in 40 mM Tris–HCl (pH 7.4), containing 60 mM NaCl, 60 mM KCl, 2 mM MgCl2, 4 mM DTT, 20 µg/ml BSA and 10 nM substrate. Fluorescence intensity was measured at 520 nm upon excitation at 490 nm. Left panel shows representative reaction progress curves obtained with 28 pM (b), 100 pM (c) and 140 pM (d) E.coli RNase H. The lowest trace (a) was obtained from a control reaction without enzyme. Right panel shows the initial rates of substrate cleavage as a function of the concentration of E.coli RNase H.
Table 1.
Hybridase activity of various enzymes
| Enzyme | kcat/Km (M−1 s−1 × 10−3) | Relative activity |
|---|---|---|
| E.coli RNase H | 2540 ± 250 | 1411 |
| RNase A | 1.8 ± 0.2 | 1 |
| hRNase-1 | 330 ± 10 | 183 |
Reaction conditions were as detailed in Figures 3 and 4. Data are the mean ± SEM of 6–11 measurements.
The specificity of the assay for RNase H activity was then assessed by testing bovine RNase A, a potent hydrolase with a known specificity for ssRNA (6). The assay of this enzyme toward the double-helical hybrid revealed a barely detectable activity with a kcat/Km of 1800 M−1 s−1 (Table 1), a value ∼1400-fold lower than that measured with E.coli RNase H. An additional control was then introduced by testing bovine DNase I, a hydrolase known to cleave efficiently both single-stranded and double-stranded DNA. With this enzyme no fluorescence enhancement could be detected even at enzyme concentrations as high as 650 nM (data not shown), giving additional support to the specificity of the assay.
Overall, these data indicate that the newly developed RNase H assay is accurate, sensitive and highly specific. Testing of hRNase-1 was subsequently undertaken.
Testing of hRNase-1 for hybridase activity
Assays were performed under physiological-like conditions, i.e. at 37°C in a solution buffered at pH 7.4 with an ionic strength of 0.15 M. As shown in Figure 4, concentrations of hRNase-1 in the low nM range were able to produce large increases in fluorescence emission. Measurement of V0 values revealed a good correlation with enzyme concentrations and yielded a kcat/Km of 330 000 M−1 s−1 (Table 1). This value is over 180-fold higher than that obtained with the homolog bovine RNase A and only 8-fold lower than that measured with E.coli RNase H.
Figure 4.

Hybridase activity of hRNase-1. Assays were performed at 37°C in 40 mM Tris–HCl (pH 7.4), containing 60 mM NaCl, 60 mM KCl, 20 µg/ml BSA and 10 nM substrate. Left panel shows representative reaction progress curves obtained with 1.8 nM (b), 2.5 nM (c), 3.2 nM (d) and 5.7 nM (e) hRNase-1. The lowest trace (a) was obtained from a control reaction without enzyme. In this set of assays, the maximum change in fluorescence intensity, obtained by incubation with a large excess of hRNase-1, was 380 fluorescence units. Right panel shows the initial rates of substrate cleavage as a function of concentration of hRNase-1.
As a control, cleavage of the substrate by hRNase-1 was also checked by a direct method. An aliquot of the double-helical hybrid was partially digested with hRNase-1 and the reaction mixture was analyzed by ion-pair reversed-phase HPLC. Comparison of the resulting elution profile with that obtained with the intact substrate (Figure 5) confirmed that only the RNA component of the hybrid was affected by the enzyme and revealed only two oligoribonucleotide products. This result rules out the possibility that the fluorescence enhancement in the above assays could be produced by (i) mere protein–nucleic acid binding or (ii) cleavage of the DNA strand by a contaminating DNase. The elution profiles also indicated that hRNase-1 cleaves the substrate at a single site. In this regard, it has to be noted that E.coli RNase H has a substrate specificity broader than that of hRNase-1 and most likely cleaves the fluorogenic substrate at many sites. Since cleavage at any of these sites will result in fluorescence enhancement, the true kcat/Km value is probably lower than that listed in Table 1, and the catalytic efficiency of hRNase-1 may actually be closer to that of the E.coli enzyme.
Figure 5.

Reaction products of hRNase-1. RNA:DNA hybrid (0.3 nmol) was incubated in 30 µl of 40 mM Tris–HCl (pH 7.4) containing 60 mM NaCl, 60 mM KCl, 20 µg/ml BSA and 3 nM hRNase-1. After 1 h incubation at 37°C, reaction products were analyzed by ion-pair reversed-phase HPLC under fully denaturing conditions (lower panel). Trace in upper panel was obtained with an aliquot of the undigested hybrid. D and R refer to the DNA and RNA strand of the hybrid, respectively. These assignments were obtained by HPLC analysis of the individual strands of the substrate. R3 and R9 refer to the 3 nt and the 9 nt RNA fragments deriving from the cleavage of the RNA strand, respectively.
For comparative purposes, the ribonucleolytic activity of hRNase-1 toward the isolated RNA strand of the substrate was also evaluated. Assays were performed by an HPLC method similar to the one originally described for human RNase-4 (32) and yielded a kcat/Km of 1 360 000 ± 70 000 M−1 s−1. This value is ∼4-fold higher than that obtained, under the same reaction conditions, with the RNA:DNA hybrid, implying that the activity toward this substrate is only 4-fold lower than that measured with the ssRNA. With reference to the activity of hRNase-1 toward dsRNA, a direct comparison of the activities toward poly(rA):poly(rU) and poly(rU), under the same reaction conditions, has in our knowledge never been reported. However, the specific activity of hRNase-1 toward dsRNA from f2 sus11 bacteriophage is known to be ∼170-fold lower than that measured with the corresponding ssRNA obtained by thermal denaturation (9). It should also be noted that the absolute value of kcat/Km for cleavage of the fluorogenic RNA:DNA hybrid reported in this article is >2-fold higher than that measured with poly(rA):poly(rU) (9).
Overall, the data reported above show that hRNase-1 is endowed with a ribonuclease H activity whose potency is in the same order of magnitude of that of E.coli RNase H.
Characterization of the hybridase activity of hRNase-1
An attempt to measure individually the kinetic parameters kcat and Km of hRNase-1 was performed by testing the enzyme at substrate concentrations between 20 and 400 nM. As shown in Figure 6, a linear correlation between V0 and [S] was obtained. This result indicates that the enzyme active site is poorly saturated at the substrate concentration tested, suggesting that the Km of hRNase-1 for the double-helical hybrid is higher than 1 µM. This implies that the affinity for the substrate of hRNase-1 is lower than that of E.coli RNase H, whose Km values for similar substrates are in the nM range. Nevertheless, the hybridase activity of hRNase-1 is expected to be highly effective even at low substrate concentration due to the relatively high kcat/Km value. For comparative purposes, the antimicrobial hRNase-3 and hRNase-7 display kcat/Km values for cleavage of tRNA of 49 000 and 2 300 000 M−1 s−1, respectively, whereas their Km are 1.8 and 2.2 µM, respectively (21). Since these values were obtained in 40 mM sodium phosphate, pH 7.0, the actual Km values at physiological ionic strength should be higher. Taken together, these data suggest that human RNases can display relevant biological activities also if they have relatively high Km values.
Figure 6.

Michaelis–Menten plot for cleavage of the double-helical substrate by hRNase-1. Assays were performed as detailed in Figure 4, except that substrate concentration was 20–400 nM and enzyme concentration was 0.14–1.4 nM.
The hybridase activity of hRNase-1 was then tested at various pHs. As shown in Figure 7, the activity profile is bell-shaped with a maximum between pH 7 and 7.5. Activities at pH 8 and 9 are 2- to 20-fold lower than maximal and that at pH 10 is undetectable. This profile is very similar to the one obtained with poly(rA):poly(rU), which is also bell-shaped with an activity maximum between pH 7 and 7.5 (33). This result shows that hRNase-1 is suited to perform its hybridase activity under physiological pH conditions, and suggests that hRNase-1 will digest double-helical substrates in human plasma better than in duodenum which exhibits pH values between 5 and 6 (34).
Figure 7.
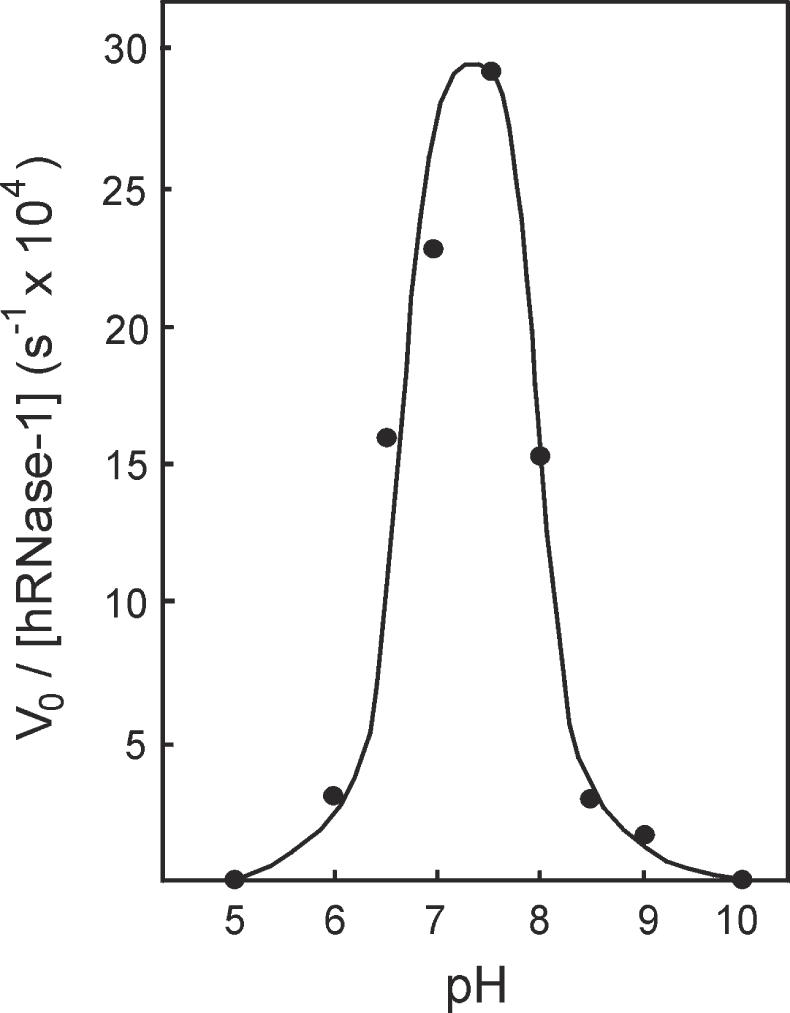
Influence of pH on the hybridase activity of hRNase-1. Assays were performed at 37°C in a multicomponent buffer system containing 50 mM sodium acetate, 50 mM 2-(4-Morpholino)ethanesulfonic acid (Mes), 50 mM 4-(2-Hydroxyethyl)-1-piperazineethanesulphonic acid (HEPES), 50 mM 2-Cyclohexylaminoethanesulphonic acid (Ches), 20 µg/ml BSA and 10 nM substrate, titrated at the indicated pH.
Human RNase-1 was then assayed in solutions at various ionic strengths. As shown in Table 2, the hybridase activity is highly dependent on the salt concentration. Activity at 270 mM is in fact 68-fold lower than that measured at 150 mM, and the activity at 30 mM is 94-fold higher than that measured at 150 mM. This activity profile is quite different from those exhibited by hRNase-1 for cleavage of single-stranded substrates and poly(rA):poly(rU). These are bell-shaped with activity maxima at 80 and 150 mM for poly(rA):poly(rU) and poly(rC), respectively (33). In contrast, the activity toward the RNA:DNA hybrid has a plateau at 30–90 mM and sharply decreases when the ionic strength is raised to 150 mM. This result may be explained by assuming that the binding of the substrate to the enzyme relies on multiple salt bridges that are known to be strongly affected by the ionic strength. These interactions may involve the negatively charged phosphates of the nucleic acid and some of the basic residues of the enzyme. An alternative explanation may be given by taking into account the stability of the double-helical substrate. It is well known that double helices are more stable at high ionic strength. Then, the increased activity of hRNase-1 at low ionic strength may be due to a lower stability of the substrate and to transiently exposed ssRNA. However, this interpretation is in contrast with the observation that the background fluorescence of the undigested substrate was the same at all the ionic strengths tested (data not shown), indicating that even at 30 mM salt concentration the substrate was stably double-stranded. This aspect was further inquired by measuring the Tm of the substrate at 30 and 150 mM salt concentration. The value of Tm at 150 mM resulted to be 61.8°C (Table 2), implying that the assays at 37°C were performed at a temperature of 25°C below the Tm. The Tm at 30 mM was found to be 58.1°C, a value only 3.7°C below the one measured at 150 mM salt. These results indicate that the stability of the substrate doesn't change much in the 30–150 mM range of ionic strength. Finally, if the increased activity at low ionic strength was due to a lower stability of the substrate and to transiently exposed ssRNA, then the activity of RNase A (that is highly specific for ssRNA) with lowering salt concentration should increase equally or even more markedly than observed with hRNase-1. In contrast, the activity of RNase A at 90 mM salt was found to be only 27-fold higher that than measured at 150 mM (Table 2), whereas the activity of hRNase-1 had increased by 97-fold. This makes the activity of hRNase-1 at 90 mM salt 685-fold higher than that of RNase A. Similar results were obtained from assays with RNase A at 30 and 210 mM salt (Table 2). Taken together, these data suggest that the markedly increased activity of hRNase-1 registered at low ionic strength is not due to a lower stability of the double-helical substrate but to a more effective substrate binding capability. It should also be noted that the hybridase activity of hRNase-1 does not change much when the salt concentration is raised from 150 to 210 mM, whereas it decreases rapidly when the ionic strength is increased to 270 mM (Table 2). The reasons for this second drop in activity with raising ionic strength (the first one was registered between 90 and 150 mM salt) are unknown and may also reflect possible effects of the high salt concentration on the catalytic efficiency of the enzyme. The data above also indicate that the hybridase activity of hRNase-1 can be more conveniently assayed at 90 mM salt. At this ionic strength the assay is, in fact, about 100-fold more sensitive than at 150 mM salt, making it possible to test hRNase-1 concentrations as low as 10 pM without affecting specificity.
Table 2.
Effect of ionic strength on hybridase activity and Tm of the double-helical substrate
| Ionic strength (mM) | V0/[E] (s−1 × 106) with | Tm (°C) | |
|---|---|---|---|
| hRNase-1 | RNase A | ||
| 30 | 178 000 ± 12 000 | 390 ± 40 | 58.1 |
| 90 | 185 000 ± 19 000 | 270 ± 10 | |
| 150 | 1900 ± 100 | 10 ± 1 | 61.8 |
| 210 | 1200 ± 100 | 5.4 ± 0.4 | |
| 270 | 28 ± 2 | ND | |
Hybridase assays were performed at 37°C in 40 mM Tris–HCl (pH 7.4) containing 20 µg/ml BSA, 10 nM substrate and variable concentrations of NaCl. Enzyme concentrations were 0.02–100 nM and 0.01–1.2 µM for the human and bovine enzyme, respectively. Activity values are the mean ± SEM of three to four measurements. Values of Tm were obtained from thermal transition profiles obtained by raising the temperature of undigested reaction mixtures from 22 to 82°C over a 90 min period. Each Tm is the mean of two values deviating from the mean by <0.6%. ND, not detectable.
Finally, the enzymatic activity of hRNase-1 was tested in the presence of the human cytosolic RNase inhibitor (cRI), a 50 kDa protein that binds tightly to most pancreatic-type RNases and inhibits competitively their ribonucleolytic activity with Ki values in the fM range (35). Human RNase-1 has already been reported to be inhibited by cRI and its Ki for cleavage of the dinucleotide CpA at pH 6.0 has a value of 200 fM (36). With regard to the hybridase activity, a 2-fold molar excess of cRI upon hRNase-1 resulted in a complete inhibition of its enzymatic activity (Figure 8).
Figure 8.

Inhibition of the hybridase activity of hRNase-1 by the cytosolic RNase inhibitor. Human RNase-1 (7 nM) was assayed in the absence (c) or the presence (b) of the cytosolic RNase inhibitor (cRI) (14 nM). This was added ∼60 s after the enzyme. The lowest trace (a) was obtained from a control reaction without enzyme. Assays were performed as described in Figure 4.
CONCLUSIONS
The data reported represent the first evidence that hRNase-1 is endowed with a ribonuclease H activity. The kcat/Km value of the human enzyme, measured under physiological-like conditions, is 330 000 M−1 s−1, a value that is 180-fold higher than that of bovine pancreatic RNase A and only 8-fold lower than that measured with E.coli RNase H, the best characterized member of the RNase H superfamily (23,24,31). The hybridase activity of hRNase-1 is maximal at neutral pH, increases with lowering ionic strength and is fully inhibited by the cytosolic RNase inhibitor.
Since RNA:DNA hybrids represent key intermediates of replication of retrovirus, the reported data provide a possible explanation for the anti-HIV activity that hRNase-1 displays in vitro (22) and thereby support a possible role in human antiviral defence, as already proposed (9,11). During evolution, hRNase-1 may have conserved the ability to cleave efficiently ssRNA and to be expressed in exocrine pancreas, as bovine RNase A, in response to the needing to digest dietary RNA. At the same time, hRNase-1 has differentiated from the bovine enzyme by acquiring a strong catalytic activity toward dsRNA and RNA:DNA hybrids. These enzymatic properties, together with an expression pattern broader than that of RNase A (10,12), may constitute the basis for a newly acquired biological function related to antiviral host defence.
Finally, it should be noted that the availability of a highly sensitive and specific assay for the hybridase activity of hRNase-1 provides the basis for the development of a new method for the determination of the blood plasma levels of hRNase-1. In this regard, preliminary tests have shown that the blood enzyme can be detected by assaying amount of plasma as little as 1–2 µl (data not shown). The refinement of this assay should hence provide with an additional tool the ongoing studies on hRNase-1 as a possible marker of pathological conditions associated with injury to the vascular system (11) and to the pancreas (37–39).
Acknowledgments
We thank the referees of Nucleic Acids Research for valuable suggestions. This work was supported by grants from Ministero dell'Istruzione, dell'Università e della Ricerca (PRIN 2002), Regione Campania (LR 5/2002) and Istituto Banco di Napoli—Fondazione. Funding to pay the Open Access publication charges for this article was provided by the Second University of Naples.
Conflict of interest statement. None declared.
REFERENCES
- 1.Weickmann J.L., Elson M., Glitz D.G. Purification and characterization of human pancreatic ribonuclease. Biochemistry. 1981;20:1272–1278. doi: 10.1021/bi00508a035. [DOI] [PubMed] [Google Scholar]
- 2.Beintema J.J., Wietzes P., Weickmann J.L., Glitz D.G. The amino acid sequence of human pancreatic ribonuclease. Anal. Biochem. 1984;136:48–64. doi: 10.1016/0003-2697(84)90306-3. [DOI] [PubMed] [Google Scholar]
- 3.Beintema J.J., Kleinedam R.G. The ribonuclease A superfamily: general discussion. Cell. Mol. Life Sci. 1998;54:825–832. doi: 10.1007/s000180050211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho S., Beintema J.J., Zhang J. The ribonuclease A superfamily of mammals and birds: identifying new members and tracing evolutionary histories. Genomics. 2005;85:208–220. doi: 10.1016/j.ygeno.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 5.Blackburn P., Moore S. Pancreatic ribonucleases. In: Boyer P.D., editor. The Enzymes. 3rd edn. Vol. 15. NY: Academic Press, Inc.; 1982. pp. 317–433. [Google Scholar]
- 6.Raines R.T. Ribonuclease A. Chem. Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 7.Ribó M., Benito A., Canals A., Nogués M.V., Cuchillo C.M., Vilanova M. Purification of engineered human pancreatic ribonuclease. Methods Enzymol. 2001;341:221–234. doi: 10.1016/s0076-6879(01)41154-2. [DOI] [PubMed] [Google Scholar]
- 8.Libonati M., Sorrentino S. Degradation of double-stranded RNA by mammalian pancreatic-type ribonucleases. Methods Enzymol. 2001;341:234–248. doi: 10.1016/s0076-6879(01)41155-4. [DOI] [PubMed] [Google Scholar]
- 9.Sorrentino S., Naddeo M., Russo A., D'Alessio G. Degradation of double-stranded RNA by human pancreatic ribonuclease: crucial role of noncatalytic basic amino acid residues. Biochemistry. 2003;42:10182–10190. doi: 10.1021/bi030040q. [DOI] [PubMed] [Google Scholar]
- 10.Futami J., Tsushima Y., Murato Y., Tada H., Sasaki J., Seno M., Yamada H. Tissue-specific expression of pancreatic-type RNases and RNase inhibitor in humans. DNA Cell Biol. 1997;16:413–419. doi: 10.1089/dna.1997.16.413. [DOI] [PubMed] [Google Scholar]
- 11.Landré J.B.P., Hewett P.W., Olivot J., Friendi P., Ko Y., Sachinidis A., Moenner M. Human endothelial cells selectively express large amounts of pancreatic-type ribonuclease (RNase 1) J. Cell. Biochem. 2002;86:540–552. doi: 10.1002/jcb.10234. [DOI] [PubMed] [Google Scholar]
- 12.Sasso M.P., Lombardi M., Confalone E., Carsana A., Palmieri M., Furia A. The differential pattern of tissue-specific expression of ruminant pancreatic type ribonucleases may help to understand the evolutionary history of their genes. Gene. 1999;227:205–212. doi: 10.1016/s0378-1119(98)00586-1. [DOI] [PubMed] [Google Scholar]
- 13.Weickmann J.L., Glitz D.G. Human ribonucleases. Quantitation of pancreatic-like enzymes in serum, urine, and organ preparations. J. Biol. Chem. 1982;257:8705–8710. [PubMed] [Google Scholar]
- 14.Morita T., Niwata Y., Ohgi K., Ogawa M., Irie M. Distribution of two urinary ribonuclease-like enzymes in human organs and body fluids. J. Biochem. 1986;99:17–25. doi: 10.1093/oxfordjournals.jbchem.a135456. [DOI] [PubMed] [Google Scholar]
- 15.Sorrentino S., De Prisco R., Libonati M. Human seminal ribonuclease. Immunological quantitation of cross-reactive enzymes in serum, urine and seminal plasma. Biochim. Biophys. Acta. 1989;998:97–101. doi: 10.1016/0167-4838(89)90125-8. [DOI] [PubMed] [Google Scholar]
- 16.Beintema J.J. The primary structure of langur (Presbytis entellus) pancreatic ribonuclease: adaptive features in digestive enzymes in mammals. Mol. Biol. Evol. 1990;7:470–477. doi: 10.1093/oxfordjournals.molbev.a040619. [DOI] [PubMed] [Google Scholar]
- 17.Cullen B.R. RNA interference: antiviral defense and genetic tool. Nature Immunol. 2002;3:597–599. doi: 10.1038/ni0702-597. [DOI] [PubMed] [Google Scholar]
- 18.Barker R.L., Loegering D.A., Ten R.M., Hamann K.J., Pease L.R., Gleich G.J. Eosinophil cationic protein cDNA. Comparison with other toxic cationic proteins and ribonucleases. J. Immunol. 1989;143:952–955. [PubMed] [Google Scholar]
- 19.Lehrer R.I., Szklarek D., Barton A., Ganz T., Hamann K.J., Gleich G.J. Antibacterial properties of eosinophil major basic protein and eosinophil cationic protein. J. Immunol. 1989;142:4428–4434. [PubMed] [Google Scholar]
- 20.Rosenberg H.F. Recombinant human eosinophil cationic protein. Ribonuclease activity is not essential for cytotoxicity. J. Biol. Chem. 1995;270:7876–7881. doi: 10.1074/jbc.270.14.7876. [DOI] [PubMed] [Google Scholar]
- 21.Harder J., Schroder J.M. RNase 7, a novel innate immune defense antimicrobial protein of healthy human skin. J. Biol. Chem. 2002;277:46779–46784. doi: 10.1074/jbc.M207587200. [DOI] [PubMed] [Google Scholar]
- 22.Lee-Huang S., Huang P.L., Kung H.F., Blithe D.L., Chen H.C. Lysozyme and RNases as anti-HIV components in beta-core preparations of human chorionic gonadotropin. Proc. Natl Acad. Sci. USA. 1999;96:2678–2681. doi: 10.1073/pnas.96.6.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein H., Hausen P. Enzyme from calf thymus degrading the RNA moiety of DNA–RNA hybrids: effect on DNA-dependent RNA polymerase. Science. 1969;166:393–395. doi: 10.1126/science.166.3903.393. [DOI] [PubMed] [Google Scholar]
- 24.Crouch R.J. Ribonuclease H: from discovery to 3D structure. New Biol. 2005;2:771–777. [PubMed] [Google Scholar]
- 25.Sela M., Anfisen C.B. Some spectrophotometric and polarimetric experiments with ribonuclease. Biochim. Biophys. Acta. 1957;24:229–235. doi: 10.1016/0006-3002(57)90186-5. [DOI] [PubMed] [Google Scholar]
- 26.Russo N., Antignani A., D'Alessio G. In vitro evolution of a dimeric variant of human pancreatic ribonuclease. Biochemistry. 2000;39:3585–3591. doi: 10.1021/bi992367q. [DOI] [PubMed] [Google Scholar]
- 27.Rizzo J., Gifford L.K., Zhang X., Gewirtz A.M., Lu P. Chimeric RNA–DNA molecular beacon assay for ribonuclease H activity. Mol. Cell. Probes. 2002;16:277–283. doi: 10.1006/mcpr.2002.0423. [DOI] [PubMed] [Google Scholar]
- 28.Kanaya S., Katsuda C., Kimura S., Nakai T., Kitakuni E., Nakamura H., Katayanagi K., Morikawa K., Ikehara M. Stabilization of Escherichia coli ribonuclease H by introduction of an artificial disulfide bond. J. Biol. Chem. 1991;266:6038–6044. [PubMed] [Google Scholar]
- 29.Kanaya S. Prokaryotic type 2 RNases H. Methods Enzymol. 2001;341:377–395. doi: 10.1016/s0076-6879(01)41165-7. [DOI] [PubMed] [Google Scholar]
- 30.Parniak M.A., Min K.L., Budihas S.R., Le Grice S.F., Beutler J.A. A fluorescence-based high-throughput screening assay for inhibitors of human immunodeficiency virus-1 reverse transcriptase-associated ribonuclease H activity. Anal. Biochem. 2003;322:33–39. doi: 10.1016/j.ab.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Nowotny M., Gaidamakov S.A., Crouch R.J., Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 32.Shapiro R., Fett J.F., Strydom D.J., Vallee B.L. Isolation and characterization of a human colon carcinoma-secreted enzyme with pancreatic ribonuclease-like activity. Biochemistry. 1986;25:7255–7264. doi: 10.1021/bi00371a002. [DOI] [PubMed] [Google Scholar]
- 33.Sorrentino S., Lavitrano M., De Prisco R., Libonati M. Human seminal ribonuclease. A tool to check the role of basic charges and glycosylation of a ribonuclease in the action of the enzyme on double-stranded RNA. Biochim. Biophys. Acta. 1985;827:135–139. doi: 10.1016/0167-4838(85)90081-0. [DOI] [PubMed] [Google Scholar]
- 34.Ovesen L., Bendtsen F., Tage-Jensen U., Pedersen N.T., Gram B.R., Rune S.J. Intraluminal pH in the stomach, duodenum, and proximal jejunum in normal subjects and patients with exocrine pancreatic insufficiency. Gastroenterology. 1986;90:958–962. doi: 10.1016/0016-5085(86)90873-5. [DOI] [PubMed] [Google Scholar]
- 35.Lee F.S., Vallee B.L. Structure and action of mammalian ribonuclease (angiogenin) inhibitor. Prog. Nucleic Acid Res. Mol. Biol. 1993;44:1–30. doi: 10.1016/s0079-6603(08)60215-9. [DOI] [PubMed] [Google Scholar]
- 36.Boix E., Wu Y., Vasandani V.M., Saxena S.K., Ardelt W., Ladner J., Youle R.J. Role of the N terminus in RNase A homologues: differences in catalytic activity, ribonuclease inhibitor interaction and cytotoxicity. J. Mol. Biol. 1996;257:992–1007. doi: 10.1006/jmbi.1996.0218. [DOI] [PubMed] [Google Scholar]
- 37.Schein C.H. From housekeeper to microsurgeon: the diagnostic and therapeutic potential of ribonucleases. Nat. Biotechnol. 1997;15:529–536. doi: 10.1038/nbt0697-529. [DOI] [PubMed] [Google Scholar]
- 38.Naskalski J.W., Kusnierz-Cabala B., Panek J., Kedra B. Poly-C specific ribonuclease activity correlates with increased concentrations of IL-6, IL-8 and sTNFR55/sTNFR75 in plasma of patients with acute pancreatitis. J. Physiol. Pharmacol. 2003;54:439–448. [PubMed] [Google Scholar]
- 39.Peracaula R., Royle L., Tabares G., Mallorqui-Fernandez G., Barrabes S., Harvey D.J., Dwek R.A., Rudd P.M., de Llorens R. Glycosylation of human pancreatic ribonuclease: differences between normal and tumor states. Glycobiology. 2003;13:227–244. doi: 10.1093/glycob/cwg019. [DOI] [PubMed] [Google Scholar]