

Abstract
The short QT syndrome is a new congenital entity associated with familial atrial fibrillation and/or sudden death or syncope. Three different gain-of-function mutations in genes encoding for cardiac potassium channels (KCNH2, KCNQ1, and KCNJ2) have been identified up to now to cause short QT syndrome. The syndrome is characterized electrocardiographically by a shortened QTc interval less than 300 to 320 milliseconds and a lack of adaptation during increasing heart rates. During programed electrical stimulation, atrial and ventricular effective refractory periods are shortened, and in a high percentage, ventricular tachyarrhythmias are inducible. Sudden cardiac death occurs in all age groups and even in newborns. Therapy for choice seems to be the implantable cardioverter-defibrillator because of the high incidence of sudden death. However, ICD therapy may be associated with an increased risk of inappropriate therapies for T wave oversensing, which, however, can be resolved by reprogramming ICD detection algorithms. The impact of sotalol, ibutilide, flecainide, and quinidine on QT prolongation has been evaluated. But only quinidine effectively suppressed gain-of-function in IKr, along with prolongation of the QT interval. Furthermore, in patients with a mutation in HERG (SQT1), quinidine rendered ventricular tachyarrhythmias noninducible and restored the QT interval/heart rate relationship toward a reference range. It may serve as an adjunct to ICD therapy or as possible alternative treatment especially for children and newborns.
Keywords: Short QT syndrome, Repolarization, Mutation, Ventricular tachyarrhythmias, Sudden death, Atrial fibrillation
1. Introduction
A short QT interval associated with familial atrial fibrillation was first described by Gussak et al [1] in 2000. The authors presented the case of a 17-year-old girl with atrial fibrillation and a QT interval of 225 milliseconds. The brother, mother, and the grandfather also displayed an extremely short QT interval, and the mother and the grandfather suffered from atrial fibrillation. Gaita et al [2] first described a familial history of sudden death associated with a short QT interval in 2 unrelated European families. This was followed by identification of a missense mutation in KCNH2 and description of the first gain-of-function mutation of the rapid component of the delayed rectifier potassium current (IKr) in the affected members of these 2 families by Brugada et al [3] (SQT1). In 2004, a further mutation in the gene KCNQ1 encoding for the slow component of the delayed rectifier potassium current (IKs) was described by Bellocq et al [4] in a single patient with a short QT interval and aborted sudden death (SQT2). The latest mutation causing the short QT-3 syndrome was recently published by Priori et al [5] in KCNJ2, another potassium channel–coding gene, resulting in a gain of function of IK1 and leading to a shortened QT interval. There are still several patients with a short QT syndrome in which genetic screening has, so far, not been able to identify a mutation. Thus, the short QT syndrome constitutes a genetically heterogeneous disease.
2. Clinical presentation
In patients with a short QT syndrome, the risk for an arrhythmic event such as syncope and/or sudden cardiac death due to ventricular tachyarrhythmias is high. Furthermore, episodes of atrial fibrillation could be documented in patients with short QT intervals at different ages even in adolescents and children. The risk for an arrhythmic event is high in patients with a short QT syndrome comprising syncope and/or sudden cardiac death due to ventricular tachyarrhythmias. Furthermore, episodes of atrial fibrillation could be documented in patients with short QT intervals at different ages. A relevant number of sudden cardiac deaths or aborted sudden cardiac deaths occurred in 3 and 4 generations, respectively, in the 2 reported families with a short QT interval [2]. The mean age at the documented events such as syncope, sudden cardiac death, and aborted sudden cardiac death in patients with a short QT syndrome is 35 ± 25 years (median, 39 years). The short QT syndrome constitutes also a cause for sudden infant death syndrome. Furthermore, it may manifest immediately with sudden cardiac death without a preceding syncope, as 4 of the reported sudden cardiac deaths had no history of prior syncope or other arrhythmic events.
The incidence of atrial fibrillation is high in patients with a short QT syndrome. Assessment of available electrocardiograms (ECGs) of 12 patients and 1 patient who died suddenly showed that 70% (9/13) of patients had either paroxysmal or permanent atrial fibrillation. The first symptomatic episode of atrial fibrillation occurred at a mean age of 41 ± 19 years. In 53% (7/13) of the patients, the first symptom of the short QT syndrome was atrial fibrillation. Thus, especially in young patients with “lone” atrial fibrillation, a short QT interval has to be excluded.
3. The ECG in short QT syndrome
The ECG in patients with a short QT syndrome is characterized by absolute QT intervals below 300 milliseconds (ranging from 220 to 300 milliseconds). The QT interval should be measured and corrected for heart rate at rates below 80 beats per minute. A second finding, which is visible in at least about half of the patients, is a tall, symmetrical peaking T wave in the right precordial leads (Fig. 1) [2-4,6]. However, this is not observed in every patient [2]. All ECGs have in common that a clear ST segment is absent and the T wave initiates immediately from the S wave. In patients with a short QT syndrome, the T waves appear often tall, peaked, and symmetrical. One further distinctive ECG feature of patients with a short QT syndrome is the relatively prolonged Tpeak-Tend interval. This may indicate an augmented transmural dispersion of repolarization, which was recently tested by Extramiana and Antzelevitch [12] in an animal model [13]. The publication with the first description of a mutation in the KCNQ1 gene (SQT2) represents a sporadic case with only 1 available ECG with a short QT interval of 290 milliseconds (QTc, 302 milliseconds) [4]. The morphology of the T waves in this patient with a gain-of-function mutation of the slow component of the cardiac delayed rectifier K+ current (IKs) (KvLQT1) appears to be comparable with the previously described patients with a gain-of-function mutation of the fast component of the delayed rectifier IKr (HERG) [3]. In contrast, in a recently published report, 2 further different ECG morphologies are described, despite that a complete 12-lead ECG of these patients is not available for a comprehensive assessment of the T-wave morphology. The 2 related patients show, in contrast to the other published patients, an asymmetrical pattern with a less steep ascending section of the T wave, which is followed by a rapid descending and terminal phase of the T wave. In contrast to the first patients with mutations of the delayed rectifier in these 2 patients, a gain-of-function mutation in the KCNJ2 gene was identified, which encoded for the strong inwardly rectifying channel protein Kir2.1, and lead to an accelerated repolarization. Functional characterization of the mutant channels was performed to test the phenotype-genotype correlation. In contrast to the HERG and KvLQT1 mutations, simulations of the cardiac action potential propagation showed a different repolarization of the KCNJ2 mutant with a sudden acceleration of the final phase of the action potential repolarization [5].
Fig. 1.

This tracing depicts the induction of ventricular fibrillation during invasive electrophysiological stimulation. Three premature stimuli induce ventricular fibrillation (HRA 1/2, HRA 3/4 bipolar electrogram from right atrial electrode, RVA 1/2 and RVA 3/4 bipolar electrogram at right ventricle).
The varying ECG morphology in patients with a short QT syndrome, together with the recently shown SQT3 subtype, underlines that we are confronted with a genetically as well as phenotypically heterogenous disease. Only after further systematic comparison of more ECGs in patients with a short QT syndrome and correlation to the genetic finding may we potentially identify genotype-specific ECG patterns compared with those already described in the long QT syndromes [7].
Furthermore, these findings underline the constant awareness on “peculiar” ECG morphologies, which, especially in combination with a personal or family history of cardiac complaints and arrhythmogenic events, should give reason for both a clinical and genetic workup. Common in all patients with a short QT syndrome and different genetic subtypes is the constant shortening in all available ECGs of these patients with a lack of significant fluctuations.
Finally, a further relevant feature in patients with short QT syndrome is the lack of adaptation of the QT interval to heart rate [8]. Compared with normals, it could be demonstrated that there is only a shallow slope of the linear decrease of QT interval with increasing heart rate. A lack of adaptation of QT interval with increasing heart rates is an additional diagnostic step in the diagnosis of the short QT syndrome.
4. Mechanisms
For most patients with a short QT syndrome, a mutation in a cardiac ion channel–encoding gene could be identified. However, several patients in whom no mutation was found after extensive genetic screening demonstrate that it constitutes a heterogeneous disease. As in the long QT syndrome or the Brugada syndrome, there are different mutations responsible for the disease, so that, as well, the malignancy and risk of each mutation may vary. The clinical presentation in 3 unrelated families with the same degree of QT shortening is different. In 2 families, there has been a high number of sudden deaths in all generations from newborns, and in the third family, there is “only” atrial fibrillation and no syncope or sudden death described [1,2].
However, the potential risk for malignant ventricular tachyarrhythmias or sudden death is high in all families. All patients, who underwent an electrophysiologic study, presented extremely short atrial and ventricular effective refractory periods, and in both families, ventricular fibrillation was inducible during programed ventricular stimulation, which reflects the high vulnerability caused by an increased dispersion of ventricular refractoriness [1,2,6]. Finally, the first episode of ventricular fibrillation in a short QT syndrome happened in a young adolescent with a mutation in HERG, who was previously asymptomatic and noninducible during programed ventricular stimulation [10]. The 3 different mutations identified to date have been investigated in vitro. A gain of function has been described leading to either a large increase in current together with failure to rectify at physiological membrane potentials, a shift in activation toward an earlier phase of the action potential, or an increased current accelerating the final phase of repolarization. Because the affected currents are active to a different extent during repolarization, the differences in shortening of the QT-interval and the difference in T-wave morphology may be explained. It is presently unknown how these mutations respond to vagal or sympathetic activation. Clinically, it is not known if the risk of dying is suddenly higher during stress or exertion vs sleep or enhanced vagal activity and other external influences [9]. The only available documentation of an onset of a spontaneous ventricular tachyarrhythmia shows a very short coupled premature beat during sleep (Fig. 3) [10]. However, the patient's father, who suffered also from a short QT syndrome, had died of ventricular fibrillation during physical activity. The grandfather and great grandmother died at rest [10].
Fig. 3.
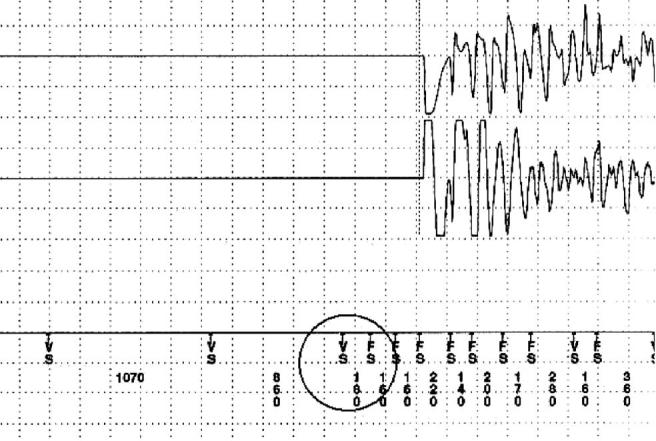
Stored endocardial electrogram derived from the ICD memory showing an episode of primary ventricular fibrillation in short QT syndrome. The ventricular tachyarrhythmia was initiated by a short coupled premature ventricular beat (VS encircled; coupling interval, 180 milliseconds) (upper electrogram strip, shock electrogram; lower electrogram strip, rate electrogram; ICD markers: FS, fibrillation sensing; FD, fibrillation detection; VS, ventricular sensing).
The common reason for susceptibility to malignant ventricular tachyarrhythmias is likely to be the large dispersion of ventricular refractoriness caused by the inhomogeneous abbreviation of the action potential within the ventricle and within different layers of the ventricular wall [1-3,11,12].
Thus, transmural dispersion of repolarization may be more pronounced low heart rates with prominent short QT intervals and, therefore, more susceptible for reentrant tachyarrhythmias initiated by premature beats.
5. Invasive electrophysiologic findings in short QT syndrome
Results of invasive electrophysiologic studies have been reported only for patients with the mutation in KCNH2 (HERG) (SQT1) [2]. All patients demonstrated very short atrial and ventricular effective refractory periods ranging from 120 to 180 milliseconds (mean, 143 ± 14 milliseconds) and 120 to 180 milliseconds (mean, 148 ± 11 milliseconds), respectively [2,6,7]. Ventricular fibrillation was inducible in 8 of 9 patients by programed ventricular stimulation (Fig. 2). In 2 patients, ventricular fibrillation was inducible in 2 separate studies, which underlines the electrical vulnerability and makes an unspecific response to programed stimulation most unlikely. Of note, atrial fibrillation was also inducible in 4 of 7 patients by a single premature stimulus stressing an increased atrial vulnerability.
Fig. 2.

This tracing depicts the induction of ventricular fibrillation during programed stimulation during the prehospital discharge test after ICD implantation. Ventricular fibrillation could be reproducibly induced by 3 premature stimuli (upper line, ventricular shock electrogram; lower electrogram strip, rate electrogram; ICD markers: S, sensing event; STIM, stimulated complex; F, fibrillation sensing; HV, high-voltage capacitor discharge).
6. Pharmacologic treatment
Because the risk of dying suddenly is high in patients with a short QT syndrome, pharmacologic therapy for the prevention of sudden death has to be seen with caution [2,3,10]. In all patients (except the 5-year-old girl) with a mutation in KCNJ2 have been proposed an implantable defibrillator, and except for 2 patients who refused ICD therapy, all other patients have been provided with an ICD.
Pharmacologic therapy may serve as an adjunct to ICD therapy to prevent ventricular tachyarrhythmias or may constitute an alternative to ICD therapy in very young patients and patients declining ICD therapy. Furthermore, in patients who suffer from symptomatic episodes of atrial fibrillation, an additional drug therapy is needed. The response to drugs has been studied only in SQT1 patients with a mutation in KCNH2 (HERG) [6,8,13].
As SQT1 represents the first ion, channelopathy, which is associated with a gain of function of IKr Sotalol as an IKr blocker, was tested but was unable to prolong QT interval in all patients. This could be reproduced in in vitro studies, where sotalol did not show an effect on the mutant current. Quinidine, however, was, in all patients, able to prolong the QT interval into the reference range, restitute rate adaptation of the QT interval, and prevent inducibility of ventricular fibrillation in 4 of 4 patients [2,3,8,13]. Furthermore, quinidine prolonged the effective ventricular refractory period in all patients. In a parallel in vitro study on the mutant current using quinidine, a reduction of the current was seen.
Ibutilide as an IKr blocker was also ineffective in 2 patients. Flecainide slightly prolonged the QT interval but did not render VF noninducible.
In 2 patients who suffered from atrial fibrillation, propafenone was able to suppress recurrences of atrial fibrillation, but did not normalize the QT interval [6].
Thus, pharmacologic treatment of patients with a short QT syndrome may be considered to suppress atrial fibrillation, a major clinical problem in short QT syndrome or as an adjunct for repetitive episodes of ventricular tachyarrhythmias. It should be reserved as a second line therapy if a defibrillator implant is denied or impossible for any reason.
Pharmacologic therapy with blockers of the affected current may be ineffective, as has been shown by in vivo and in vitro data for a mutation in KCNH2 (HERG).
Pharmacologic testing should finally include investigation of refractory periods and rate adaptation of the QT interval because an agent that is able to prolong QT interval at low heart rates potentially fails at higher heart rates, and prolongation of ventricular effective periods went along with noninducibility of ventricular fibrillation.
7. ICD therapy
Because of the high risk for ventricular tachyarrhythmias, the implantable cardioverter defibrillator is, to date, the therapy for choice in patients with a short QT syndrome. For 8 patients with a short QT syndrome, it is known that they received an ICD for primary or secondary prophylaxis.
In 4 of the 5 initially implanted patients, T wave oversensing and inappropriate ICD shock therapy occurred during follow-up. Short coupled intracardiac T waves with high amplitudes led to inappropriate therapies because of double counting of the R and T waves. In 1 patient, adapting the sensitivity to lower values prevented initially further inappropriate shock therapies (Medtronic Inc, Minneapolis, Minn) [14]. In the other 3 patients, reprogramming of multiprogrammable sensitivity parameters (start threshold, decay delay, and sensitivity) prevented effectively additional inappropriate therapies (St Jude Medical Inc, St Paul, Minn). Three further patients who received an ICD did not experience inappropriate therapies during short-term follow-up (Guidant, Indianapolis, Ind) [6].
References
- 1.Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94:99. doi: 10.1159/000047299. [DOI] [PubMed] [Google Scholar]
- 2.Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: a familial cause of sudden death. Circulation. 2003;108:965. doi: 10.1161/01.CIR.0000085071.28695.C4. [DOI] [PubMed] [Google Scholar]
- 3.Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short QT syndrome linked to mutations in HERG. Circulation. 2004;109:30. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- 4.Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109:2394. doi: 10.1161/01.CIR.0000130409.72142.FE. [DOI] [PubMed] [Google Scholar]
- 5.Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96:800. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 6.Bjerregaard P, Gussak I. Atrial fibrillation in the setting of familial short QT interval. Heart Rhythm. 2004;1:S165. abstract. [Google Scholar]
- 7.Zhang L, Timothy KW, Vincent GM, et al. Spectrum of ST–T-wave patterns and repolarization parameters in congenital long-QT syndrome ECG findings identify genotypes. Circulation. 2000;102:2849. doi: 10.1161/01.cir.102.23.2849. [DOI] [PubMed] [Google Scholar]
- 8.Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol. 2005;16:54. doi: 10.1046/j.1540-8167.2005.04470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gussak I, Liebl N, Nouri S, et al. Deceleration-dependent shortening of the QT interval: a new electrocardiographic phenomenon? Clin Cardiol. 1999;22:124. doi: 10.1002/clc.4960220213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schimpf R, Bauersfeld U, Gaita F, et al. Short QT syndrome: successful prevention of sudden cardiac death in an adolescent by implantable cardioverter defibrillator treatment for primary prophylaxis. Heart Rhythm. 2005;2:416. doi: 10.1016/j.hrthm.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 11.Antzelevitch C. Cellular basis and mechanism underlying normal and abnormal myocardial repolarization and arrhythmogenesis. Ann Med. 2004;36:5. doi: 10.1080/17431380410032553. [DOI] [PubMed] [Google Scholar]
- 12.Extramiana F, Antzelevitch C. Amplified transmural dispersion of repolarization as the basis for arrhythmogenesis in a canine ventricular-wedge model of short-QT syndrome. Circulation. 2004;110:3661. doi: 10.1161/01.CIR.0000143078.48699.0C. [DOI] [PubMed] [Google Scholar]
- 13.Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43:1494. doi: 10.1016/j.jacc.2004.02.034. [DOI] [PubMed] [Google Scholar]
- 14.Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT-syndrome and ICD treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol. 2003;14:1273. doi: 10.1046/j.1540-8167.2003.03278.x. [DOI] [PubMed] [Google Scholar]