

Abstract
Background—The early phase of action potential (AP) repolarization is critical to impulse conduction in the heart because it provides current for charging electrically coupled cells. In the present study we tested the impact of heart failure-associated electrical remodeling on AP propagation.
Methods and Results—Subepicardial, midmyocardial, and subendocardial myocytes were enzymatically dissociated from control and pressure-overload failing left ventricle (LV), and APs were recorded. A unique coupling-clamp technique was used to electrically couple 2 isolated myocytes with a controlled value of coupling conductance (Gc). In sham-operated mice, AP duration manifested a clear transmural gradient, with faster repolarization in subepicardial myocytes than in subendocardial myocytes. AP propagation from subendocardial to subepicardial myocytes required less Gc compared with conduction in the opposite direction. In failing heart, AP morphology was dramatically altered, with a significantly elevated plateau potential and prolonged AP duration. Spatially nonuniform alteration of AP duration in failing heart blunted the transmural gradient of repolarization. Furthermore, increased pacing rate prolonged AP duration exclusively in myocytes from failing heart, and the critical conductance required for successful AP propagation decreased significantly at high frequencies. Finally, in failing heart, asymmetry of transmural electrical propagation was abolished.
Conclusions—In failing heart, preferential conduction from subendocardial to subepicardial myocytes is lost, and failing myocytes manifest facilitated AP propagation at fast rates. Together, these electrical remodeling responses may promote conduction of premature impulses and heighten the risk of malignant arrhythmia, a prominent feature of heart failure.
Keywords: arrhythmia, conduction, electrophysiology, heart failure, remodeling
Each year in the United States, >300 000 people die of sudden cardiac death stemming from malignant ventricular arrhythmia. Among the mechanisms contributing to this disease presentation, prolongation of action potential duration (APD) in ventricular myocytes is a hallmark feature of both cardiac hypertrophy and heart failure (HF). However, mechanisms underlying the contribution of APD prolongation to cardiac arrhythmogenesis are incompletely characterized.
Clinical Perspective p 1856
Studies in several mammalian species have revealed the existence of 3 distinct layers of myocardial cell types within the left ventricular (LV) wall: subepicardial myocytes, midmyocardial myocytes, and subendocardial myocytes.1 In normal tissue, a transmural gradient in the kinetics of action potential (AP) repolarization exists among these cell types. In a variety of heart diseases, altered transmural dispersion of repolarization (TDR) contributes to ventricular tachycardia by promoting phase 2 reentry.1
Recent studies in an intact porcine model have tested the hypothesis that the time lag between the peak of the ECG T wave and its end (Tpeak-Tend) is a direct reflection of TDR. These investigators found that Tend fits well to the end of repolarization recorded at the endocardium.2 These data, along with others,3,4 point to myocytes in subepicardial and subendocardial zones as key determinants of the kinetics of electrical excitability recovery. Little is known, however, about disease-related changes in impulse conductivity between cells at the 2 extremes, namely, subepicardial and subendocardial myocytes, or their role in the transmural gradient of AP conductivity in intact heart.
We have previously uncovered a critical role of transient outward K+ current (Ito) in AP propagation at fast pacing rate in rabbit atrial myocytes.5 Increasing stimulation frequency significantly facilitated AP propagation, which was a consequence of frequency-dependent suppression of Ito. Suppression of Ito slowed the early phase of repolarization (prolonged APD30), thereby providing the leader cell with the capacity to deliver greater coupling current to the follower cell, promoting attainment of the AP threshold in the follower cell. In other words, the early phase of repolarization is the energy source that provides coupling current required to maintain successful AP conduction. Thus, Ito, a prominent target of pathological electrical remodeling in HF, is critical for successful propagation of myocyte excitation.
We recently developed a model of pressure-overload HF in mice,6 which provides an experimental platform to systematically study AP remodeling and propagation in the failing ventricle. In the present study we report significant remodeling of AP profiles in subepicardial and subendocardial myocytes and demonstrate that this remodeling greatly facilitates discontinuous AP conduction in the failing heart. At the same time, asymmetrical conduction between subepicardial and subendocardial myocytes is lost. We speculate that these mechanisms contribute significantly to ventricular arrhythmogenesis in this common syndrome.
Methods
Severe Thoracic Aortic Banding
Pressure-overload HF was induced by severe thoracic aortic banding6 in mice (Charles River Labs, Wilmington, Mass), according to protocols approved by the institution’s Animal Care and Use Committee.
Dissociation of Single Myocytes
Myocytes were dissociated enzymatically from LV as described7 with modifications. In brief, after retrograde perfusion with Krebs-Ringer solution at 2 mL/min (5 minutes), the heart was perfused with a fresh solution containing 0.8 mg/mL collagenase (Worthington type II, 15 minutes), and the apex was then removed. A small 90-degree curved forceps was used to carefully tear off a thin layer of endomyocardium and then isolate and collect the midregion myocardium. Endomyocardium, midmyocardium, and epimyocardium were separately minced into small pieces in KB solution. After trituration, subepicardial, midmyocardial, and subendocardial myocytes were studied within 4 to 6 hours. All steps were performed at 36°C with gassing of 95% O2/5% CO2.
Electrophysiological Recordings
Isolated myocytes were studied in a continuously superfused (1.5 mL/min) recording chamber fixed to an inverted microscope. Junctional potentials were corrected, the cell membrane was ruptured by application of further suction, and cell capacitance and series resistance were electrically compensated. Ito was induced by a 100-ms test pulse from -50 to +60 mV, and INa was inactivated by a 30-ms prepulse to -50mV from a holding potential of -80 mV. CdCl2 (0.2 mmol/L) was added to the external solution to block Ca2+ currents and the calcium-dependent transient outward current (Ito2)8 while recording Ito. AP and Ito data were collected after 5 to 10 minutes of equilibration between pipette solution and intracellular contents. Recording protocols were as described previously.9
Coupling Experiments
We used a novel coupling-clamp technique with 2 cells perfused in 2 separate chambers. Here, an electrical circuit provides variable coupling conductance (Gc) between 2 isolated myocytes that are not in physical contact with each other5 (Figure 1). We define Vm,1 as a time-varying membrane potential of cell 1 and Vm,2 as a time-varying membrane potential of cell 2. If the 2 cells were coupled together by an intercellular conductance Gc, there would be a time-varying current IC from cell 1 to cell 2 given by IC=(Vm,1-Vm,2)×Gc. We used a computer equipped with fast A/D and D/A converters (Digidata 1200, Molecular Devices, Sunnyvale, Calif) to compute IC from the sampled values of Vm,1 and Vm,2 and a selected value of Gc at time intervals of <25 μs. This value of IC is then added to cell 2 and subtracted from cell 1 to produce the desired value of Gc.
Figure 1.
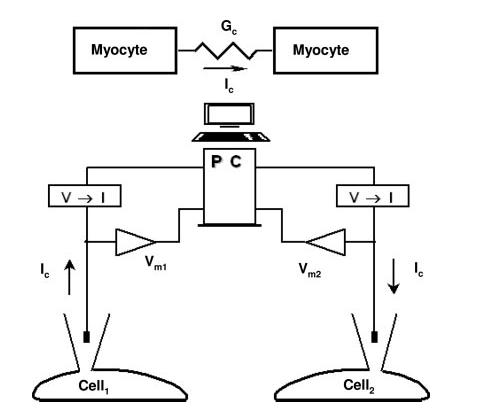
Schematic diagram of the coupling-clamp model.
Solutions
Krebs-Ringer solution was used for cell isolation (mmol/L): NaCl 35, KCl 4.75, KH2PO4 1.19, Na2HPO4 16, sucrose 134, NaCO3 25, glucose 10, HEPES 10, pH 7.4 with NaOH. KB solution was used for cell storage (mmol/L): taurine 10, glutamic acid 70, KCl 25, KH2PO4 10, glucose 22, EGTA 0.5, pH adjusted to 7.2 with KOH. Tyrode’s solution was used for AP and Ito recording (mmol/L): NaCl 135, MgCl2 1.1, CaCl2 1.8, KCl 5.4, HEPES 10, and glucose 10, pH 7.4 with NaOH. Pipette solution was used for AP recording (mmol/L): KCl 135, MgCl2 1, HEPES 10, Mg-ATP 5, Na2-creatine phosphate 5, pH 7.2 with KOH. Pipette solution was used for Ito recording (mmol/L): KCl 130, MgCl2 1, HEPES 10, EGTA 10, Mg-ATP 5, Na2-creatine phosphate 5, pH 7.2 with KOH.
Statistical Analysis
Ito amplitude was measured as the difference between the peak current and the sustained current remaining at the end of the 100-ms test pulse. Currents were normalized to cell capacitance and expressed as current density (pA/pF). For statistical purposes, each animal was considered an independent experiment and each cell a replicate observation. Statistical analysis was performed with the use of Sigmastat for Windows (Jandel Scientific, San Rafael, Calif). The paired and unpaired t test was used in most of the comparisons, and P<0.05 was regarded as significant. Mann-Whitney rank sum test was performed if either the normality or equal variance test failed.
The authors had full access to the data and take full responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
To study cell-cell coupling in HF, animals were subjected to severe thoracic aortic banding, a procedure that induces pressure-overload HF.6 Animals subjected to severe thoracic aortic banding manifested clinical features of HF, including lethargy, impaired mobility, and edema. Hearts dissected from a total of 25 mice 3 weeks after surgery were markedly enlarged and dilated with increased heart mass (heart weight to body weight ratio increased from 5.5±0.3 to 9.4±0.5 mg/g; 71% increase; P<0.05). Evidence of pulmonary venous congestion, a hallmark of HF, was noted with lung weight to body weight ratios increased from 7.3±1.1 to 13±1.4 mg/g; 78% increase; P<0.05). Ventricular myocytes isolated from these hearts were significantly hypertrophied: Subepicardial cell capacitance increased from 114.2±2.5 pF (n=134) in sham-operated control heart to 235±5.2 (n=64; P<0.01). Subendocardial cell capacitance increased from 119.1±2.7 (n=130) to 233.9±7.7 pF (n=52; P<0.01).
Transmural Differences in AP Repolarization in Mouse LV
In numerous mammalian species, electrically distinct “M cells” exist in the midregion of LV.10 To test whether M cells exist in mouse LV, we recorded APs at 1 Hz from subendocardial, midmyocardial, and subepicardial myocytes. As reported previously,7 subepicardial myocytes (n=44) manifested significantly shorter APD30 (P<0.05) and APD90 (P<0.01) (Table 1,Figure 2). APD30 and APD90 in midregion myocytes (n=11) were intermediate: 2.3±0.1 and 34.2±3.7 ms, respectively (P<0.05 compared with subepicardial myocytes). Thus, AP repolarization in midregion myocytes fell into a range between subendocardial and subepicardial myocytes, strongly suggesting that M cells do not exist in mouse LV. Because changes in AP repolarization in mouse LV are graded and monotonic, we chose to focus on cells at the 2 extremes (subepicardial and subendocardial) to study the effects of a transmural gradient of repolarization on AP conduction.
TABLE 1.
AP Characteristics in Myocytes From Mouse LV
| Subepicardial Myocytes (n 44) | Midmyocardial Myocytes (n 11) | Subendocardial Myocytes (n 22) | |
|---|---|---|---|
| RMP, mV | -77.1±1.3 | -77.4±1.1 | -77.6±1.3 |
| Peak, mV | 32.2±1.1 | 31.2±1.3 | 33.0±1.4 |
| dV/dt, V/s | 157.8±2.8 | 156.3±3.3 | 159.3±4.3 |
| APD30, ms | 2.0±0.2 | 2.3±0.1 | 2.5±0.2* |
| APD90, ms | 29.6±3.0 | 34.2±3.7* | 47.3±6.1* |
P<0.05 compared with subepicardial myocytes.
Figure 2.
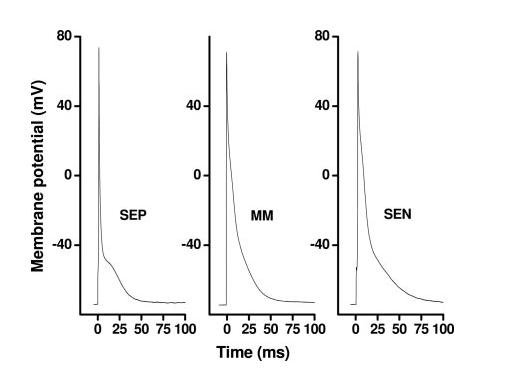
Representative AP waveforms recorded in subepicardial (SEP), midmyocardial (MM), and subendocardial (SEN) myocytes from normal mouse LV.
In mouse, with its rapid physiological heart rate, Ito is the predominant current governing ventricular myocyte repolarization.11 Thus, a transmural gradient of Ito density is likely to contribute to the differences in AP morphology between subepicardial and subendocardial myocytes. Consistent with this, the Ito channel blocker 4-aminopyridine (4-AP; 2 mmol/L) prolonged AP duration in both subepicardial and subendocardial myocytes (n=10) (Figure 3). Indeed, when Ito was blocked by 4-AP, APD in subepicardial and subendocardial cells became similar (P>0.05 for both APD30 and APD90).
Figure 3.
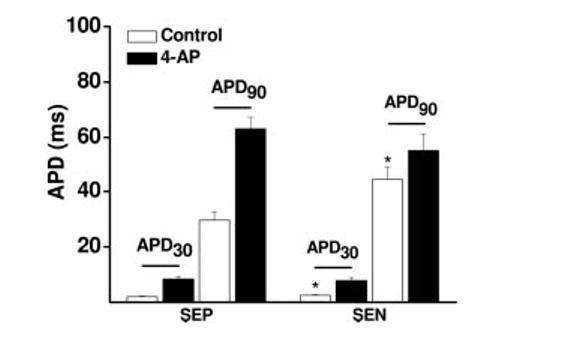
Changes in repolarization kinetics in mouse ventricular myocytes (n=10) in the absence and presence of 4-AP. Inhibition of Ito attenuates the transmural gradient of APD. APD30 indicates APD at 30% repolarization; APD90, APD at 90% repolarization. *P<0.05 compared with subepicardial myocytes (SEP). SEN indicates subendocardial myocytes.
HF-Associated AP Remodeling
Lengthening of the ventricular APD is commonly observed in both cardiac hypertrophy12 and HF.13 In many models of HF, diminished outward, repolarizing current secondary to down-regulated K+ channel expression (particularly Ito) is observed.13 Consistent with this, we observed that both APD30 and APD90 were significantly prolonged in both subepicardial and subendocardial myocytes from failing heart. However, prolongation of APDs in failing myocytes was disproportionate; HF-associated prolongation of APD in subepicardial cells was greater than that observed in subendocardial myocytes, a response that diminishes the normal, intrinsic transmural gradient of APD. No changes in peak potential of the AP upstroke (peak) or maximal upstroke velocity (dV/dt) were observed (Table 2).
TABLE 2.
Frequency-Dependent Changes in APD in Myocytes From Sham-Operated and Failing LV
| Mouse Myocytes and AP Parameters | SEP |
SEN |
||
|---|---|---|---|---|
| 1 Hz | 3 Hz | 1 Hz | 3 Hz | |
| Control (SEP=44, SEN=22) | ||||
| APD30, ms | 2.2±0.23 | 2.2±0.25 | 2.5 ±0.25 | 2.6±0.27 |
| APD90, ms | 29.6±3.0 | 31.6±3.8 | 47.3±6.1 | 51.3±6.7 |
| Peak, mV | 32.2±1.1 | 31.6±1.3 | 33.0±1.4 | 32.6±1.8 |
| dV/dt, V/s | 157.8±2.8 | 156.6±3.3 | 159.3±4.3 | 158.6±3.5 |
| HF (SEP= 12, SEN= 13) | ||||
| APD30, ms | 4.6±0.2 | 6.6±0.3* | 5.2±0.7 | 7.3±1.3* |
| APD90, ms | 63±4.6 | 75±6.4* | 70.4±6.2 | 82.1±6.2* |
| Peak, mV | 35.0±1.4 | 34.8±0.6 | 35.1±1.8 | 34.6±1.3 |
| dV/dt, V/s | 159.0±6.3 | 161.2±4.6 | 160.1±6.0 | 158.6±5.3 |
SEP indicates subepicardial myocytes; SEN, subendocardial myocytes.
P<0.05 compared with 1 Hz.
As heart rate increases and the duty cycle of each heartbeat shortens, the AP shortens in concert. However, frequency-dependent AP shortening is variable across species owing to the different ways in which AP repolarization is accomplished. For example, in human atrial myocytes, increasing pacing rate from 1 to 3 Hz significantly shortened APD90 but did not change APD30, whereas dramatic prolongation of both APD30 and APD90 was seen in rabbit atrial myocytes.14 In failing ventricular myocytes from both humans and rabbits, however, AP duration was prolonged at long cycle lengths, but AP prolongation relative to healthy controls was substantially diminished at shorter, more physiological cycle lengths.15
To test this in a model of HF in mice, we measured frequency-dependent changes in AP profiles in myocytes isolated from sham-operated and failing hearts. In sham-operated control heart, increasing stimulation rate from 1 to 3 Hz had no effect on AP morphology or duration. In contrast, rapid pacing in failing ventricular myocytes prolonged both APD30 and APD90 significantly, and these frequency-dependent changes were similar in subepicardial and subendocardial myocytes (Figure 4,Table 2). These data suggest that HF-associated remodeling of repolarization is amplified at near-physiological pacing rates.
Figure 4.
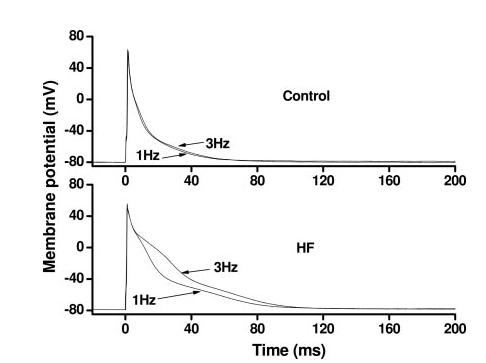
Frequency-dependent changes in AP profile differ between myocytes isolated from sham-operated control LV and from failing LV. Depicted here are representative, frequency-dependent AP waveform changes from 2 subendocardial myocytes.
Transmural Asymmetrical AP Conduction Is Abolished in Failing Heart
Given the critical role played by early phases of the AP in impulse conduction5 and the marked transmural gradient in APD30 in mammalian ventricle (Figure 2,Table 1), we hypothesized that transmural electrical conduction would be asymmetrical, with conduction from subendocardial to subepicardial myocytes being facilitated relative to the opposite direction. Furthermore, because HF-associated remodeling of the ventricular AP results in blunted differences in APD30 and APD90 between subepicardial and subendocardial cells, we hypothesized that the directional preference in conduction would be abolished. To test this, we coupled a subepicardial cell to a subendocardial cell to quantify the conductance required for successful AP propagation.
Consistent with our hypotheses, we found that in sham-operated, control hearts, conduction from subendocardial to subepicardial myocytes (n=23 cell pairs) required 1.2±0.1 nS of coupling conductance (Gc), whereas AP propagation from subepicardial to subendocardial myocytes required significantly greater Gc (3.1±0.2 nS; P<0.05). In other words, impulse conduction from subendocardial to subepicardial myocytes was relatively favored (Figure 5). Over a broad range of Gc, AP conduction occurred from subendocardial to subepicardial myocytes but was blocked from subepicardial to subendocardial myocytes. These data reveal substantial asymmetry in the facility with which impulses propagate across the normal LV wall and point to transmural gradients of Ito as a contributing mechanism.
Figure 5.
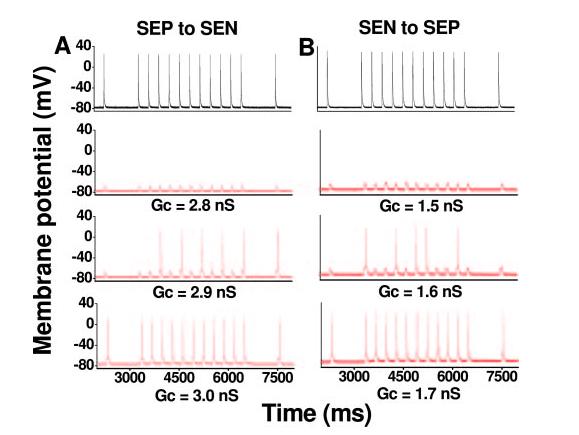
AP propagation in a coupled-cell pair from control mouse LV. Representative recordings from the leader cell are depicted in black, and recordings from the follower cell are in red. AP conduction from a subendocardial myocyte (SEN) to a subepicardial myocyte (SEP) (B) failed at Gc 1.5 nS. Increasing Gc to 1.6 nS led to partial impulse propagation. By further increasing Gc to 1.7 nS, all APs propagated successfully at both pacing frequencies (1 Hz, 3 Hz). For the same cell pair, however, AP propagation from SEP to SEN (A) required more coupling conductance: Gc 2.9 for partial propagation and Gc 3.0 for full AP propagation. Thus, there is unidirectional conduction block (from SEP to SEN) over a wide range of Gc (from 1.7 to 2.8).
As a control, we also tested conduction in myocytes from the same region of LV. AP conduction within the same region of LV required similar Gc: The critical Gc for conduction between subendocardial cell pairs (n=10) was 1.38±0.05 nS, and Gc required for subepicardial-subepicardial conduction (n=12) was 1.60±0.06. Thus, consistent with a requirement for early repolarization current in impulse propagation, Gc required for conduction between subendocardial cells was significantly less (P<0.05) than Gc between subepicardial cells. These data suggest the presence of uniform conductivity within a single region of ventricular myocardium with uniform AP characteristics.
In contrast to healthy ventricle, AP propagation in failing heart manifested no directional preference (Figure 6). In failing ventricle, where APD is significantly prolonged and the transmural gradient of repolarization is attenuated, the critical Gc for AP conduction from subendocardial to subepicardial myocytes (3.2±1.0 nS) was not significantly different (P>0.05) from that required for subepicardial to subendocardial conduction (3.5±1.2 nS; n=5 cell pairs for both directions). In control ventricle, unidirectional conduction block existed over a wide range of Gc (eg, 1.7 to 2.8 nS for cell pair of Figure 5), whereas in failing heart, directional preference of AP propagation was absent (Figure 6).
Figure 6.
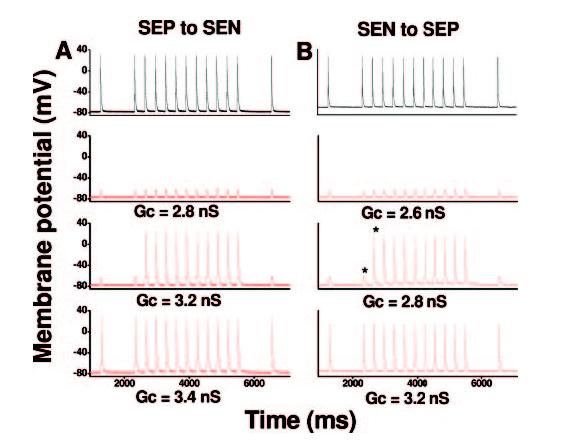
AP propagation in a coupled-cell pair from failing LV. Representative recordings from the leader cell are depicted in black, and recordings from the follower cell are in red. Critical Gc for AP propagation from subendocardial myocyte (SEN) to subepicardial myocyte (SEP) was 3.2 nS (B), and Gc required for AP propagation from SEP to SEN was 3.4 nS (A).
Successful AP conduction depends on the amount of current provided by the source (ie, leader cell) as well as the excitability of the follower cell. To test whether asymmetrical conduction in normal ventricle stems from differential intrinsic excitability of subepicardial and subendocardial myocytes, we injected current into isolated ventricular myocytes and measured the current required to achieve AP threshold. In sham-operated myocytes, the critical current (2-ms duration) for AP initiation was not different in subendocardial versus subepicardial myocytes: 1.70±0.3 nA in subendocardial cells (n=22) and 1.68±0.3 nA in subepicardial myocytes (n=44; P>0.05). In HF, the critical current required to achieve AP threshold in failing myocytes was greater than in control cells but did not differ between subendocardial and subepicardial myocytes: 2.70±0.13 nA in subendocardial cells (n=12) and 2.86±0.17 nA in subepicardial myocytes (n=11; P>0.05). Together, these data suggest that differing degrees of myocyte excitability do not account for the directionality of AP conductance in either control or HF conditions.
Frequency-Dependent Facilitation of AP Conduction in the Failing Heart
We observed frequency-dependent prolongation of AP duration in myocytes from failing heart but not in cells from sham-operated, control heart (Figure 4,Table 2). Given this, we predicted that fast pacing would facilitate AP propagation in the failing heart. To test this, we coupled 2 myocytes isolated from failing LV and determined the critical Gc for AP propagation at different pacing frequencies. In response to rapid pacing (3 Hz), the critical Gc required for successful AP propagation in failing myocytes decreased from 3.3±0.14 to 2.7±0.12 nS (n=10; P<0.01). In contrast, rapid pacing did not alter the conductance required for AP propagation in myocytes from sham-operated, control hearts (data not shown). Together, these data suggest that fast pacing facilitates AP conduction exclusively in failing heart.
We next hypothesized that the frequency-dependent facilitation of AP conduction in failing myocytes results from frequency-dependent prolongation of APD due to inhibition of Ito in those cells. To test this, we replotted APs illustrated in Figure 6B (third row) at a faster time scale (Figure 7). Here we observed that increased stimulation frequency from 1 to 3 Hz provoked an immediate prolongation of APD30 in the pacing cell. This slowing of early repolarization renders the pacing cell capable of providing additional coupling current to the follower cell to induce membrane depolarization, which triggers an AP in the follower cell.
Figure 7.
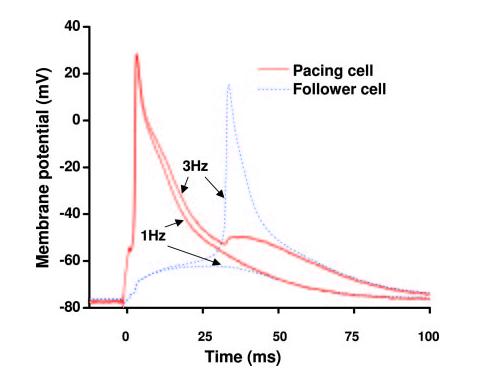
A replot of APs from Figure 6B (asterisks, third row) at a faster time scale. In failing myocytes, early repolarization of the AP is delayed immediately on increasing stimulation frequency, contributing to facilitated cell-cell electrotonic coupling.
Finally, we hypothesized that frequency-dependent prolongation of APD30 is a consequence of inhibition of Ito. To test this, we compared Ito recorded from failing myocytes with Ito recorded from healthy, control myocytes. In the present study we found that increasing stimulation frequency significantly inhibited Ito amplitude in myocytes isolated from failing LV but had no effect on Ito in myocytes from sham-operated LV (Figure 8). In a total of 8 myocytes from failing LV, increasing stimulation frequency decreased Ito from 8.3±1.5 to 5.3±1.3 pA/pF (39±5% decline; P<0.05). In contrast, no significant frequency-dependent inhibition of Ito was observed in ventricular myocytes from sham-operated LV (data not shown). These findings are consistent with frequency-dependent inhibition of Ito as a mechanism underlying frequency-dependent APD30 prolongation and consequent loss of impulse conduction asymmetry in failing ventricle.
Figure 8.
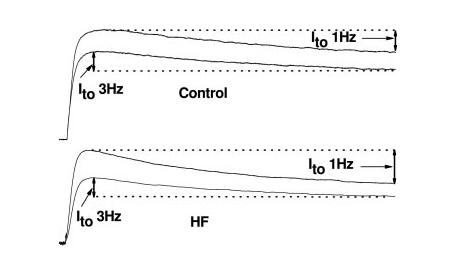
Frequency-dependent responses of Ito recorded in myocytes from control (top) and failing (bottom) LV. Representative recordings demonstrate that increasing stimulation frequency from 1 to 3 Hz significantly inhibited Ito amplitude in failing ventricular myocytes but had no effect on Ito in control myocytes.
Discussion
Lengthening of the ventricular AP and perturbations in impulse conduction are each hallmarks of electrical remodeling in HF. Little is known, however, about how HF-related AP remodeling contributes to altered propagation of electrical excitability. In this study we demonstrate dramatic prolongation of the AP in ventricular myocytes from failing, murine ventricle, confirming prior studies and extending them to the mouse. We also report dramatic frequency dependence of this electrical remodeling response. Furthermore, we demonstrate that the facility of electrical propagation in the ventricle depends on the direction of charge movement, with propagation in the physiological direction (endocardium to epicardium) being preferred in normal ventricle. In HF, however, this asymmetry is abolished as a result of disordered repolarization in failing myocytes. Together, these findings shed light on mechanisms of pathologically enhanced electrical excitability in HF and consequent arrhythmogenesis.
Transmural Gradient of AP Repolarization
Ito density varies across the thickness of the ventricular wall in numerous mammalian species.12 As a result, AP profiles are significantly different in myocytes isolated from different regions of the ventricular wall, which leads to transmural heterogeneity of the recovery of electrical excitability. In addition, AP propagation in canine heart traverses a zone of midmyocardial cells, which are morphologically indistinguishable from other myocytes but display much longer APD at slow pacing rates or in the presence of APD-prolonging agents.16 Whereas these M cells have been observed in dissociated myocytes from pig heart,17 they appear to be absent in intact pig hearts.18 The existence and location of M cells are modulated by anemone toxin, suggesting that they represent a functionally defined rather than anatomically defined entity.19
The presence and distribution of ventricular M cells have been investigated in detail only in canine LV; indeed, to date, existence of M cells in murine ventricle has not been reported. Consistent with this, we failed to identify such cells in our study. The prolonged APD of M cells has been attributed to relatively small IKs and relatively large sodium-calcium exchanger current (INCX) compared with other cells.20 Mouse LV does not have IKs,21 and INCX is less relevant in shaping mouse APD, which we propose may be the reason why M cells have not been detected in mouse LV.
In any event, the extent to which M cells contribute to the transmural dispersion of ventricular repolarization in intact ventricle remains unclear. For example, an intramural zone of prolonged activation recovery intervals or repolarization times has not been observed in intact human heart.3,4 Rather, transmural dispersion of repolarization in intact heart is marked primarily by the differences in AP duration between subepicardial and subendocardial myocytes.22 Thus, although subepicardial and subendocardial myocytes are never directly coupled, AP propagation between them provides an accurate reflection of impulse propagation in intact ventricle, where AP duration shortens progressively from endocardial to epicardial zones of tissue.
Asymmetry of Transmural AP Propagation in Normal LV
Using a unique, computer-based cell-cell coupling paradigm,5 we have uncovered significant asymmetry in AP propagation across the mouse ventricle, where conduction is favored in the direction from endocardium to epicardium, the direction of normal, physiological depolarization. Owing to the longer APD30 in subendocardial myocytes, AP propagation from subendocardial cells to subepicardial cells is privileged (requires less coupling conductance than propagation in the opposite direction). Importantly, this asymmetry in normal tissue favors physiological conduction but disfavors abnormal, retrograde conduction such as that arising from a site of focal automaticity in the subepicardium.
Our data suggest that differential intrinsic excitability of subepicardial and subendocardial myocytes cannot account for the directionality of AP conductance in either control or HF conditions. However, the current required for AP initiation and the critical Gc required for successful AP conduction in failing myocytes were both significantly larger in failing cells. It is likely that increased cell size (and accordingly decreased input resistance) contributes to this requirement. According to Ohm’s law, to depolarize the follower cell membrane to a level (ΔV) sufficient to trigger an AP, the leader cell must provide more coupling current under conditions in which cellular input resistance R is decreased. Although the fast inward Na+ current is an important determinant for the excitability of cardiac myocytes, our observations of unchanged maximal upstroke velocity and AP peak potential in failing heart (Table 2) suggest that decreases in inward Na+ current, if any, cannot account for a requirement of increased excitation current in failing myocytes.
HF-Associated Abolition of Asymmetrical Conduction
Most prior studies have focused on HF-associated prolongation of the entire AP (APD90) owing to its association with effective refractory period and dispersion of repolarization. Much less attention has been centered on early phases of the AP, those governed by Ito. In addition, the majority of prior investigations studying remodeling of cardiac conductivity in HF have focused on decreased membrane excitability and increased intercellular coupling resistance. However, a critical aspect affecting AP conduction between myocytes, the cellular energy source, has been less well studied. Successful impulse conduction in HF, in which the intercellular coupling resistance is increased because of interstitial fibrosis, disordered extracellular matrix, and alterations in gap junctions, hinges critically on the electrical energy provided by the depolarized myocyte functioning as the leader cell. By electrically coupling cells in isolation, the specific contribution of changes in early repolarization to pathophysiological conduction in HF can be analyzed, apart from the contributions of other factors.
Normally, electrical activation of the ventricle initiates in the subendocardial Purkinje network and spreads outward through the ventricular wall. Although the epicardium is activated last, it repolarizes faster than the endocardium. The combination of this activation sequence plus repolarization gradient produces an ECG T wave with polarity similar to the QRS. In other words, in intact heart, the major determinants of TDR are the repolarization characteristics of subepicardial and subendocardial myocytes. In contrast, when an AP conducts from subepicardium to subendocardium, TDR is greatly amplified owing to the fact that the subendocardial cell, with its long APD, is activated last. The heightened TDR favors reentry, an important mechanism for ventricular tachyarrhythmia. This fact is corroborated by the high incidence of ventricular tachycardia in HF patients subjected to epicardial-site pacing.23 This proarrhythmic mechanism, however, is counterbalanced by the fact that AP propagation from subepicardial to subendocardial myocytes is disfavored (Figure 5).
In failing heart, this “protective” asymmetry is lost, which we speculate contributes to the propensity to ventricular rhythm disturbances in these patients. In our model of pressure-overload HF, remodeling of the ventricular AP attenuated the transmural gradient of repolarization in LV. As a result, preferential directionality of AP propagation in the failing heart is lost, and abnormal impulses generated in the subepicardium are capable of spreading through the ventricle.
The fact that the critical Gc required for successful AP conduction is higher in HF is a result of increased myocyte size and, accordingly, decreased cellular input resistance. In HF in situ, intercellular coupling resistance, a resistance that impedes charge flow from leader to follower cell, is increased. Together, these 2 effects greatly reduce AP conductivity and heighten the requirement for electrical energy that occurs during early phases of AP repolarization.
Findings reported here confirm and extend a recent study in canine, pacing-induced cardiomyopathy.24 These in vivo investigations demonstrated faster impulse conduction in subendocardium relative to subepicardium. Furthermore, these investigators observed facilitated conduction in HF at rapid stimulation rates relative to control. In the present study we provide evidence that a greater amount of coupling current provided by leader cells during early phases of repolarization is a major underlying mechanism. Furthermore, we have uncovered a role for frequency-dependent suppression of Ito as a mechanism underlying the facilitated conduction in HF at rapid stimulation rates.
Limitations
Mouse heart is electrophysiologically different from human heart. For instance, Ito is the major current underlying AP repolarization in mouse. Furthermore, the L-type calcium current and INCX are less important in mouse ventricle than in human.25 In addition, human ventricular myocytes respond to increased rates of stimulation with a shortened APD90,26 whereas APD in normal murine ventricular myocytes does not change. Despite these differences, these 2 species share numerous physiological properties that contribute to cardiac conduction, including the transmural gradient of APD30 in healthy LV and the attenuation of this gradient in failing LV.26,27 Furthermore, the currents (and channels) that mediate early repolarization (Ito) are highly conserved between mice and humans. In other words, even though APD is much shorter in mice than in humans, the currents that determine the early repolarization phase in mouse ventricular myocytes, and hence govern cell-cell coupling, are very similar to those in humans. Thus, the mouse is an excellent model in which to study electrotonic cell-cell coupling and the effects of HF thereon. Finally, frequency-dependent slowing of repolarization is similar in human failing LV26,27 and mouse. In light of these facts, abnormal conduction in mouse LV as reported here may have significant implications for human HF.
Implications for Arrhythmogenesis
In the present study we have demonstrated 2 interacting types of electrical remodeling in failing ventricle: loss of asymmetrical conduction and facilitated impulse propagation at rapid stimulation rates. Loss of asymmetrical conduction would be expected to allow premature excitations generated from an epicardial site, which is known to be proarrhythmic,23 to propagate easily through the entire failing heart, a response that would be relatively disfavored in normal LV. Furthermore, facilitated conduction at rapid stimulation rates would tend to favor propagation of those premature impulses that are frequently observed in HF. We speculate that, together, these effects combine to promote arrhythmia propagation in failing heart and the suggested proarrhythmic effects associated with epicardial pacing, such as with biventricular devices.28
CLINICAL PERSPECTIVE
Patients with heart failure are at markedly increased risk of ventricular arrhythmia, ranging from benign premature beats to malignant rhythm disturbances that culminate in sudden cardiac death. Among the mechanisms contributing to this phenotype are alterations in repolarization of the myocyte action potential. In fact, disease-related remodeling of action potential repolarization is an established cause of ventricular tachycardia. In the present study we report that disease-associated changes in the early phases of action potential repolarization are a major mechanism of proarrhythmia. We show that remodeling of early repolarization alters the cell-cell coupling energy source, such that the normal preferentiality of impulse conduction from endocardium to epicardium is abolished. As a result, conduction of premature electrical events is abnormally supported, which is likely to contribute to arrhythmia development. These data are consistent with a proarrhythmic effect of epicardial-site pacing, a suggestion that has been supported by clinical observations. Together, our findings reveal mechanisms whereby disease-related remodeling of the ventricular action potential–distinct from other pathological changes, such as fibrosis–promotes a proarrhythmic substrate. As a consequence, these data point to disease-related molecular changes in cardiomyocyte repolarization as potential targets of novel therapeutic intervention in heart failure.
Acknowledgments
This work was supported by grants (to Dr Hill) from the Donald W. Reynolds Cardiovascular Clinical Research Center, the National Institutes of Health (HL-075173), and the American Heart Association (EI 0640084N).
Footnotes
Disclosures
References
- 1.Antzelevitch C, Fish J. Electrical heterogeneity within the ventricular wall. Basic Res Cardiol. 2001;96:517–527. doi: 10.1007/s003950170002. [DOI] [PubMed] [Google Scholar]
- 2.Xia Y, Liang Y, Kongstad O, Liao Q, Holm M, Olsson B, Yuan S. In vivo validation of the coincidence of the peak and end of the T wave with full repolarization of the epicardium and endocardium in swine. Heart Rhythm. 2005;2:162–169. doi: 10.1016/j.hrthm.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 3.Taggart P, Sutton PM, Opthof T, Coronel R, Trimlett R, Pugsley W, Kallis P. Transmural repolarisation in the left ventricle in humans during normoxia and ischaemia. Cardiovasc Res. 2001;50:454–462. doi: 10.1016/s0008-6363(01)00223-1. [DOI] [PubMed] [Google Scholar]
- 4.Conrath CE, Wilders R, Coronel R, de Bakker JM, Taggart P, de Groot JR, Opthof T. Intercellular coupling through gap junctions masks M cells in the human heart. Cardiovasc Res. 2004;62:407–414. doi: 10.1016/j.cardiores.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 5.Wang YG, Wagner MB, Kumar R, Goolsby WN, Joyner RW. Fast pacing facilitates discontinuous action potential propagation between rabbit atrial cells. Am J Physiol. 2000;279:H2095–H2103. doi: 10.1152/ajpheart.2000.279.5.H2095. [DOI] [PubMed] [Google Scholar]
- 6.Rothermel BA, Berenji K, Tannous P, Kutschke W, Dey A, Nolan B, Yoo KD, Demetroulis E, Gimbel M, Cabuay B, Karimi M, Hill JA. Differential activation of stress-response signaling in load-induced cardiac hypertrophy and failure. Physiol Genomics. 2005;23:18–27. doi: 10.1152/physiolgenomics.00061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z, Kutschke W, Richardson KE, Karimi M, Hill JA. Electrical remodeling in pressure-overload cardiac hypertrophy: role of calcineurin. Circulation. 2001;104:1657–1663. doi: 10.1161/hc3901.095766. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Feng J, Shi H, Pond A, Nerbonne JM, Nattel S. Potential molecular basis of different physiological properties of the transient outward K+ current in rabbit and human atrial myocytes. Circ Res. 1999;84:551–561. doi: 10.1161/01.res.84.5.551. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Xu H, Kumar R, Tipparaju SM, Wagner MB, Joyner RW. Differences in transient outward current properties between neonatal and adult human atrial myocytes. J Mol Cell Cardiol. 2003;35:1083–1092. doi: 10.1016/s0022-2828(03)00200-1. [DOI] [PubMed] [Google Scholar]
- 10.Sicouri S, Antzelevitch C. A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle: the M cell. Circ Res. 1991;68:1729–1741. doi: 10.1161/01.res.68.6.1729. [DOI] [PubMed] [Google Scholar]
- 11.Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 alpha subunit. Circ Res. 1998;83:560–567. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- 12.Hill JA. Electrical remodeling in cardiac hypertrophy. Trends Cardiovasc Med. 2003;13:316–322. doi: 10.1016/j.tcm.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Armoundas AA, Wu R, Juang G, Marban E, Tomaselli GF. Electrical and structural remodeling of the failing ventricle. Pharmacol Ther. 2001;92:213–230. doi: 10.1016/s0163-7258(01)00171-1. [DOI] [PubMed] [Google Scholar]
- 14.Fermini B, Wang Z, Duan D, Nattel S. Differences in rate dependence of transient outward current in rabbit and human atrium. Am J Physiol. 1992;263:H1747–H1754. doi: 10.1152/ajpheart.1992.263.6.H1747. [DOI] [PubMed] [Google Scholar]
- 15.Vermeulen JT, McGuire MA, Opthof T, Coronel R, de Bakker JM, Klopping C, Janse MJ. Triggered activity and automaticity in ventricular trabeculae of failing human and rabbit hearts. Cardiovasc Res. 1994;28:1547–1554. doi: 10.1093/cvr/28.10.1547. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade des pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038–2047. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- 17.Stankovicova T, Szilard M, De SI, Sipido KR. M cells and transmural heterogeneity of action potential configuration in myocytes from the left ventricular wall of the pig heart. Cardiovasc Res. 2000;45:952–960. doi: 10.1016/s0008-6363(99)00418-6. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Sinovas A, Cinca J, Tapias A, Armadans L, Tresanchez M, Soler-Soler J. Lack of evidence of M-cells in porcine left ventricular myocardium. Cardiovasc Res. 1997;33:307–313. doi: 10.1016/s0008-6363(96)00205-2. [DOI] [PubMed] [Google Scholar]
- 19.Ueda N, Zipes DP, Wu J. Functional and transmural modulation of M cell behavior in canine ventricular wall. Am J Physiol. 2004;287:H2569–H2575. doi: 10.1152/ajpheart.00526.2004. [DOI] [PubMed] [Google Scholar]
- 20.Antzelevitch C. Modulation of transmural repolarization. Ann N Y Acad Sci. 2005;1047:314–323. doi: 10.1196/annals.1341.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brunet S, Aimond F, Li H, Guo W, Eldstrom J, Fedida D, Yamada KA, Nerbonne JM. Heterogeneous expression of repolarizing, voltage-gated K+ currents in adult mouse ventricles. J Physiol. 2004;559:103–120. doi: 10.1113/jphysiol.2004.063347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antzelevitch C. Transmural dispersion of repolarization and the T wave. Cardiovasc Res. 2001;50:426–431. doi: 10.1016/s0008-6363(01)00285-1. [DOI] [PubMed] [Google Scholar]
- 23.Medina-Ravell VA, Lankipalli RS, Yan GX, Antzelevitch C, Medina-Malpica NA, Medina-Malpica OA, Droogan C, Kowey PR. Effect of epicardial or biventricular pacing to prolong QT interval and increase transmural dispersion of repolarization: does resynchronization therapy pose a risk for patients predisposed to long QT or torsade de pointes. Circulation. 2003;107:740–746. doi: 10.1161/01.cir.0000048126.07819.37. [DOI] [PubMed] [Google Scholar]
- 24.Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res. 2004;95:717–725. doi: 10.1161/01.RES.0000144125.61927.1c. [DOI] [PubMed] [Google Scholar]
- 25.Hasenfuss G. Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res. 1998;39:60–76. doi: 10.1016/s0008-6363(98)00110-2. [DOI] [PubMed] [Google Scholar]
- 26.Nabauer M, Beuckelmann DJ, Uberfuhr P, Steinbeck G. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation. 1996;93:168–177. doi: 10.1161/01.cir.93.1.168. [DOI] [PubMed] [Google Scholar]
- 27.Nabauer M, Beuckelmann DJ, Erdmann E. Characteristics of transient outward current in human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:386–394. doi: 10.1161/01.res.73.2.386. [DOI] [PubMed] [Google Scholar]
- 28.Bristow MR, Saxon LA, Boehmer J, Krueger S, Kass DA, De Marco T, Carson P, DiCarlo L, DeMets D, White BG, DeVries DW, Feldman AM. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med. 2004;350:2140–2150. doi: 10.1056/NEJMoa032423. [DOI] [PubMed] [Google Scholar]