

Abstract
Computation of LOD scores is a valuable tool for mapping disease-susceptibility genes in the study of Mendelian and complex diseases. However, computation of exact multipoint likelihoods of large inbred pedigrees with extensive missing data is often beyond the capabilities of a single computer. We present a distributed system called “SUPERLINK-ONLINE,” for the computation of multipoint LOD scores of large inbred pedigrees. It achieves high performance via the efficient parallelization of the algorithms in SUPERLINK, a state-of-the-art serial program for these tasks, and through the use of the idle cycles of thousands of personal computers. The main algorithmic challenge has been to efficiently split a large task for distributed execution in a highly dynamic, nondedicated running environment. Notably, the system is available online, which allows computationally intensive analyses to be performed with no need for either the installation of software or the maintenance of a complicated distributed environment. As the system was being developed, it was extensively tested by collaborating medical centers worldwide on a variety of real data sets, some of which are presented in this article.
Computation of LOD is a valuable tool for mapping disease-susceptibility genes in the study of Mendelian and complex diseases. Computation of the LOD score—defined as 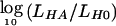 , where LH0 is the likelihood under the null hypothesis of no linkage between markers and a disease locus and LHA is the likelihood of linkage—requires efficient methods, especially for large pedigrees and many markers. A linkage analysis is performed by placing a trait locus at various positions along a map of markers, to locate regions that show evidence of linkage and that merit further study. To extract full linkage information from pedigree data, it is desirable to perform multipoint likelihood computations with the joint use of all available relevant data.
, where LH0 is the likelihood under the null hypothesis of no linkage between markers and a disease locus and LHA is the likelihood of linkage—requires efficient methods, especially for large pedigrees and many markers. A linkage analysis is performed by placing a trait locus at various positions along a map of markers, to locate regions that show evidence of linkage and that merit further study. To extract full linkage information from pedigree data, it is desirable to perform multipoint likelihood computations with the joint use of all available relevant data.
There are two main approaches for computing multipoint likelihoods: the Elston-Stewart1 and the Lander-Green2 algorithms. The complexity of the Elston-Stewart algorithm is linear in the number of individuals but exponential in the number of markers. On the other hand, the complexity of the Lander-Green algorithm increases linearly in the number of markers but exponentially in the number of individuals in the pedigree. A recently proposed approach is to combine and generalize the previous two methods by using the framework of Bayesian networks as the internal representation of linkage analysis problems.3 Using this representation enables the efficient handling of a wide variety of likelihood computations by automatic choice of computation order, according to the problem at hand.
The computation of exact multipoint likelihoods of large inbred pedigrees with extensive missing data is often beyond the computational capabilities of a single computer. Two complementary approaches can facilitate more-demanding linkage computations: designing more-efficient algorithms and parallelizing computation to use multiple computers. Both approaches have been pursued over the years. Several algorithmic improvements of exact likelihood computations have been reported.3–10 For example, efficient implementation of the Lander-Green algorithm by the GENEHUNTER program allows multipoint analysis of medium-sized pedigrees with large numbers of markers7,11; VITESSE v.2 implements optimization for the Elston-Stewart algorithm, extending its computational boundaries by orders of magnitude5,9; and SUPERLINK applies enhanced optimization techniques for finding better orders of computation in Bayesian networks, making it possible to perform multipoint analysis of larger inbred families.3,12,13 Several parallel algorithms for linkage analysis have been reported.14–21 Parallel computing was successfully applied to improve the performance of LINKAGE and FASTLINK packages, speeding up the computations with the use of a set of dedicated processors.14,15,17–19 Efficient parallel implementations of GENEHUNTER, which involve dividing the computations over high-performance processors, achieve significant speedups compared with the serial version and allow for the analysis of larger pedigrees.20
Despite the advantages of parallel computations, the use of parallel programs for linkage analysis is quite limited by their dependency on the availability of high-performance execution environments, such as a cluster of high-performance, dedicated machines or a supercomputer. Such hardware can usually be found only in specialized research centers because of its high cost and operational complexity.
In this article, we introduce a distributed system for exact linkage analysis called “SUPERLINK-ONLINE,” which is capable of analyzing inbred families of several hundreds of individuals with extended missing data, thereby outperforming all existing tools for exact linkage computations on such inputs. The system is based on the parallelization of SUPERLINK, which is a stateof-the-art program for parametric linkage analysis of dichotomous/binary traits with large inbred pedigrees. Our approach, made possible by recent advances in distributed computing,22 eliminates the need for expensive hardware by distributing the computations over thousands of nondedicated personal computers, using their idle cycles. The main algorithmic challenge has been to efficiently split a large linkage analysis task for execution in a dynamic, distributed running environment. Such environments are characterized by the presence of many computers with different capabilities and operating systems, frequent failures, and extreme fluctuations of the number of computers available for execution.
SUPERLINK-ONLINE delivers the newest information technology to geneticists via a simple Internet interface that completely hides the complexity of the underlying distributed system. The system allows for the concurrent submission of linkage tasks by multiple users, dynamically adapting the parallelization strategy in accordance with the current load and the number of computers available to perform the computations. As the system was being developed, it was extensively tested by collaborating medical centers worldwide on a variety of real data sets, some of which are presented in this article. Our results show improved running times of >2 orders of magnitude versus the serial version of the program. This allows users to avoid the undesirable breakup of larger pedigrees into smaller pieces, which often weakens the linkage signal.
Background
Exact Parametric Linkage Analysis Software
Linkage analysis tests for cosegregation between alleles at markers in a chromosomal region and at a trait locus of interest. In parametric linkage analysis, the likelihood for evidence of linkage L(θ) is computed under a given model of disease-allele frequency, penetrances, recombination fraction between markers, and recombination fraction (θ) between a disease locus and a reference locus. Computations of L(θ) in this article assume Hardy-Weinberg equilibrium, linkage equilibrium, and no interference, which are common assumptions in most linkage analyses performed to date. It is common to consider 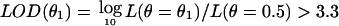 as indication of linkage.23
as indication of linkage.23
Consider a pedigree and let xi denote the observations that include affection status and marker information at one or multiple loci of the ith pedigree member. The likelihood is the probability of the observations defined by L(θ)=P(x1,x2,…,xm|θ). Elston and Stewart have shown that, for simple pedigrees without loops, the likelihood may be represented as the telescoping sum
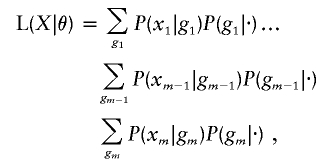 |
where the individuals are ordered such that parents precede their children and where P(gi|·) represents either the ith child’s multilocus genotype, given the parental multilocus genotypes, or the probability that a founder individual (with no parents in the pedigree) has a multilocus genotype gi.1
The Elston-Stewart algorithm and its extensions have been employed in many linkage analysis programs. Whereas the early implementations (i.e., LIPED24 and LINKAGE25,26) allow for computation of two-point and multipoint LOD scores of small pedigrees with the use of few markers, their successors—such as later versions of LINKAGE and FASTLINK4—extend their capabilities considerably. The current serial version of FASTLINK improves the analysis of complex pedigrees by efficient loop-breaking algorithms.27 Further versions of FASTLINK use parallel computing to achieve higher performance.15,17–19 Another example of efficient optimizations is VITESSE v.1, which raises the computational boundaries of the Elston-Stewart algorithm and allows for computation of multipoint LOD scores for several polymorphic markers with many unknown genotypes via “set recoding.”5
The complexity of the Elston-Stewart algorithm grows linearly with the size of the pedigree but exponentially with the number of markers in the analysis, since the algorithm essentially performs summation over all possible genotype vectors. Also, in the extension for the analysis of complex pedigrees, the loop-breaking method grows exponentially in the number of different loop breakers to be considered.
Exact multipoint linkage computations involving many markers for small-to-medium–sized families was first made practical with the introduction of the GENEHUNTER software,11 which uses the Lander-Green algorithm.2 Whereas, in the Elston-Stewart algorithm, the unobserved quantity is the genotype, the Lander-Green algorithm proposes to condition observations on inheritance vectors for each locus, where the inheritance vector is a binary vector indicating the parental source of each allele for each nonfounder in the pedigree.
The Lander-Green algorithm can potentially compute multipoint likelihoods on practically unbounded numbers of markers. However, the computational capabilities of the algorithm are restricted to the analysis of pedigrees of moderate size, since its computing time and memory requirements scale exponentially in the number of nonfounders, which is translated into the number of inheritance vectors to be considered. Later versions of GENEHUNTER optimized the performance by applying the Fast Fourier Transform for matrix multiplication7 and by considering only the set of inheritance vectors compatible with the observed genotypes.28 ALLEGRO8 and MERLIN10 present alternative implementations of the Lander-Green algorithm that show speedups of up to 2 orders of magnitude versus GENEHUNTER, achieved through further reduction of the inheritance vector space and application of software optimization techniques. Advances in parallel computing allowed parallelization of GENEHUNTER for execution on clusters of high-performance workstations, enabling faster computations.20 Also, GENEHUNTER-TWOLOCUS29 has been parallelized.21
Another approach is presented by SUPERLINK, in which the pedigree data are represented as a Bayesian network, allowing one to significantly improve the performance by optimizing the order in which variables are eliminated.3,12 As opposed to the Elston-Stewart and Lander-Green algorithms, in which variables are eliminated in a predetermined order, SUPERLINK finds the order of variable elimination at run time according to the problem at hand, enabling multipoint-likelihood computations of large inbred families that cannot be performed by other current linkage analysis software.
Bayesian Networks
Bayesian networks,30,31 also known as directed graphical models, are a knowledge representation formalism that offers a powerful framework to model complex multivariate problems, such as the ones posed by genetic analysis. A Bayesian network is defined via directed acyclic graphs (DAGs). A DAG is a directed graph with no directed cycles. Each node v has a set of parents Pav—namely, a set of vertices from which there are edges leading into v in the DAG. A Bayesian network is a DAG, where each vertex v corresponds to a discrete variable Xv∈X with a conditional probability distribution P(Xv=xv|Pav=pav), and the joint probability distribution P(X) is the product of the conditional probability distributions of all variables. In other words,
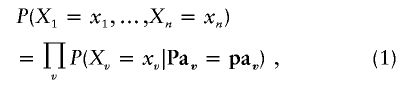 |
where pav is the joint assignment {Xi=xi|Xi∈Pav} to the variables in Pav. When Pav=∅, the respective term in equation (1) reduces to P(Xv=xv). Note that each missing edge represents a conditional independence assertion. This is the key factor for using Bayesian networks to efficiently handle joint probability distributions for large numbers of variables.
An example is shown in figure 1. This example shows a Bayesian network for three-point analysis (two markers and one disease locus) of a nuclear family with two typed children. The genetic loci variables of individual i at locus j are denoted by Gmi,j and Gpi,j for maternal and paternal alleles, respectively. Variables Ei,j, Smi,j, and Spi,j denote the marker phenotypes (unordered pair of alleles) and the maternal and the paternal selector variables, respectively, of individual i at locus j. Variable Ei denotes the affection status of individual i. Individual 4 is affected, as indicated by node E4. Highlighted nodes represent evidence variables, including available marker phenotypes and affection status. Marker phenotypes of the parents are unknown in this example. The quantities P(Gmi,j) and P(Gpi,j) represent allele frequencies, P(Smi,j|Smi,j-1) and P(Spi,j|Spi,j-1) represent recombination probabilities, and P(Ei|Gmi,j,Gpi,j) represent penetrances. The joint distribution is the product of all probability tables. The assumptions of no interference and of Hardy-Weinberg and linkage equilibria are encoded by edges missing in the Bayesian network. More details are given elsewhere.32
Figure 1.

A Bayesian network for three-point analysis
Bayesian networks are used in this article for computing the probability of evidence, which is represented as a joint assignment e={Xe1=e1,Xe2=e2,…,Xem=em}, to a subset of variables E={Xe1,Xe2,…,Xem}. It is computable from the joint probability distribution table by summing over all variables X1,…,Xk not in E—namely,
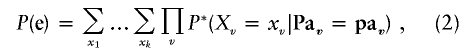 |
where P* is obtained from P by the assignment of the observed values {e1,e2,…,em} to the respective variables in E. In figure 1, evidence variables such as marker phenotypes and affection status are highlighted.
The straightforward approach to compute P(e)—by first multiplying all conditional probability tables and then computing all sums—is infeasible because of the exponential size of the joint probability distribution. Instead, it is possible to interleave summations and multiplications, summing variables one after another at early stages of the computation by pushing the summation sign in equation (2) as far to the right as possible.
When summing over all values of a variable X, it suffices to compute the product of only those probability tables that contain X, yielding an intermediate table over the variables of the tables being multiplied. The summation over variable X eliminates it from the product, reducing the dimensions of the intermediate table by a factor equal to the number of values of X. This technique of computing equation (2) is called “variable elimination” in the Bayesian network literature.33
Variable elimination alone is often inapplicable because of the prohibitively large size of intermediate tables generated during the computations, which exceed the physical memory of contemporary computers. An alternative approach to computing equation (2) is to simplify a given problem by first assigning values to some subset of variables C⊆X and then performing the computation for every joint assignment c to the variables in C. Assigning a value to variables in C decreases the size of the corresponding probability tables and, consequently, the size of intermediate tables, reducing the original problem and fitting it for computation via variable elimination. Equation (2) can then be rewritten as
 |
where 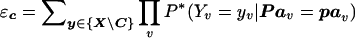 represents the computation of the problem for specific joint assignment c to the variables in C. The variables in C are called the “conditioning variables,” and this method is called “conditioning.”30 It has been used to extend the Elston-Stewart algorithm to looped pedigrees.34
represents the computation of the problem for specific joint assignment c to the variables in C. The variables in C are called the “conditioning variables,” and this method is called “conditioning.”30 It has been used to extend the Elston-Stewart algorithm to looped pedigrees.34
Conditioning, as described above, is inefficient because of the repetitive evaluation of identical subexpressions when computing ɛc for different joint assignments c to the conditioning variables. A more efficient algorithm, called “constrained variable elimination,” significantly reduces the amount of redundant computations by interleaving conditioning and elimination. This algorithm is the basis of the genetic linkage software SUPERLINK.12
The constrained elimination algorithm applies variable elimination until no more variables can be eliminated without exceeding the specified memory constraints. Conditioning is then performed on the smallest subset of variables, which suffices to reduce the size of intermediate tables to meet the specified memory constraints. The steps of elimination and conditioning are interleaved until all products and sums have been computed.
Both time and space complexity of the constrained variable elimination algorithm depend on the order in which variables are conditioned or eliminated. The running time of the algorithm applied to the same problem can range many orders of magnitude, depending on the chosen order of computations. Finding an optimal combined conditioning and elimination order that minimizes the computation time for general graphs is computationally hard.13 In fact, the Elston-Stewart and the Lander-Green algorithms can be viewed as instances of the variable elimination method that uses predetermined elimination orders—namely, the Elston-Stewart algorithm eliminates one nuclear family after another, whereas the Lander-Green algorithm eliminates one locus after another.13
SUPERLINK implements a stochastic, greedy algorithm for determining the combined conditioning and elimination order that strives to minimize the execution time under given memory constraints.13 The next variable for elimination or conditioning is selected from the set of variables ranked highest by some choice criteria. The specific elimination variable is selected at random from this set. A single iteration of the algorithm computes an elimination and conditioning order and its elimination cost, defined as the sum of sizes of all intermediate tables generated during the computation. This procedure is invoked multiple times with the use of various choice criteria, producing a set of orders. Finally, the order with the lowest elimination cost is chosen. The order found is optimized for the linkage problem at hand, automatically handling pedigrees of any topology and size as well as missing data and multiple markers.
This algorithm, called “Find-Order,” is crucial for the efficient parallelization developed in the “Methods” section and is specified in appendix A.
Grid Computing
Grid computing offers a model for employing the unused computational resources, such as central processing unit (CPU) cycles, of a large number of computers via a distributed network infrastructure. As opposed to the traditional approach of high-performance computations via expensive, dedicated hardware, grid computing focuses on the ability to support demanding computations, such as linkage analysis, on desktop computers and on computers with a variety of architectures and capabilities that are owned by different parties.
Although grid computing can provide potentially unbounded low-cost computation power, there are steps to take to allow applications to realize this potential. The simplest scenario includes finding the appropriate computer(s) for executing the task, enabling the task’s remote invocation by creating the execution environment necessary to run it on the remote machine, and collecting the results back to the originating computer after its completion. These steps must be performed securely, reliably, and under various constraints. It is particularly challenging to accomplish that in a dynamic environment in which computers can fail, can be shut down or disconnected from the network, or can be removed from the computation by their owners without any prior notification. Finally, the grid environment is a multiuser system that requires special measures to avoid contention over resources.
These and other issues are handled by grid middleware, such as Condor,22 which is designed to hide the complexities of using grid computing. A typical scenario of using such middleware is to request execution of N tasks by placing them in a queue. The system will attempt to run the tasks by allocating available computers that match the specific requirements of memory size, operating system, etc. However, the system guarantees neither the total amount of allocated machines nor their uninterrupted operation. Each task is executed on a single computer, independently of others. The number of computers allocated for a single user depends on the system load created by other users and on the total number of vacant computers available, and it may change over time. Computer failures during task execution are detected and result in an automatic attempt to find an alternative computer for completing the interrupted task. As long as there are incomplete tasks in the queue, the system tries to find additional computers for running them, to ensure that all tasks are completed.
Not all applications can benefit from execution in grid environments, because of high network delays and unpredictable resource failures. For example, parallel applications requiring periodic synchronization between subtasks running on different computers are not likely to obtain performance gains. However, grid environments with a sufficient number of computers allow for high speedups for problems that can be parallelized into many independent subtasks without needed synchronization, as we do for linkage analysis computations.
Methods
The main mechanisms that empower SUPERLINK-ONLINE are parallelization of multipoint linkage analysis for grid environments, handling of multiple linkage tasks, choice of parallelization strategy according to the complexity of the problem at hand, reliable execution, and notification of progress and completion. The working system employs thousands of computers from several universities across the globe. These issues are described below.
Parallelization of Multipoint Linkage Analysis
Multipoint LOD score computations are performed in three phases:
-
1.
Phase I. Each pedigree in the input is transformed to a Bayesian network representation,32 such as the one given in figure 1. A new Bayesian network is constructed for every position of the disease locus.
-
2.
Phase II. The Find-Order algorithm (see appendix A) is applied for each Bayesian network, yielding an elimination order that strives to optimize the computations under given memory constraints.
-
3.
Phase III. Likelihood computations are performed via equation (2) for each Bayesian network by elimination of the variables according to the specified order, which yields the likelihood of the data for a specific disease-locus position (see appendix A).
Several levels of parallelism are readily available. First, each Bayesian network created in phase I is processed concurrently. This alone is insufficient for enabling LOD score computations of large pedigrees for a given disease locus position. Thus, parallelization is performed even in computing the LOD score for one locus. This change yields a significant algorithmic improvement over serial computations.
Parallelization of phase II
The algorithm Find-Order (see appendix A) yields better elimination orders as more iterations are executed. The execution time of computing an optimized order of likelihood computations should constitute a small fraction of the total running time and has been restricted to 5% in the serial implementation.12 Clearly, any speedup of such a small part of the computations would have only minimal direct impact on the overall performance. However, since the complexity of the order found is crucial for the entire likelihood computations, parallelization can be used to considerably increase the number of iterations of Find-Order, yielding orders of significantly lower elimination costs.
The algorithm Parallel-Find-Order, presented below, provides significant improvement that is far beyond the speedup of the optimization phase alone. The input to the algorithm is a Bayesian network N and a threshold T. The threshold represents the amount of memory available for likelihood computations on a single computer and currently ranges between 108 and 1010 bytes. Failure to fit the available memory leads to performance degradation and is avoided. In grid environments with many computers of various capabilities, where it is impossible to predict which computer will be allocated for execution, the threshold T is determined dynamically among the computers available for execution according to a computer with the minimum amount, but above some predefined value, of memory.
Parallel-Find-Order Algorithm
Input: A Bayesian network N and a threshold of T bytes.
Output: An elimination order Π and its cost, cost(Π).
-
1.
Quick complexity evaluation on one computer; takes seconds.
-
a.
Run Find-Order (N,T,L) for L=5 iterations and report the best elimination order Π1.
-
b.
Set T1 to be the average run time of one iteration.
-
c.
If cost(Π1)<C1, then output the order Π1 and the corresponding elimination cost, cost(Π1), and exit.
-
d.
If cost(Π1)>C′1, then output “task too complex” and exit.
-
a.
-
2.
Refined complexity evaluation on one computer; takes minutes.
-
a.
Set I1 to be the number of iterations for the elimination cost, cost(Π1), via a conversion table for serial execution.
-
b.
Run Find-Order (N,T,L) for L=I1 iterations and report the best elimination order Π2.
-
c.
If cost(Π2)<C2, then output the order Π2 and the corresponding elimination cost, cost(Π2), and exit.
-
d.
If cost(Π2)>C′2, then output “task too complex” and exit.
-
a.
-
3.
Final complexity evaluation on several computers in parallel; takes minutes to hours.
-
a.
Set k to be the value corresponding to cost(Π2) via a conversion table for parallel execution.
-
b.
Run k Find-Order (N,T,L) tasks in parallel, each for L=I1 iterations, using at most k computers, and report the best evaluation order Π3 found.
-
c.
If cost(Π3)>C′3, then output “task too complex” and exit.
-
d.
Output the order Π3 and the corresponding evaluation cost, cost(Π3), and exit.
-
a.
The Parallel-Find-Order algorithm comprises three steps, each of which refines the optimization results of the previous step by applying more iterations of the procedure Find-Order (see appendix A). The first step runs five iterations of the Find-Order procedure, often yielding a far-from-optimal elimination order, especially for high-complexity problems. However, if the complexity of the order is below the threshold C1, the total running time of likelihood computation is small, and further optimization is not needed.
For problems with higher complexity, the second step is executed. The number of iterations is now determined using a conversion table, which associates the complexity cost of the elimination order found in the first step with the total expected running time of the likelihood computations. The number of iterations to be executed in this step is determined so as to allocate 5% of the total expected computing time for the execution of the optimization algorithm, with the assumption that the average time for a single iteration is the same as that for the execution of the first step. For complex problems, several thousands of iterations of the Find-Order procedure are executed in this step. When this step is completed, the elimination cost of the order found in this step is reevaluated, to determine whether additional optimization is required. Indeed, if the elimination cost is below the threshold C2, further optimization is unlikely to yield any significant improvement in the running time, and the optimization algorithm stops. On the other hand, if the complexity found is above a threshold C′2, then the task is rejected.
The third optimization step is executed in parallel on several computers, and the total number of iterations depends both on the problem complexity and on the number of computers available at the moment of execution. Each computer is assigned to perform the same number of iterations as was performed in the second step, but the total number of computers can reach 100 for problems that are on the edge of system capabilities. If, however, the number of available computers drops during the execution, as is common in grid environments, the number of iterations to perform is dynamically decreased, so that the execution time of the parallel-ordering phase does not dominate the total revised execution time. The effect of the parallel ordering for large pedigrees is demonstrated by our results, where a speedup of 2 orders of magnitude is obtained solely because of this phase. Note that a similar increase in the number of iterations in the serial version of the algorithm leads to performance degradation, since the speedup due to better order is outweighed by long running times of the optimization stage.
Parallelization of phase III
The choice of a parallelization strategy is guided by two main requirements. First, subtasks are not allowed to communicate or to synchronize their state during the execution. This factor is crucial for the performance of parallel applications in a grid environment, where communication between computers is usually slow or even impossible because of security constraints. Second, parallel applications must tolerate frequent failures of computers during the execution by minimizing the impact of failures on the overall performance. These requirements have not been considered in previous parallelization approaches for Bayesian networks software.35,36
Our approach is to divide a large complex task into several smaller independent subproblems that can be processed concurrently. As noted previously, the serial algorithm computes the result of equation (2) by eliminating variables one by one and by using conditioning when the intermediate results do not fit the memory constraints. However, we can apply conditioning before eliminating any variable, as shown in equation (3). We calculate ɛx for every joint assignment to the variables in C concurrently on several computers and then obtain the final result by summation, as in equation (3). Since each subproblem ɛx is simpler than the initial problem by a factor of up to the number of joint assignments, parallel computation is expected to significantly reduce the running time. To further divide the problem to take advantage of more computers, more variables are used for conditioning.
We incorporate these ideas in the Parallel-Constrained-Elimination algorithm.
Parallel-Constrained-Elimination Algorithm
Input: A Bayesian network N, an elimination order Π, and a threshold C.
Output: Likelihood of data.
-
1.
P←∅.
-
2.
While cost(Π)>C,
-
a.
choose a conditioning variable X from Π and add it to the set P,
-
b.
remove X from order Π, and
-
c.
adjust the Bayesian network N by setting X=x in all probability tables in which X appears.
-
a.
-
3.
Obtain the total number of parallel subtasks L by multiplying the number of values of all variables in P.
-
4.
Create L Bayesian networks Ni and L elimination orders Πi, one for every joint assignment of the variables in P.
-
5.
Run L Constrained-Elimination (Ni,Πi) tasks in parallel using SUPERLINK.
-
6.
Output the likelihood by summing all partial results.
The input to the algorithm is a Bayesian network N, elimination order Π found by the Parallel-Find-Order procedure, and a threshold C. The threshold C defines the maximum complexity of each subtask, and it is proportional to the running time of a single subtask. The algorithm begins by selecting conditioning variables to be used for parallelization by iteratively adding variables to a set P. In each iteration, a new variable is selected from the set of all conditioning variables in the order Π, and the next unused conditioning variable is chosen according to Π. The order Π and the Bayesian network N are adjusted as follows: the selected variable is removed from Π, and the Bayesian network N is modified by setting that variable to a specific value in all probability tables in which it appears. Thus, every iteration further simplifies the Bayesian network and reduces the cost of the elimination order. The process continues as long as the elimination cost of the modified elimination order exceeds the maximum allowed complexity threshold C. The number of subtasks L created by this step equals the number of joint assignments to all variables added to P. After L subtasks are created, they are executed in parallel with the use of the serial Constrained-Elimination procedure (see appendix A). The final result is obtained by summing the partial results of all subtasks.
The choice of the number of subtasks L and their respective maximum size C is crucial for efficiency of the parallelization. The inherent overheads of distributed environments, such as scheduling and network delays, often become a dominating factor inhibiting meaningful performance gains, suggesting that long-running subtasks should be preferred. On the other hand, performance degradation as a result of computer failures will be lower for short subtasks, which suggests that the amount of computations per subtask should be reduced. Furthermore, decreasing the amount of computations per subtask increases the number of subtasks generated for computing a given problem, which improves load balancing and utilization of available computers.
Our algorithm controls the subtask size by specifying the maximum allowable complexity threshold C. Specifying lower values of C increases the number of subtasks L, which decreases the subtask complexity and, consequently, its running time. The value of C for a given problem is determined as follows. We initially set C so that a subtask’s running time does not exceed the average time a task can execute without interruption on a computer in the grid environment being used. If such value of C yields the result that the number of subtasks is below the number of available computers, then C is iteratively reduced to allow division into more subtasks. The lower bound on C is set so that overheads due to scheduling and network delays constitute <1% of the subtask’s running time.
Using conditioning for parallelization results in repetitive evaluation of identical subexpressions by multiple computers, thus reducing the efficiency of the parallel algorithm. The amount of such redundant computations depends on the number of conditioning variables used for parallelization. We found that, even for the most complex problems that are split into thousands of subtasks, the overhead does not exceed 10% because of the small number of conditioning variables required for parallelization. Such an overhead is far outweighed by the performance gains due to splitting a task into a set of independent subtasks, completely avoiding communication and synchronization between them, and, by that, allowing their execution in an opportunistic grid environment.
Multiuser Online System for Linkage Analysis
The SUPERLINK-ONLINE system for submission of linkage analysis tasks via the Internet frees users from numerous technical issues incurred in distributed environments; parallelization, load balancing, fault tolerance, progress monitoring, and other technical issues are fully automated behind the scene. The user is notified about the availability of results by e-mail and can access them via the Internet. The user’s data, as well as the results, are protected from unauthorized access. These and other mechanisms were implemented to allow easy, reliable, and secure use of this service, providing access to thousands of computers located at several universities.
Preventing overload via staged complexity evaluation
Geneticists are not always aware of the computational load induced by a linkage analysis task. Addition of a single marker to the analysis of a large pedigree may increase the running time from only few seconds to months (as demonstrated in the “Results” section), making the results of little value from a practical perspective and rendering the system inaccessible to other users.
To prevent unintentional overload caused by high-complexity tasks, the system rejects tasks exceeding the maximum complexity threshold. To reach the conclusion about task feasibility during early stages of execution, the task complexity is assessed after every step of the Parallel-Find-Order algorithm, as explicated above. The complexity thresholds C′1, C′2, and C′3 in the algorithm are set in accordance with the amount of computational power available in the grid environment being used.
Minimizing response time via multiple queues
The ability to efficiently handle concurrent computations of tasks of markedly different complexities is crucial for providing adequate performance when servicing multiple execution requests. To allow efficient handling of small tasks while simultaneously serving complex ones, the system classifies the tasks according to their complexity, as determined by the Parallel-Find-Order procedure. Each range of complexities forms its own queue that is handled independently of others and uses a different set of computational resources, providing the shortest response time possible under a given system load. The higher the task complexity, the more computational power is employed. Whereas very short tasks are executed on a single dedicated computer without any scheduling delay, the tasks of higher complexity are parallelized and are invoked in a pool of a few dozen computers with low remote-invocation overhead. Very complex tasks are transferred to queues of their own, which allows the potential use of several hundreds of computers but causes higher delays in servicing the request. If, for some reason, the task is executed in its queue for longer than is allowed by the queue specification, it is migrated to a queue for longer tasks, with all the available partial results preserved.
The feature of migrating partially completed tasks among a set of queues, without losing the computations already finished, is a novel addition to the grid-computing paradigm that was introduced because of the high computational demands of SUPERLINK-ONLINE.
Increasing throughput via multiple pools of computers
To accommodate high loads, we implemented mechanisms to allow expansion of the system beyond the boundaries of a single Condor pool of computers. Currently, SUPERLINK-ONLINE spans five pools with a total of 2,700 computers, including two pools at the Technion-Israel Institute of Technology in Haifa, and three large pools at the University of Wisconsin in Madison. Tasks are first submitted to the Technion pools and then migrate between pool sites, according to the availability of computers and other load-balancing criteria.
We implemented mechanisms to further extend the computational power of SUPERLINK-ONLINE by augmenting it with additional pools expected to be contributed by users and institutions worldwide and to be used on a free-cycle basis without affecting the contributing owners' loads.
Results
We demonstrate the system capabilities of performing exact LOD-score computations. We use the grid environment of ∼2,700 computers of various performance characteristics, though only computers having >500 MB of random access memory (RAM) and providing performance >300 million floating-point operations per s (MFLOPs) were used for the execution, which reduced the overall number to ∼2,000. The computer characteristics are provided by Condor.22 The specified characteristics correspond roughly to Intel Pentium IV, 1.7 GHz.
Experiment A: Testing Correctness
We ran SUPERLINK-ONLINE on all 146 data sets used elsewhere.3,12 For all these data sets—which differ in size, number of typed persons, and degree of consanguinity—SUPERLINK-ONLINE computed correct LOD scores, validating our implementation.
Experiment B: Published Disease Data Set
The pedigree in figure 2 was used for studying cold-induced sweating syndrome in a Norwegian family.37 The pedigree consists of 93 individuals, 2 of whom are affected, and only 4 were typed. The original analysis was done using FASTLINK. The maximum LOD score of 1.75 was reported using markers D19S895, D19S566, and D19S603, with the analysis limited to only three markers because of computational constraints. According to the authors, using more markers for the analysis was particularly important in this study since, “in the absence of ancestral genotypes, the probability that a shared segment is inherited IBD from a common ancestor increases with the number of informative markers contained in the segment; that is, a shared segment containing only three markers has a significant probability of being simply identical by state, whereas a segment containing a large number of shared markers is much more likely to be IBD from a common ancestor. Thus, the maximum four-point LOD score of 4.22 for the two families combined, on the basis of only three markers from within an interval containing 13 shared markers, is an underestimate of the true LOD score.”37 Study of another pedigree, as well as the application of additional statistical methods, were employed to confirm the significance of the findings.
Figure 2.

Pedigree reprinted from Knappskog et al.37
Using SUPERLINK-ONLINE, we computed sixpoint LOD scores with the markers D19S895, M4A, D19S566, D19S443, and D19S603, yielding LOD=3.10 at marker D19S895, which would facilitate the linkage conclusion based on the study of this pedigree alone.
Experiment C: Number of Computers for Multipoint Analysis
This experiment explicates the exponential growth of the required amount of computational resources when the number of markers used for the analysis is increased. We performed our computations on the pedigree presented in figure 3, which consisted of 105 individuals. It comes from the study of brittle hair syndrome in a large consanguineous Amish kindred38 and originally was analyzed using two-point analysis.
Figure 3.

Pedigree reprinted from Seboun et al.38
We performed two-, three-, and four-point analyses, using 10-allelic polymorphic markers D7S484, D7S2497, and D7S510, with respective interval distances of 3.3 and 0.5 cM. Table 1 summarizes LOD scores obtained at marker DS2497 and the corresponding amount of computational resources required to perform the computations within the specified time. We were unable to perform five-point analysis because of the very high complexity of the computations. Consequently, the respective entries in the table were calculated in accordance with the problem-complexity estimation provided by SUPERLINK-ONLINE when the task was processed.
Table 1.
| No. of Markers |
LOD Score |
Running Time |
No. of Computers |
| 1 | 7.16 | 6 s | 1 |
| 2 | 9.08 | 180 s | 1 |
| 3 | 9.66 | 47 min | 82 |
| 4 | NA | ∼100 h | ∼20,000 |
Note— Five-point analysis could not be completed, and the values are estimated. NA = not available.
Experiment D: Impact of Parallelized Ordering
We performed two-point analysis of several pedigrees derived from a single large genealogy of thousands of individuals, using PEDHUNTER.39 The pedigree was shrunk by a user of SUPERLINK-ONLINE until its complexity permitted the performance of exact computations.
This example demonstrates a sophisticated use of a large genealogy by employing PEDHUNTER and SUPERLINK-ONLINE in sequel. Such use of a genealogy reduces pedigree errors, which may be prevalent when large pedigrees are elicited.40
The pedigree was initially reduced to contain 231 individuals, with 89% untyped and 10% affected. The analysis was performed using a 13-allelic locus, yielding a LOD score of 2.49. Increasing the pedigree size to 266 individuals, with 87% untyped and 8% affected, and performing two-point analysis with the use of the same marker yielded LOD=3.65, which indicated that fine mapping is worthy of pursuit. This pedigree is depicted in figure 4.
Figure 4.

Pedigree with 266 individuals (produced by OPediT)
Detailed analysis of the execution trace revealed that parallelizing the task of finding the order of computation played a significant role in the overall system performance. Initial estimation of complexity for the pedigree with 266 individuals produced complexity that would require ∼11 CPU years for the analysis to complete. SUPERLINK v1.5 reduced the complexity by 2 orders of magnitude, which would still require ∼200 h to compute on our system, given that 1,000 personal computers are available. Finally, the application of the parallelized-ordering algorithm yielded an additional reduction in complexity of 2 orders of magnitude, which allowed the system to complete the computations in <7 h.
Experiment E: Evaluation of Performance
We measured the total run time for computing the LOD score at one disease-locus position for large pedigrees. The results reflect the time a sole user would wait for a task to complete, from submission via a Web interface to receipt of an e-mail notification about task completion. Results are summarized in table 2.
Table 2.
Summary of Experiments on Large Pedigrees
|
Running Timeb |
No. of Computers |
||||||
|
No. of Personsa |
No. of Markers |
Typed (%) |
LOD Score | SUPERLINK v1.5 | SUPERLINK-ONLINE | Averagec | Maximum |
| 33 | 5 | 50 | 5.5 | 5,000 s | 1,050 s | 10 | 10 |
| 68 | 3 | 17 | .7 | 5,600 s | 520 s | 11 | 11 |
| 84 | 4 | 25 | 7.2 | 20 h | 2 h | 23 | 30 |
| 105* | 3 | 20 | 9.7 | 450 min | 47 min | 82 | 83 |
| 115 | 2 | 26 | 10.7 | ∼300 h | 7.1 h | 38 | 91 |
| 248 | 1 | 12 | 2.5 | 297 min | 27 min | 82 | 100 |
| 266** | 1 | 13 | 3.7 | ∼138 d | 6.2 h | 349 | 450 |
| 325† | 1 | 38 | 3.0 | ∼2,092 s | 1,100 s | 7 | 8 |
| 336† | 1 | 39 | 3.0 | ∼231 h | 3 h | 139 | 500 |
| 362† | 1 | 45 | 5.9 | ∼160 d | 8 h | 310 | 360 |
Runs for the pedigrees in figures 3 and 4 are marked by an asterisk (*) and double asterisk (**), respectively. Runs marked by a dagger (†) are performed on the same genealogy, by increasing the number of individuals in the analysis.
Running times of SUPERLINK-ONLINE include network delays and resource failures (handled automatically by the system).
Computed from the number of computers sampled every 5 min during the run.
We also compared the running time with that of the newest serial version of SUPERLINK, invoked on a 64-bit Intel Pentium Xeon processor (3.0 GHz, 2 gigabytes RAM). The entries of the running time exceeding 2 d were obtained by measuring the portion of the problem completed within 2 d, as made available by SUPERLINK, and by extrapolating the remaining running time, with the assumption of similar progress. The time savings with the online system versus that with a single computer ranged from a factor of 10 to 700.
In a large multiuser grid environment used by SUPERLINK-ONLINE, the number of computers employed in computations of a given task may fluctuate during the execution from only a few to several hundred. Table 2 presents the average and the maximum number of computers used during execution. We note that the performance can be improved significantly if the system is deployed in a dedicated environment.
Discussion
SUPERLINK-ONLINE has been designed to dynamically balance between performance optimization for a single task and better service for all users. These contradictory requirements lead to degradation of performance of a single task when several tasks are being concurrently served by the system. To allow for top performance, we made available an initial downloadable version that allows a user to duplicate the entire online system onto the user's own set of computers. Alternatively, users can contribute pools of computers to enhance the online system for everyone’s benefit.
Our system prevents unauthorized access via the Internet to the user-provided data and the results of the analysis. A unique password is generated for each linkage analysis task and is sent to the user’s e-mail address provided during the submission. However, the input data are processed by multiple remote computers that are not under the system’s control and, thus, rely on the security settings configured by their system administrators. Despite this potential data exposure, a user can easily obscure the input by removing all identifying information, such as names of persons and markers, before submission. This would still allow performance of the linkage analysis, while making the data useless for any unauthorized observer.
In summary, with the advancement of free middleware such as Condor,22 it is becoming feasible to perform exact analysis of pedigree information with the use of many computers, as is evident by SUPERLINK-ONLINE. Furthermore, the methods presented will make it feasible to compute exact LOD scores for polygenic diseases on large pedigrees, which may advance genetic research on such complex diseases, even though only monogenic diseases have been analyzed in this article.
Acknowledgments
This research is supported by the Israeli Science Ministry and the Israeli Science Foundation. We thank our colleagues Ohad Birk, Helge Boman, Tzipi Falik, Jacek Majewski, Rivki Ophir, Alejandro Schäffer, and Eric Seboun for providing us with data that enabled realistic evaluation of our system, and we thank Alejandro Schäffer for valuable user feedback and comments on earlier drafts of this article.
Appendix A
SG(N,T,E) Procedure13
Input: A Bayesian network N with the set of variables V, a threshold T, and elimination choice criterion E.
Output: An elimination order Π, such that the elimination cost of each variable is ⩽T (elimination cost is the size of the intermediate table created by eliminating a variable).
-
1.
While V≠∅,
-
a.
pick three variables {Ve1,Ve2,Ve3} from V, according to the elimination choice criterion E;
-
b.
choose at random Ve∈{Ve1,Ve2,Ve3};
-
c.
compute the elimination cost E(Ve); and
-
d.
decide whether to perform conditioning or to eliminate Ve.
If E(Ve)>T, then
-
i.
pick Vc∈V, according to the conditioning choice criterion C;
-
ii.
add Vc to Π as a conditioning variable;
-
iii.
remove Vc from V; and
-
iv.
update the Bayesian network N after conditioning by reducing all tables containing Vc by the factor equal to the number of values of Vc.
Else,
-
i.
add Ve to Π as an elimination variable,
-
ii.
remove Ve from V, and
-
iii.
update the Bayesian network N after elimination by producing a single table with variables from all tables containing Ve and removing these tables from the Bayesian network.
-
i.
-
a.
-
2.
Return Π.
Constrained-Elimination Procedure (N,Π)13
Input: A Bayesian network N, a combined conditioning, and elimination order Π.
Output: Probability of evidence P.
-
1. While Π≠∅,
-
a. pick next variable V from Π.
-
b. Π←Π∖V.
-
c. If V is an elimination variable,
-
i. multiply all tables containing V and
-
ii. sum over V to obtain the result Pt.
-
iii. If Pt is a number, no further summation is needed, and P←P×Pt.
-
i.
-
d. Otherwise, for each value v of V,
-
i. create Nv by assigning V=v in all tables containing V.
-
ii. P←P+ Constrained-Elimination (Nv,Π).
-
i.
-
a.
-
2.
Return P.
Find-Order Procedure (N,T,L)13
Input: A Bayesian network N, threshold T, number of iterations L, and a set of l choice criteria C1,…,Cl used to choose the next variable to eliminate. For example, criterion C1 is used to choose a variable that produces the smallest intermediate table. Other criteria are described elsewhere.13
Output: An elimination order Π, such that the elimination cost of each variable is ⩽T (elimination cost is the size of the intermediate table created by eliminating a variable).
-
1.
j←1, Cost←∞, Found←false.
-
2. For i←1 to L,
-
a. run Lmin iterations with the use of choice criterion Cj to compute a candidate elimination order. For k←i to i+Lmin,
-
i. Πtemp←SG(N,T,Cj),
-
ii. compute the sum of all tables created when the order Πtemp is used—namely, Costtemp←Cost(Πtemp)—and
-
iii. update the best order.If Costtemp < Cost, then Π←Πtemp; Cost←Costtemp; Found←true.
-
i.
-
b. Switch to the next choice criterion if no order is found. This way of skipping between criteria enhances the Find-Order Procedure. If Found is false, then j=(j+1)mod l.
-
c. Found←false; i←i+Lmin.
-
a.
-
3.
Return Π.
Web Resources
URLs for data presented herein are as follows:
- OPediT, http://www.circusoft.com/content/product/5 (for drawings of the pedigree in experiment D)
- SUPERLINK-ONLINE, http://bioinfo.cs.technion.ac.il/superlink-online (for download, http://bioinfo.cs.technion.ac.il/superlink-online/download)
References
- 1.Elston R, Stewart J (1971) A general model for the analysis of pedigree data. Hum Hered 21:523–542 [DOI] [PubMed] [Google Scholar]
- 2.Lander E, Green P (1987) Construction of multilocus genetic maps in humans. Proc Natl Acad Sci 84:2363–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fishelson M, Geiger D (2004) Optimizing exact genetic linkage computations. J Comput Biol 11:263–275 10.1089/1066527041410409 [DOI] [PubMed] [Google Scholar]
- 4.Cottingham RW, Idury RM, Schäffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed] [Google Scholar]
- 5.O’Connell J, Weeks D (1995) The VITESSE algorithm for rapid exact multilocus linkage analysis via genotype set-recoding and fuzzy inheritance. Nat Genet 11:402–408 10.1038/ng1295-402 [DOI] [PubMed] [Google Scholar]
- 6.Schäffer AA (1996) Faster linkage analysis computations for pedigrees with loops or unused alleles. Hum Hered 46:226–235 [DOI] [PubMed] [Google Scholar]
- 7.Kruglyak L, Lander E (1998) Faster multipoint linkage analysis using Fourier transform. J Comput Biol 5:1–7 [DOI] [PubMed] [Google Scholar]
- 8.Gudbjartsson F, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13 10.1038/75514 [DOI] [PubMed] [Google Scholar]
- 9.O’Connell J (2001) Rapid multipoint linkage analysis via inheritance vectors in the Elston-Stewart algorithm. Hum Hered 51:226–240 10.1159/000053346 [DOI] [PubMed] [Google Scholar]
- 10.Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101 10.1038/ng786 [DOI] [PubMed] [Google Scholar]
- 11.Kruglyak L, Daly M, Reeve-Daly M, Lander E (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- 12.Fishelson M, Geiger D (2002) Exact genetic linkage computations for general pedigrees. Bioinformatics 18:S189–S198 [DOI] [PubMed] [Google Scholar]
- 13.Fishelson M, Dovgolevsky N, Geiger D (2005) Maximum likelihood haplotyping for general pedigrees. Hum Hered 59:41–60 10.1159/000084736 [DOI] [PubMed] [Google Scholar]
- 14.Miller P, Nadkarni P, Gelernter G, Carriero N, Pakstis A, Kidd K (1991) Parallelizing genetic linkage analysis: a case study for applying parallel computation in molecular biology. Comput Biomed Res 24:234–248 10.1016/0010-4809(91)90046-Y [DOI] [PubMed] [Google Scholar]
- 15.Dwarkadas S, Schäffer A, Cottingham R, Cox A, Keleher P, Zwaenepoel W (1994) Parallelization of general linkage analysis problems. Hum Hered 44:127–141 [DOI] [PubMed] [Google Scholar]
- 16.Matise T, Schroeder M, Chiarulli D, Weeks D (1995) Parallel computation of genetic likelihoods using CRI-MAP, PVM, and a network of distributed workstations. Hum Hered 45:103–116 [DOI] [PubMed] [Google Scholar]
- 17.Gupta S, Schäffer A, Cox A, Dwarkadas S, Zwaenepoel W (1995) Integrating parallelization strategies for linkage analysis. Comput Biomed Res 28:116–139 10.1006/cbmr.1995.1009 [DOI] [PubMed] [Google Scholar]
- 18.Rai A, Lopez-Benitez N, Hargis J, Poduslo S (2000) On the parallelization of Linkmap from the LINKAGE/FASTLINK package. Comput Biomed Res 33:350–364 10.1006/cbmr.2000.1547 [DOI] [PubMed] [Google Scholar]
- 19.Kothari K, Lopez-Benitez N, Poduslo S (2001) High-performance implementation and analysis of the Linkmap program. J Biomed Inform 34:406–414 10.1006/jbin.2002.1031 [DOI] [PubMed] [Google Scholar]
- 20.Conant G, Plimpton S, Old W, Wagner A, Fain P, Pacheco T, Heffelfinger G (2003) Parallel Genehunter: implementation of a linkage analysis package for distributed-memory architectures. J Parallel Distrib Comput 63:674–682 10.1016/S0743-7315(03)00080-7 [DOI] [Google Scholar]
- 21.Dietter J, Spiegel A, an Mey D, Pflug HJ, al Kateb H, Hoffmann K, Wienker T, Strauch K (2004) Efficient two-trait-locus linkage analysis through program optimization and parallelization: application to hypercholesterolemia. Eur J Hum Genet 12:542–550 10.1038/sj.ejhg.5201196 [DOI] [PubMed] [Google Scholar]
- 22.Thain D, Livny M (2003) Building reliable clients and servers. In: Foster I, Kesselman C (eds) The grid: blueprint for a new computing infrastructure. Morgan Kaufmann, San Francisco, pp 285–318 [Google Scholar]
- 23.Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 10.1038/ng1195-241 [DOI] [PubMed] [Google Scholar]
- 24.Ott J (1976) A computer program for linkage analysis of general human pedigrees. Am J Hum Genet 28:528–529 [PMC free article] [PubMed] [Google Scholar]
- 25.Lathrop G, Lalouel J, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 81:3443–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lathrop GM, Lalouel JM (1984) Easy calculations of Lod scores and genetic risks on small computers. Am J Hum Genet 36:460–465 [PMC free article] [PubMed] [Google Scholar]
- 27.Becker A, Geiger D, Schäffer A (1998) Automatic selection of loop breakers for genetic linkage analysis. Hum Hered 48:49–60 10.1159/000022781 [DOI] [PubMed] [Google Scholar]
- 28.Markianos K, Daly M, Kruglyak L (2001) Efficient multipoint linkage analysis through reduction of inheritance space. Am J Hum Genet 68:963–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strauch K, Fimmers R, Kurz T, Deichmann KA, Wienker TF, Baur MP (2000) Parametric and nonparametric multipoint linkage analysis with imprinting and two-locus-trait models: application to mite sensitization. Am J Hum Genet 66:1945–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pearl J (1988) Probabilistic reasoning in intelligent systems. Morgan Kaufmann, San Francisco [Google Scholar]
- 31.Lauritzen SL (1996) Graphical models. Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 32.Friedman N, Geiger D, Lotner N (2000) Likelihood computation with value abstraction. In: Proceedings of the 16th Conference on Uncertainty in Artificial Intelligence (UAI). Morgan Kaufmann, San Francisco, pp 192–200 [Google Scholar]
- 33.Dechter R (1998) Bucket elimination: a unifying framework for probabilistic inference. In: Jordan M (ed) Learning in graphical models. Kluwer Academic Press, Dordecht, the Netherlands, pp 75–104 [Google Scholar]
- 34.Lange K, Elston R (1975) Extentions to pedigree analysis. I. Likelihood calculations for simple and complex pedigrees. Hum Hered 25:95–105 [DOI] [PubMed] [Google Scholar]
- 35.Kozlov A, Jaswinder P (1994) A parallel Lauritzen-Spiegelhalter algorithm for probabilistic inference. In: Proceedings of the Supercomputing '94 Conference, IEEE Computer Society, Washington, DC, pp 320–329 [Google Scholar]
- 36.Madsen A, Jensen F (1999) Parallelization of inference in Bayesian networks. Tech rep R-99-5002, Department of Computer Science, Aalborg University, Denmark [Google Scholar]
- 37.Knappskog P, Majewski J, Livneh A, Nilsen P, Bringsli J, Ott J, Boman H (2003) Cold-induced sweating syndrome is caused by mutations in the CRLF1 gene. Am J Hum Genet 72:375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seboun E, Lemainque A, Jackson C (2005) Amish brittle hair syndrome gene maps to 7p14.1. Am J Med Genet A 134:290–294 [DOI] [PubMed] [Google Scholar]
- 39.Agarwala R, Biesecker L, Hopkins K, Francomano C, Schäffer A (1998) Software for constructing and verifying pedigrees within large genealogies and an application to the Old Order Amish of Lancaster County. Genome Res 8:211–221 [DOI] [PubMed] [Google Scholar]
- 40.Zlotogora J, Bisharat B, Barges S (1998) Can we rely on the family history? Am J Med Genet 77:79–80 [DOI] [PubMed] [Google Scholar]