

Abstract
In a large kindred of German descent, we found a novel allele that segregates with deafness when present in trans with the 35delG allele of GJB2. Qualitative polymerase chain reaction–based allele-specific expression assays showed that expression of both GJB2 and GJB6 from the novel allele is dramatically reduced. This is the first evidence of a deafness-associated regulatory mutation of GJB2 and of potential coregulation of GJB2 and GJB6.
Mutations in GJB2, the gene encoding gap junction protein connexin 26 (Cx26), are the most common cause of prelingual-onset, recessively inherited, nonsyndromic, sensorineural hearing loss (SNHL) in humans. GJB2 and GJB6, which encodes connexin 30 (Cx30), comprise the DFNB1/A3 locus at 13q12 (fig. 1). In the vertebrate cochlea, Cx26 and Cx30 are coexpressed as heteromeric connexons in nonsensory cells of the organ of Corti as well as in subsets of cells of the spiral ligament and stria vascularis1–3 and are implicated in the maintenance of cochlear K+ gradients necessary for proper hair-cell function.4,5
Figure 1. .

Map encompassing haplotyped region of 13q11-12, showing locations of GJB2, GJB6, genotyped microsatellite and SNP markers, and breakpoints of del(GJB6-D13S1830) and del(GJB6-D13S1854), all shown approximately to scale. The locations of the variants used for allele-specific expression assays—GJB2 35delG, GJB2 +94, and rs7333214—are also indicated. Boxes indicate exons, and the hatched boxes indicate the coding regions of the two genes. Vertical lines show approximate locations of genotyped microsatellites (in bold) and SNPs. D13S232 is the most proximal microsatellite showing recombination of the novel haplotype in affected individuals. The transcriptional start sites of GJB2 and GJB6 are indicated by right-angled arrows.
More than 80 recessive mutations of GJB2, nearly all affecting proper translation of the Cx26 protein, and several dominant missense mutations are implicated in DFNB1/A3 hearing loss. Others cause skin disease with accompanying SNHL, including Vohwinkel syndrome (MIM 124500), Bart-Pumphrey syndrome (MIM 149200), palmoplantar keratoderma, with deafness (PPK [MIM 148350]), and keratitis-ichthyosis-deafness (KID [MIM 148210]). Three dominant missense mutations of GJB6 have been shown to cause the skin disorder Clouston syndrome (hidrotic ectodermal dysplasia [MIM 129500]).
In contrast to the abundance and diversity of GJB2 hearing-loss mutations, only a single dominant mutation of GJB6 causing nonsyndromic hearing loss had been published6 before the identification of two large deletions—of 309 kb and 232 kb: del(GJB6-D13S1830) and del(GJB6-D13S1854), respectively—truncating the 5′ end of GJB6 (fig. 1). These deletions segregate with hearing loss when present homozygously or heterozygously with each other or in trans with a recessive GJB2 mutation.7–11 Investigators have suggested, but have not demonstrated, that loss of appropriate regulation of GJB2 from chromosomes bearing these deletions may underlie the hearing loss in these individuals,7–11 which may be exacerbated by loss of one GJB6 allele. Common et al.12 showed immunohistochemical evidence that Cx26 expression is disrupted in certain skin-cell types in an individual bearing the larger deletion in trans with GJB2 35delG, suggesting disruption of a GJB2 cis-regulatory element located within this deleted interval. Mutation screening of GJB2 in individuals whose hearing loss and family history are consistent with GJB2 deafness reveals a significant number of subjects with only one identified mutation. Additional screening for GJB6 deletions explains only some of these heterozygotes.
We report here evidence of a novel pathogenic DFNB1 allele for which we demonstrate reduction in or loss of detectable expression of message from both GJB2 and GJB6. Figure 2 shows a small portion of the pedigree of MSU-DF5, a large American kindred of mainly German descent. After obtaining written informed consent, we collected DNA and performed audiological testing on >200 family members. We screened all samples for GJB2 35delG and for SLC26A4 L445W. Ten MSU-DF5 family members are hearing impaired due to homozygosity of 35delG (one, DF5-68, is shown in fig. 2); one deaf family member has received a diagnosis of Pendred syndrome (MIM 274600) and is homozygous for SLC26A4 L445W (not shown in pedigree). Microsatellite and SNP genotyping across the GJB2/GJB6 genomic region (fig. 1 and table 1) allowed us to define a number of haplotypes. Three distinct haplotypes bearing 35delG exist within the family, indicating more than one founder for 35delG in this community (two 35delG haplotypes appear in fig. 2). Notably, four deaf family members, DF5-20, -70, -122, and -194, are heterozygous for 35delG and share a common haplotype (indicated in fig. 2 by a black bar with a star) that is longer than 600 kb and shorter than 3.1 Mb on their non-35delG chromosome. These individuals are profoundly hearing impaired, in contrast to DF5-68, who is homozygous for 35delG and the father of DF5-70 (fig. 3). Of the 14 family members who carry the novel allele, all those with 35delG on their other chromosome (4 individuals) have hearing loss, whereas all those with any other allele (10 individuals) have normal hearing (P<.001, by Fisher’s exact test). We also performed a linkage analysis (easyLINKAGE Plus v.3.01RC1, SuperLink v1.4) at the DFNB1 locus, using 28 individuals (shown in the boxed areas in fig. 2). We assigned the same mutant status to both the novel haplotype and the 35delG allele, and all other haplotypes were considered nonmutant. The resulting LOD score of 2.08 indicates that there is a good likelihood that the novel haplotype harbors a DFNB1 mutation.
Figure 2. .

A small portion of the MSU-DF5 pedigree, with DFNB1 haplotypes indicated for selected individuals. Five individuals with congenital SNHL are represented by blackened symbols. One of these, DF5-68, is hearing impaired because of homozygosity of GJB2 35delG (ΔG). The other four are heterozygous for GJB2 35delG and bear the same haplotype on their non-35delG chromosome (black bar with star). Individuals contained within the boxed areas were included in the linkage analysis.
Table 1. .
Selected Haplotype Assignments Based on Genotypes at Microsatellites and SNPs in a 620-kb Region of Chromosome 13 Including GJB2 and GJB6[Note]
| Marker | Location (bp) |
Novel DFNB1 Allele |
A | B | C | D | E |
| D13S1316 | 19,580,089-442 | 6 | 6 | 6 | 5 | 5.5 | 6 |
| rs3751385 | 19,660,956 | C | T | T | C | C | C |
| 35delG | 19,661,685 | + | ΔG | ΔG | + | + | + |
| rs9552098 | 19,662,895 | C | T | T | C | C | C |
| rs7994748 | 19,664,130 | T | C | C | T | C | T |
| GJB2 +94 | 19,664,944 | C | C | C | A | C | C |
| SNP1245 | 19,665,308 | T | C | C | T | C | T |
| rs9509086 | 19,665,957 | C | A | ND | C | A | ND |
| rs9552102 | 19,668,529 | T | A | ND | T | T | ND |
| A21445T | 19,673,593 | A | A | A | A | T | A |
| rs9552104 | 19,673,997 | C | G | ND | C | C | C |
| A19277T | 19,675,761 | A | ND | A | ND | T | A |
| rs9315384 | 19,682,018 | A | G | G | A | G | A |
| rs7333214 | 19,694,497 | C | C | C | C | A | C |
| D13S175 | 19,746,380-734 | 4 | 3 | 3 | 2 | 4 | 2 |
| rs13379006 | 19,795,790 | T | C | C | T | C | C |
| rs7337318 | 19,795,911 | C | G | G | C | G | G |
| rs13378957 | 19,795,932 | A | C | C | A | C | C |
| rs12873647 | 19,796,279 | T | C | C | T | C | C |
| rs9509139 | 19,796,316 | A | G | G | A | G | G |
| rs11616264 | 19,796,330 | A | T | T | A | T | T |
| rs944014 | 19,837,040 | G | A | G | G | G | G |
| rs9552241 | 20,025,569 | ND | ND | G | C | G | G |
| rs2772175 | 20,198,712 | T | T | ND | A | A | A |
Note.— The “A” and “B” columns indicate delG (ΔG) alleles carried by DF5-20 and DF5-70, respectively. The “C” column corresponds to the haplotype shown by a box with stippling, “D” to the haplotype shown by a box with horizontal lines, and “E” to the haplotype shown by a white box in figures 2, 5, and 6. Locations are based on UCSC Genome Browser, May 2004 assembly. + = wild type; ND = not determined.
Figure 3. .

Audiograms of four MSU-DF5 family members (identified by ID/sex/age). dB HL= decibels hearing level; ANSI = American National Standards Institute. DF5-20, -70, and -122 are GJB2 35delG (35ΔG) heterozygotes bearing the novel pathogenic allele. Note that all three have profound SNHL at all tested frequencies. By contrast, DF5-68, homozygous for GJB2 35delG and the father of DF5-70, has significantly more residual hearing, particularly in his left ear. Although only pure-tone air-conduction thresholds are shown (circle = right ear; × = left ear; arrows = threshold beyond tested limit), bone conduction thresholds and immittance measures (not shown) indicate that the hearing loss in all four family members is sensorineural.
We sequenced the entire coding regions and 5′ and 3′ UTRs of both GJB2 and GJB6 in three of our affected probands, including splice sites, alternative exons and promoter regions, the entire single intron of GJB2, and several kilobases of sequence around each gene, and found no sequence variants unique to the novel haplotype. Heterozygosities found in sequencing also showed that GJB2 and GJB6 are intact on both chromosomes and that the affected individuals do not bear either del(GJB6-D13S1830) or del(GJB6-D13S1854). Southern blotting indicated no rearrangements or unusual methylation around GJB2 (fig. 4).
Figure 4. .
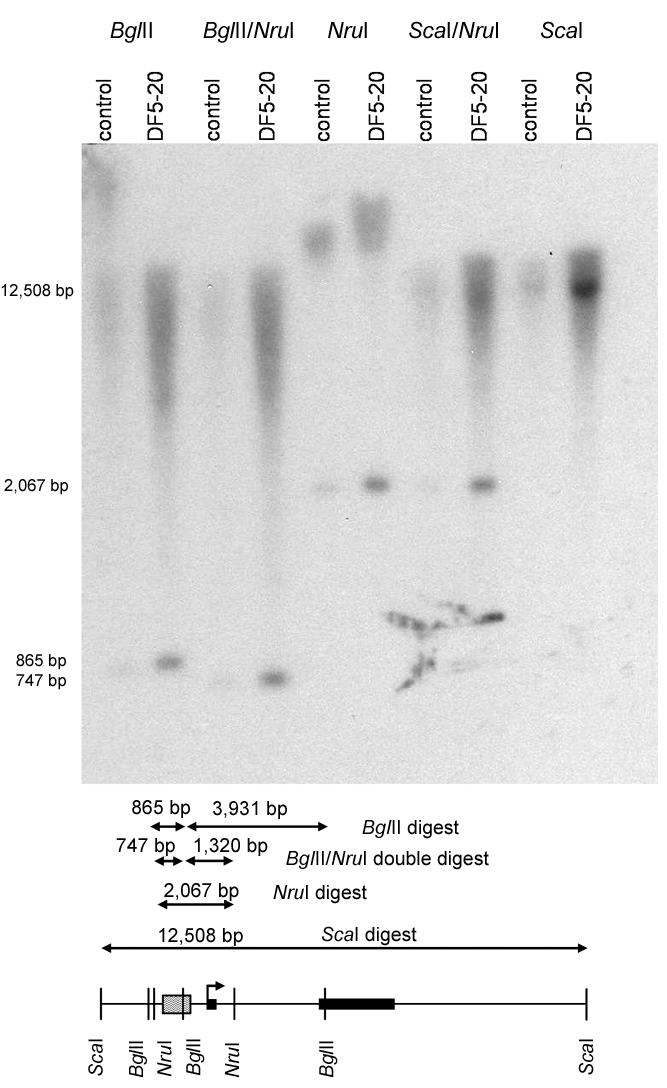
Southern blot of region around GJB2, in DF5-20 and an unrelated control. Transcriptional start site is indicated by a right-angled arrow, exons by boxes, and 32P-labeled probe by a shaded box. The detected probe is associated with expected digestion-product sizes, and results are similar for both DF5-20 and the control. Since NruI is a methylation-sensitive restriction enzyme, these results also preclude unusual methylation on either allele of DF5-20. Five-microgram aliquots of DNA were digested with 1 U of BglII, NruI, or ScaI (all New England Biolabs) for single digests or with 1 U of BlgII or ScaII in combination with 1 U of NruI for double digests. Each digestion was incubated for 20 h at 37°C in the appropriate concentration of NEB Buffer 3 (New England Biolabs) in 50-μl reactions. Of each digested sample (after addition of 5 μl loading dye to each tube), 45 μl was run on a 0.7% agarose gel with Markers II and III (Roche) and was transferred to a nylon membrane by use of a standard protocol. A 618-bp PCR product, amplified from genomic sequence 500 to 1,000 bp upstream of the transcriptional start site of GJB2, was radioactively labeled with ∼50 μCi [α-32P]dATP, by use of the protocol provided with the Invitrogen Random Primers DNA Labeling System, and was purified by Chroma Spin column (BD Biosciences). The probe was hybridized to the membrane overnight at 65°C by use of a standard protocol; the membrane was washed and exposed on a PhosphoImager overnight and then on x-ray film for 4 d (film image is shown).
We hypothesized that an unidentified cis-regulatory element of GJB2 exists within the DFNB1 locus and is disrupted on our novel pathogenic allele. Given the impracticality of searching for candidate variants across the entire locus, we first sought to acquire evidence of loss of GJB2 expression from this allele. We developed three allele-specific PCR assays to assess the relative abundance of transcript from each allele, using tissue both easily available and expected to express Cx26. To isolate RNA, buccal cells from several family members were collected on Cytosoft brushes and were immediately stored in 1 ml Trizol (Gibco/Life Technologies) on ice. Some samples were stored at −20°C for one to several days. RNA was isolated after the Trizol protocol, was resuspended in 20 μl H2O, and was stored at −80°C. We followed a modified Superscript II (Invitrogen) reverse-transcription protocol for cDNA synthesis; 5–8 μl template RNA, 1 μl of 0.5 μg/μl oligo(dT) primer, 1 μl of 10-mM dNTP, and 2 μl H2O were heated to 70°C for 10 min, were chilled on ice, and then were mixed and incubated at 42°C for 2 min after 4 μl of 5× first-strand buffer and 2 μl of 0.1-M dithiothreitol was added. Addition of 1 μl Superscript II reverse transcriptase was followed by incubation at 42°C for 50 min and then incubation at 70°C for 15 min. A final incubation at 37°C for 20 min followed the addition of 1 μl RNaseH. A negative control to test for genomic DNA contamination was generated for each sample by omitting the Superscript II and replacing it with 1 μl H2O. These negative controls were carried through the assays side by side with the corresponding sample. In no instance was product observed for these controls (data not shown).
We designed a cDNA-specific PCR assay to assess the relative abundance of 35delG and non-35delG product amplified from GJB2 cDNA synthesized from 35delG heterozygotes (fig. 5A). A standard PCR-based assay for the identification of 35delG in genomic DNA13 is based on the introduction of a restriction site for BstNI by a 3′ mismatch primer (5′-GCTGGTGGAGTGTTTGTTCACACCCGC-3′) that overlaps the run of Gs between nt 30 and nt 35. The BstNI restriction site is present in PCR product amplified from wild-type (non-35delG) alleles only and is absent in PCR product amplified from 35delG alleles. We paired a 5′ primer located in exon 1 (5′-CGCAGAGACCCCAACGCCGAGA-3′) with the 3′ mismatch primer to generate a 139-bp PCR product from cDNA template only, which, when incubated with BstNI, will yield digestion products of 110 bp and 29 bp only if the 35delG mutation is not present. For each individual we assayed, we amplified from cDNA template and from the corresponding negative control. A 20-μl volume PCR (1–5 μl template cDNA, 2 μl 10× Qiagen buffer, 4 μl Q solution (Qiagen), 0.16 μl of 25-mM dNTP (Invitrogen), 1 μl of each 20-μM primer, 5 U Invitrogen Taq DNA polymerase, and remainder H2O) was denatured for 3 min at 94°C, followed by 40 cycles at 94°C for 30 s, 66°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 5 min. Of each PCR, 10 μl was digested with ∼10 U BstNI (New England Biolabs) for >2 h, and 3 μl of each reaction was run on a 3.5% NuSieve 3:1 agarose gel containing 0.3 μg/ml ethidium bromide.
Figure 5. .

Allele-specific expression assays for GJB2. A, Schematic showing design of BstNI (which assays for 35delG [35ΔG] genotype) and BseYI (which assays for +94 genotype) digestion assay performed using 139-bp PCR product amplified from cDNA only. A forward primer (F) located in the first, noncoding exon of GJB2 is separated on genomic DNA from the reverse mismatch primer (R), located in the coding region of GJB2, by a >3-kb intron (F’ = forward primer for genomic 35delG assay). The BstNI site is introduced into product amplified from wild-type (non-35delG [indicated by “+”]) template. B and D, Pedigrees including DF5-70 and DF5-20, heterozygous for both GJB2 35delG and the novel pathogenic allele (black bar with star). Also indicated are the 35delG and +94 genotypes. C and E, Results of cDNA-specific assay for GJB2 expression based on 35delG genotype. Products of 139 bp were amplified from all tested family members (ud = undigested, no enzyme added; d = addition of restriction enzyme). DF5-20, -70, and -71 (normal-hearing sibling of DF5-70) are all heterozygous for 35delG; however, after digestion with BstNI, only DF5-71 clearly shows both 139-bp and 110-bp products, indicating representation of two different alleles at the cDNA level. DF5-20 and DF5-70, who both bear the novel pathogenic non-35delG allele, show either no or barely detectable 110-bp product, indicating underrepresentation of this allele in the pool of amplified product (M = Marker V [Roche]). F, Results of cDNA-specific assay for GJB2 expression based on genotype at GJB2 +94. Products of 139 bp (ud = undigested) were amplified from both tested family members, who are both heterozygous (A/C) at +94. Although the digested product (d) amplified from DF5-72 cDNA clearly indicates heterozygosity at the cDNA level, lack of any detectable 139-bp product in the digested DF5-67 sample indicates underrepresentation of the novel (C) allele among the amplified product.
The results (fig. 5B–5E) for the two deaf 35delG heterozygotes that we assayed, DF5-20 and DF5-70, indicate that, although they are heterozygous for 35delG at the genomic DNA level, the barely detectable or absent 110-bp product indicates underrepresentation of GJB2 transcript from the novel (non-35delG) allele. Results from the parents and sibling of DF5-70 are consistent with their genotypes and provide appropriate controls. Unbiased amplification and digestion of both 35delG and non-35delG alleles is shown by the result for DF5-71, the sibling of DF5-70, who is also heterozygous for 35delG but carries a normal non-35delG chromosome. Sequencing of genomic DNA across the 139-bp target sequence (not shown) confirmed that there are no differences between these two individuals that would give rise to a bias in either amplification or digestion.
To demonstrate that loss of expression of this allele is not unique to individuals carrying the 35delG mutation on their other chromosome 13, we developed a second assay to determine allele-specific GJB2 expression in a normal-hearing adult offspring of DF5-20 bearing the novel haplotype (fig. 5A and 5D). DF5-67 carries a previously unreported exon 1 SNP at position +94. BseYI digestion of the 139-bp product amplified with the same primers as described above will yield 102-bp and 27-bp products only if an A is present at position +94 (10 μl of each PCR product was digested with 5 U BseYI enzyme [New England Biolabs] for >2 h, 5 μl of 1% SDS was added, and electrophoresis was performed as above). Although both DF5-67 and -72 are heterozygous for +94 at the genomic level, all of the product amplified from DF5-67 cDNA was digested (fig. 5F), indicating that no or very little of the PCR product had been amplified from template derived from the novel allele that bears the common variant, C. Again, sequencing of genomic DNA across the target sequence shows no differences between DF5-67 and DF5-72 that would yield biased amplification or digestion of the resulting amplimers.
We hypothesized that loss of expression of GJB2 might be accompanied by loss of expression of GJB6, since these two genes lie within 30 kb of each other and their products are coexpressed in the cochlea. DF5-65, a normal-hearing adult offspring of DF5-20, bears the novel haplotype and is heterozygous for rs7333214 in the 3′ UTR of GJB6, allowing us to test this hypothesis (fig. 6A and 6B). Using a forward primer located in the fourth noncoding exon (5′-CACCATTGGCTTCTAGGCAC-3′) and a reverse primer located close to the 3′ end of the 3′ UTR (5′-CCACACTGTTCCGTCTACAT-3′), we amplified a 1,550-bp product from cDNA template only, in a 20-μl PCR under the following conditions: 2 μl of 10× Qiagen buffer, 4 μl Q solution (Qiagen), 0.16 μl of 25-mM dNTP (Invitrogen), 1 μl of each 20-μM primer, 5 U Taq DNA polymerase (Invitrogen), and remainder H2O were denatured for 5 min at 94°C, followed by 50 cycles at 94°C for 30 s, 57°C for 30 s, and 72°C for 2 min, with a final extension at 72°C for 5 min. Digestion of this product for ∼2 h at 37°C with ∼10 U HpyCH4IV (New England Biolabs) yields digestion products of 537, 460, 338, and 214 bp if an A (the rare variant) is present at rs7333214; the 460-bp product is digested to 292- and 168-bp products if a C is present at rs7333214 (the common variant, carried, in this case, on the novel allele). Two microliters of each reaction was run on a 1.5% agarose gel (10 μg ethidium bromide/30 ml) in 0.5× Tris-borate-EDTA buffer. Comparison of the result for DF5-65 with that for a control (DF5-72, his mother) who is also heterozygous for rs7333214 (fig. 6B) and whose DNA is identical across the target sequence, shows that the 292- and 168-bp products, derived from amplification of the novel allele, are significantly underrepresented in DF5-65 (fig. 6C). This is consistent with the interpretation that expression of GJB6 as well as that of GJB2 is diminished from the novel allele. Additionally, since the cDNA specificity of each assay depends on the amplification of PCR product from only cDNA from which a long intron has been properly spliced out, failure to accumulate PCR product might also be expected from an allele that is mutant for proper splicing. It is highly unlikely that GJB2 and GJB6 on this chromosome both contain unidentified splice mutations.
Figure 6. .

Allele-specific expression assay for GJB6. A, Schematic of design of HpyCH4IV digestion assay for allele-specific expression of GJB6 mRNA performed using 1,550-bp PCR product amplified from cDNA only. A forward primer (F) located in exon 5 of GJB6 is separated on genomic DNA from the reverse primer (R), located in the GJB6 3′ UTR, by a >6-kb intron in addition to ∼1,500 bp of the final exon. B, Pedigree showing rs7333214 genotypes of assayed subjects. DF5-65 bears the novel pathogenic allele (black bar with star) and is heterozygous at rs7333214. C, Results of cDNA-specific assay for GJB6 expression. Digestion of product amplified from DF5-72 yields 460-bp as well as 292- and 168-bp products, indicating that both GJB6 alleles are expressed. Digestion of product amplified from DF5-65 yields a robust 460-bp band, but the band at 292 bp is only barely visible. This indicates underrepresentation of the novel allele among the amplified product.
This study provides evidence that the MSU-DF5 family is segregating a novel DFNB1 allele that is characterized by significant reduction in expression of both GJB2 and GJB6. This loss of expression is heritable, cis-acting, and not due to a simple parent-of-origin epigenetic modification. We sought and found no coding-region mutations, splice-site mutations, or any other sequence variant unique to the novel allele within or close to GJB2 or GJB6, suggesting that loss of function of an as-yet-unidentified cis-regulatory element(s) is responsible for our observations. It is possible that a locus-control region (LCR) regulates the coexpression of GJB2 and GJB6. An LCR was initially described in the β-globin locus, where it is absolutely required for expression of any of the genes in this cluster. Other examples include the growth hormone and TH2 cytokine loci.14 Such an element(s) may be located within the common interval deleted on chromosomes bearing del(GJB6-D13S1830) or del(GJB6-D13S1854). The severity of hearing loss in the individuals in our kindred who are heterozygous for 35delG and this novel allele is similar to that in individuals who are del(GJB6-D13S1830)/35delG, who as a group have been shown to have more severe hearing loss than 35delG homozygotes.15 Beyond the identification of the basal promoters of both genes,16–19 nothing is known of cis-acting elements that regulate GJB2 and GJB6. The common observation of an excess of deaf individuals bearing only a single identified GJB2 mutation strongly suggests that GJB2 regulatory mutations remain to be identified.
Acknowledgments
We thank the members of the MSU-DF5 family, for their generous participation, and Dr. Thomas Friedman, Dr. Robert Morell, and Dr. Patrick Venta, for helpful discussion and critical reading of the manuscript. This research was supported by the Michigan State University Foundation and by National Institute on Deafness and Other Communication Disorders grant DC004568 (to K.H.F.).
Web Resource
The URL for data presented herein is as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for Vohwinkel syndrome, Bart-Pumphrey syndrome, PPK, KID, hidrotic ectodermal dysplasia, and Pendred syndrome)
References
- 1.Forge A, Becker D, Casalotti S, Edwards J, Marziano N, Nevill G (2003) Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessment of connexin composition in mammals. J Comp Neurol 467:207–231 10.1002/cne.10916 [DOI] [PubMed] [Google Scholar]
- 2.Forge A, Marziano N, Casalotti SO, Becker DL, Jagger D (2003) The inner ear contains heteromeric channels composed of cx26 and cx30 and deafness-related mutations in cx26 have a dominant negative effect on cx30. Cell Commun Adhes 10:341–346 [DOI] [PubMed] [Google Scholar]
- 3.Ahmad S, Chen S, Sun J, Lin X (2003) Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochem Biophys Res Commun 307:362–368 10.1016/S0006-291X(03)01166-5 [DOI] [PubMed] [Google Scholar]
- 4.Kikuchi T, Kimura RS, Paul DL, Takasaka T, Adams JC (2000) Gap junction systems in the mammalian cochlea. Brain Res Rev 32:163–166 10.1016/S0165-0173(99)00076-4 [DOI] [PubMed] [Google Scholar]
- 5.Wangemann P (2002) K+ cycling and the endocochlear potential. Hear Res 165:1–9 10.1016/S0378-5955(02)00279-4 [DOI] [PubMed] [Google Scholar]
- 6.Grifa A, Wagner CA, D’Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, Rabionet R, Arbones ML, Monica MD, Estivill X, Zelante L, Lang F, Gasparini P (1999) Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet 23:16–18 [DOI] [PubMed] [Google Scholar]
- 7.Lerer I, Sagi M, Ben-Neriah Z, Wang T, Levi H, Abeliovich D (2001) A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: a novel founder mutation in Ashkenazi Jews. Hum Mutat 18:460 10.1002/humu.1222 [DOI] [PubMed] [Google Scholar]
- 8.del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, Menendez I, Moreno F (2002) A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 346:243–249 10.1056/NEJMoa012052 [DOI] [PubMed] [Google Scholar]
- 9.Pallares-Ruiz N, Blanchet P, Mondain M, Claustres M, Roux AF (2002) A large deletion including most of GJB6 in recessive non syndromic deafness: a digenic effect? Eur J Hum Genet 10:72–76 10.1038/sj.ejhg.5200762 [DOI] [PubMed] [Google Scholar]
- 10.del Castillo FJ, Rodriguez-Ballesteros M, Alvarez A, Hutchin T, Leonardi E, de Oliveira CA, Azaiez H, et al (2005) A novel deletion involving the connexin-30 gene, del(GJB6-D13S1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet 42:588–594 10.1136/jmg.2004.028324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.del Castillo I, Moreno-Pelayo MA, del Castillo FJ, Brownstein Z, Marlin S, Adina Q, Cockburn DJ, et al (2003) Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. Am J Hum Genet 73:1452–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Common JE, Bitner-Glindzicz M, O’Toole EA, Barnes MR, Jenkins L, Forge A, Kelsell DP (2005) Specific loss of connexin 26 expression in ductal sweat gland epithelium associated with the deletion mutation del(GJB6-D13S1830). Clin Exp Dermatol 30:688–693 10.1111/j.1365-2230.2005.01878.x [DOI] [PubMed] [Google Scholar]
- 13.Wilcox SA, Osborn AH, Dahl HH (2000) A simple PCR test to detect the common 35delG mutation in the connexin 26 gene. Mol Diagn 5:75–78 10.1016/S1084-8592(00)00014-X [DOI] [PubMed] [Google Scholar]
- 14.Dean A (2006) On a chromosome far, far away: LCRs and gene expression. Trends Genet 22:38–45 10.1016/j.tig.2005.11.001 [DOI] [PubMed] [Google Scholar]
- 15.Snoeckx RL, Huygen PLM, Feldmann D, Marlin S, Denoyelle F, Waligora J, Mueller-Malesinska M, et al (2005) GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet 77:945–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tu ZJ, Kiang DT (1998) Mapping and characterization of the basal promoter of the human connexin26 gene. Biochim Biophys Acta 1443:169–181 [DOI] [PubMed] [Google Scholar]
- 17.Kiang DT, Jin N, Tu ZJ, Lin HH (1997) Upstream genomic sequence of the human connexin26 gene. Gene 199:165–171 10.1016/S0378-1119(97)00365-X [DOI] [PubMed] [Google Scholar]
- 18.Essenfelder GM, Larderet G, Waksman G, Lamartine J (2005) Gene structure and promoter analysis of the human GJB6 gene encoding connexin30. Gene 350:33–40 10.1016/j.gene.2004.12.048 [DOI] [PubMed] [Google Scholar]