

Abstract
Osseous abnormalities, including long-bone dysplasia with pseudarthrosis (PA), are associated with neurofibromatosis type 1 (NF1). Prospectively acquired tissue from the PA site of two individuals with NF1 was used for immunohistochemical characterization and genotype analysis of the NF1 locus. Typical immunohistochemical features of neurofibroma were not observed. Genotype analysis of PA tissue with use of four genetic markers (D17S1863, GXALU, IN38, and 3NF1-1) spanning the NF1 locus demonstrated loss of heterozygosity. These results are the first to document double inactivation of NF1 in PA tissue and suggest that the neurofibromin-Ras signal transduction pathway is involved in this bone dysplasia in NF1.
Neurofibromatosis type 1 (NF1 [MIM +162200]) is a common autosomal dominant genetic disorder that affects 1 in 3,500 individuals worldwide. It is a genetic condition with a high degree of variability of clinical expression, although it is fully penetrant in adults. The primary skeletal abnormalities associated with NF1, reported in 38% of patients,1 include long-bone dysplasia, sphenoid-wing dysplasia, and scoliosis. Long-bone dysplasia, seen in 5% of patients with NF1,2,3 typically involves the tibia and frequently presents with anterolateral bowing that may progress to fracture and nonunion. Tibial dysplasia is most often unilateral, evident in the first year of life, and usually not associated with a neurofibroma.4
The biologic basis, pathogenesis, and molecular causes of pseudarthrosis (PA) and tibial dysplasia are not known. The unilateral nature of tibial dysplasia implicates a random molecular event, which then predisposes the abnormal bone to a progressive sequence of bowing, followed by fracture and subsequent poor healing that may be inherent in the bone itself. In neurofibromas, there is biallelic inactivation of NF1; however, studies that examine alterations in the neurofibromin-Ras signal transduction pathway in the osseous dysplasias of NF1 have not been conducted elsewhere.
We evaluated two unrelated individuals with NF1 who had long-bone dysplasia and PA. Patient 1 is a 42-year-old man with a significant family history: a father with NF1 and a brother with NF1 and lower-extremity PA (status postamputation). Clinical findings included >10 café-au-lait macules, axillary freckling, multiple cutaneous and subcutaneous neurofibromas, tibial PA, and mild lumbar scoliosis. Anterolateral bowing of the right lower extremity presented at birth. He fractured his right tibia at age 1 year and underwent a tibia-fibula syndesmosis, and union was achieved. At age 41 years, he fractured his right tibia while attempting to sit down. Radiographs taken 8 mo later showed PA between the middle and distal third of the tibia, with prominent anterior bowing in the setting of severe osteopenia. Surgical intervention was attempted with a resection of the right tibial PA, tibial osteotomy with realignment and internal fixation with plating, and iliac-crest bone grafting. There was a great deal of scarring and neovascularity, with several centimeters of very poor bone. The synostosis of the tibia and fibula was osteotomized together, with removal of a couple of centimeters of bone. Because of the procurvatum deformity, a realignment osteotomy was done secondarily, which revealed healthy bone on either end of the PA tissue. The specimens consisted of multiple coarse, irregular portions of cortical and cancellous bone, in aggregate, measuring 6.0×5.0×1.8 cm. At 2 mo and 4 mo after surgery, the PA site was not completely healed, but partial healing was noted at 6 mo postsurgery (fig. 1).
Figure 1. .
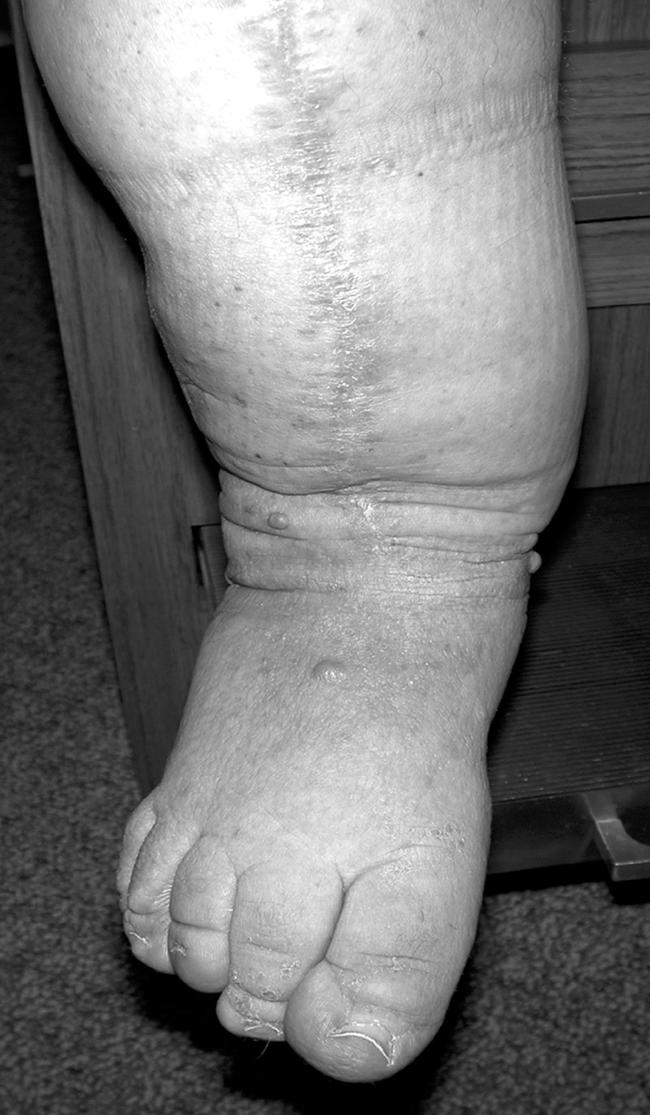
Lower extremity of patient 1
Patient 2 is a 2-year-old boy whose tibial and fibular bowing presented at birth, with subsequent fibular fracture at age 2 wk. Clinical findings consistent with NF1 include more than five café-au-lait macules and tibial PA (fig. 2). Family history was significant: a mother with NF1. Radiographs taken at age 4 mo confirmed tibial and fibular PA, and a brace was applied. Just before surgical intervention, radiographs showed tibial and fibular PA, without callus formation, with 64° of anterior angulation (fig. 3). At age 2 years and 3 mo, the boy underwent left tibial and fibular PA take-down, with open reduction and internal fixation and autogenous as well as demineralized bone matrix grafting. The tibial PA was resected en bloc to bleeding bone ends proximally and distally (fig. 4). The fibular PA was also resected in a similar fashion. Williams rodding of the left tibia and fibula was performed, with left iliac crest bone grafting supplemented with demineralized bone matrix. Radiographs 3 mo after surgery showed callus formation around the PA site, with overall diminished bone density.
Figure 2. .
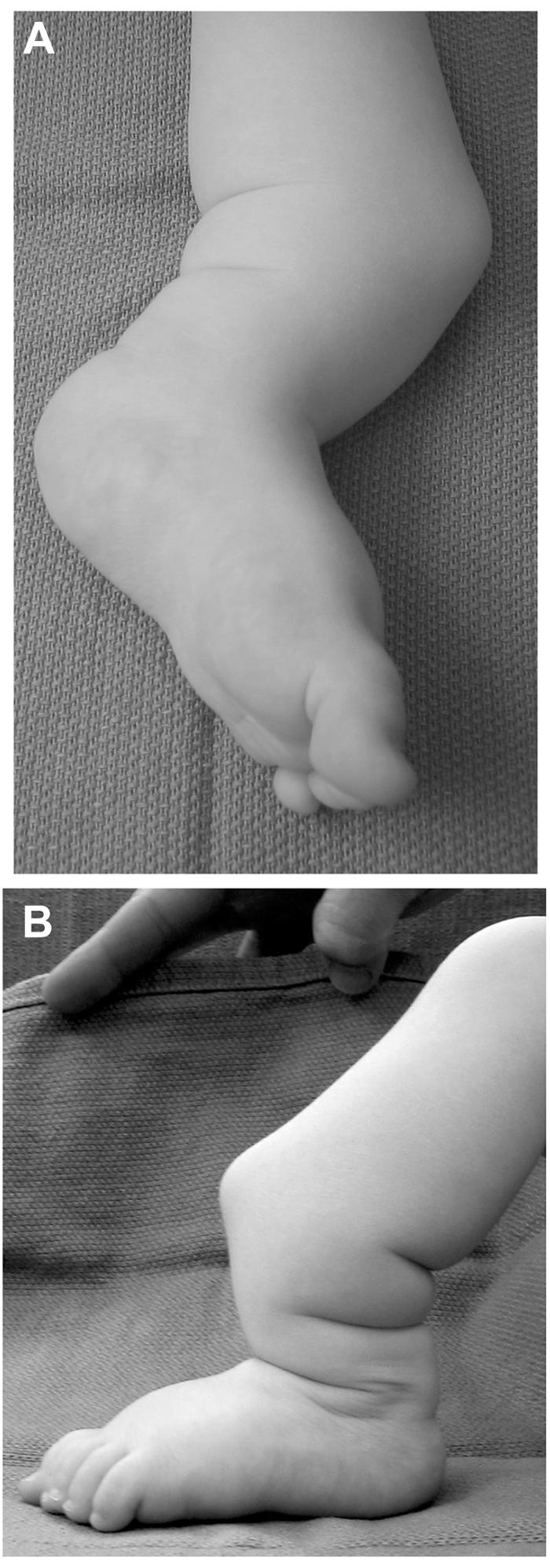
Lower extremity of patient 2
Figure 3. .
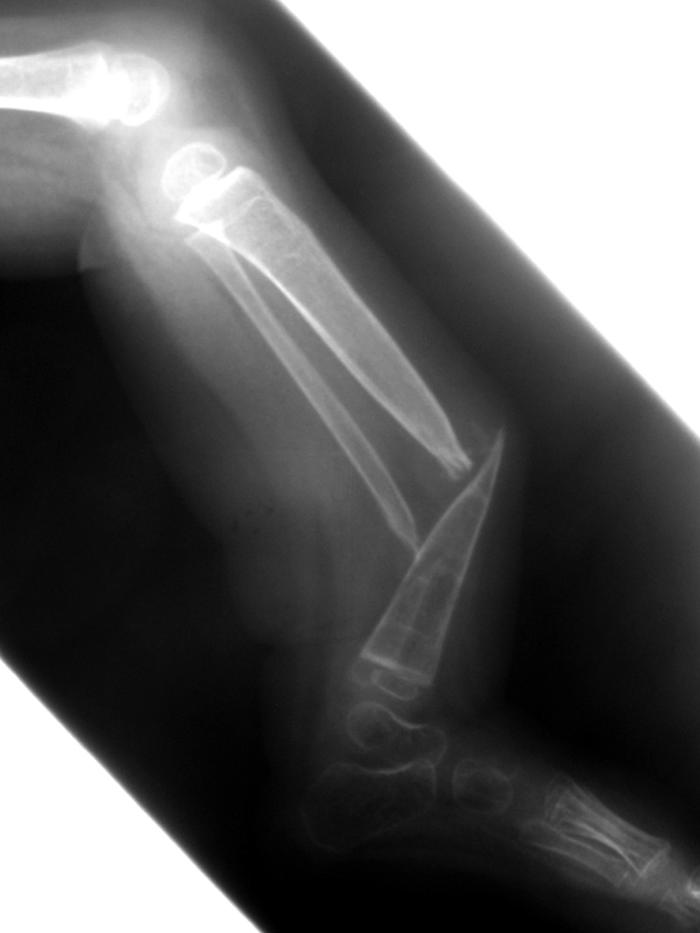
Radiographs of patient 2 showing tibial and fibular bowing, with nonunion of fracture.
Figure 4. .

Intraoperative images of PA site of patient 2. A, PA site prior to resection. B, Operative site after resection of tibial PA.
Informed consent was obtained from the study subjects, and the study was approved by the Institutional Review Board at the University of Utah. Tissue from the PA site was obtained prospectively from patient 2 during surgery and was obtained retrospectively from paraffin blocks for patient 1. Both tissues were evaluated for immunohistochemical characterization (fig. 5) and genotype analysis of the NF1 locus.
Figure 5. .

Area where DNA was extracted for genotyping. A, In patient 1, hematoxylin and eosin stain of a paraffin-embedded section from a tibial PA specimen. B, In patient 2, hematoxylin and eosin stain of a paraffin-embedded section from a tibial PA specimen.
Well-defined antibody stains were used for routine immunohistochemistry. Samples from both patients demonstrated lack of S100 staining, a marker of Schwann cells typically seen in neurofibromas. In both patients, the PA tissues were composed of cellular soft tissue between bone ends of the fracture gaps and lacked a bony callus seen in normal fracture healing. In patient 1, the cellular tissue was associated with granulation tissue, fibrocartilage, and hyaline cartilage with minimal endochondral ossification. Samples from patient 2 showed lamellar and woven bone with “fibrosis” and osteoclastic giant cells and focal fibrocartilage. The periosteum was markedly thickened with rare foci of reactive bone. No endochondral ossification was observed. Cellular tissue adjacent to bone did not have a distinctive appearance, and typical features of neurofibroma were not observed.
The histologically distinct, hypercellular portions of surgically resected PA tissue from patient 1 were identified using formalin-fixed, paraffin-embedded serial sections stained with hemotoxylin and eosin. Additional serial, unstained sections were microdissected, and DNA was isolated for genome amplification5 before specific amplification of the NF1 locus by PCR. For patient 2, DNA was extracted from fresh-frozen PA tissue by use of standard protocols. Constitutional DNA from white blood cells was used for comparison with tissue DNA, to assess allelic imbalance of the NF1 locus.
Amplified products were subjected to genotype analysis with use of four genetic markers (D17S1863, GXALU, IN38, and 3NF1-1) spanning the NF1 locus, to detect allelic imbalance and loss of heterozygosity (LOH). D17S1863 and 3NF1-1 are extragenic markers flanking the NF1 locus (UCSC Genome Browser; May 2004 assembly). Marker 3NF1-1 is a dinucleotide-repeat polymorphic marker (∼218 kb 3′ of NF1) with a product size of 245 bp (forward primer: CTTCCATGGCTGCTAACATC; reverse primer: CCCTGTGGTGTAGTTCAACA).6 GXALU is an intragenic STR polymorphism (AAAT)n in intron 27b.7 IN38 is an intragenic STR polymorphism (CA)n in intron 38.8 This methodology has been used extensively to identify LOH at the NF1 locus in NF1-related tumor tissue.9,10 After PCR, the PCR product was run on the Applied Biosystems 3130xl Genetic Analyzer with Applied Biosystems POP-7 Polymer, 36-cm capillary, and GeneScan500 LIZ size standard. The data were analyzed and scored using Applied Biosystems GeneMapper software (Applied Biosystems). Genotype analysis of the amplified DNA products from the histologically distinct, hypercellular portions of surgically resected PA tissue showed clear LOH at D17S1863, GXALU, and 3NF1-1 in patient 1 and showed D17S1863, IN38, and 3NF1-1 in patient 2 (IN38 was noninformative in patient 1, and GXALU was noninformative in patient 2). This was confirmed on separate analysis. Comparison of allele ratios of PCR products of the informative genetic loci from both blood and tissue is shown in figures 6 and 7.
Figure 6. .

Allele imbalance genotype analysis of blood DNA and tissue DNA for patient 1, with use of three genetic markers. A, D17S1863, which lies centrometric of the NF1 locus. B, GXALU, intragenic marker (STR in intron 27b). C, 3NF1-1, which lies just telomeric of the NF1 locus.
Figure 7. .
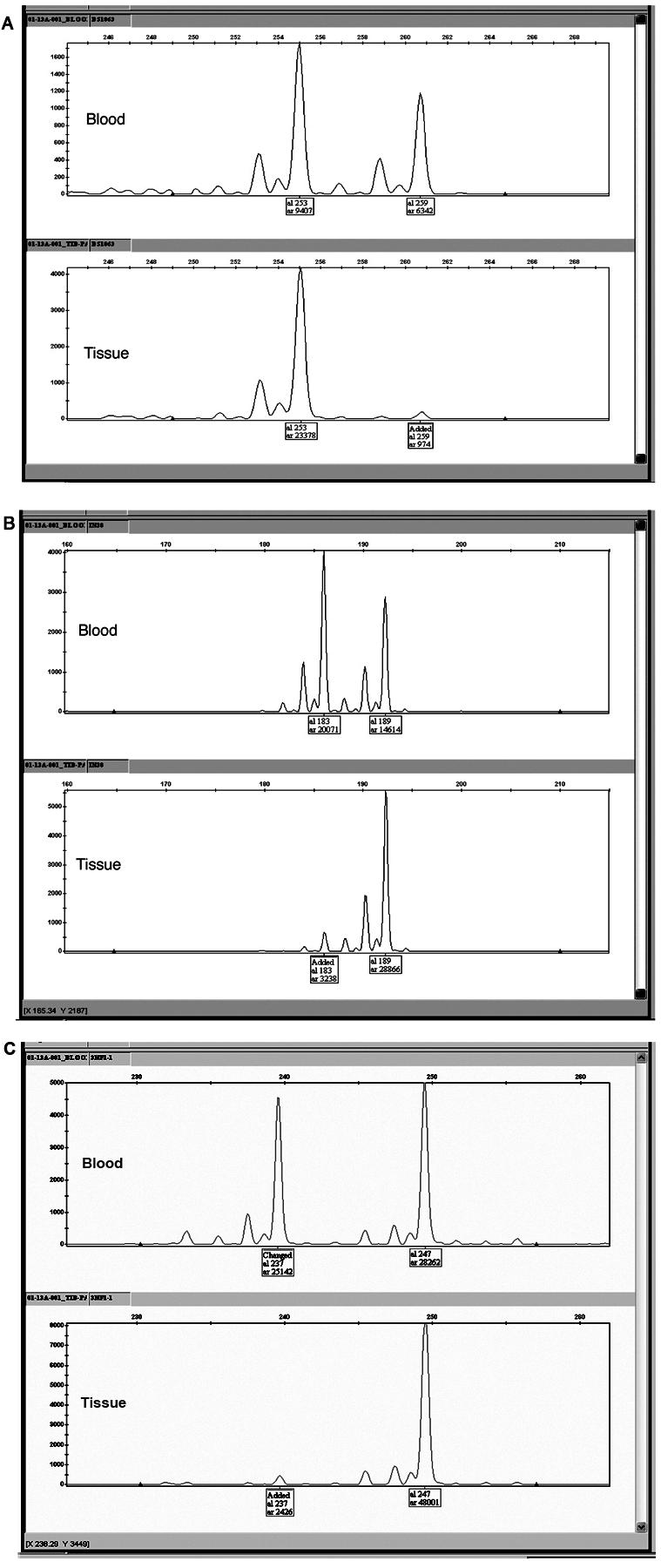
Allele imbalance genotype analysis of blood DNA and tissue DNA for patient 2, with use of three genetic markers. A, D17S1863, which lies centrometric of the NF1 locus. B, IN38, intragenic marker (STR in intron 38). C, 3NF1-1, which lies just telomeric of the NF1 locus.
Peripheral blood was collected from both patients for constitutional mutation analysis with use of protocols published elsewhere.11 The constitutional NF1 gene mutation from peripheral lymphocytes was identified as a nonsense mutation in exon 16 (c.2446C→T; p.R816X) in patient 1 and as a nonsense mutation in exon 45 (c.7846C→T; p.R2616X) in patient 2 (fig. 8). The mutant allele was retained in the PA tissue in patient 2, which documents double inactivation of NF1. We were unable to identify which allele was retained in patient 1 because of limited and poor-quality DNA extracted from the paraffin-embedded sample of the PA tissue.
Figure 8. .

Sequencing profile of blood (A) and tissue (B) of patient 2.
To our knowledge, this is the first documentation of double inactivation of NF1 in PA tissue. These results suggest that rare double inactivation of NF1 by somatic mutation of the NF1 gene, in a population of cells that depend on appropriate neurofibromin-regulated Ras signaling to maintain normal bone, contributes to the development or progression of PA in NF1. Still, the role neurofibromin plays in the growth and development of bone is poorly understood. Neurofibromin, the NF1 gene product, has “tumor suppressor” activities through its interactions with Ras and may converge with other biochemical pathways that involve bone, such as bone morphogenetic protein-signal transduction.
Previous histological evaluations of bone and the surrounding tissue have failed to determine the pathogenesis of PA.12–16 The primary histologic finding was the presence of abnormal, highly cellular, fibromatosis-like tissue, commonly associated with a thickened periosteum surrounding the PA site. Most investigators concluded that there was no evidence of an intraosseous or periosteal neurofibroma, and comparison between those with and without NF1 did not identify histological differences.
In our immunohistochemical study, the cellular tissues were negative for S100, a marker for Schwann cells that is invariably seen in benign neurofibroma tissue, which supports the general consensus that neurofibromas rarely occur at the site of PA. The lack of association of neurofibromas with long-bone dysplasia and PA in this and previous reports suggests that intrinsic bone pathology, caused by loss of a functional NF1 gene and aberrant Ras signaling, may play a primary role in the skeletal abnormalities in NF1.
Several recent studies of animal models have provided evidence that NF1 plays an important role in regulating osteoprogenitors and the composition of the bone matrix during ossification. In heterozygous Nf1+/− mice, Nf1 haploinsufficiency deregulated Ras signaling in bone marrow–inducible osteoprogenitors, induced premature osteoblast apoptosis, and altered osteoprogenitor cell proliferation and differentiation.17 Phosphorylated p42/p44 MAP kinases (MAPKs) were found to be elevated in hypertrophic chondrocytes of Nf1+/− rodent embryos.18 In a study of fracture healing of mouse tibia and experimental PA in rat,19 NF1 gene expression was present in maturing and hypertrophic cartilage in both models. In the same study, phosphorylated p44/42 MAPK was detected in a subpopulation of the hypertrophic chondrocytes. Kuorilehto et al.19 concluded that NF1 gene expression and neurofibromin activity are needed for normal fracture healing, possibly by restraining excessive Ras-MAPK pathway activation.
In normal fracture healing, bony callus develops with new bone formation through membranous (periosteal) and endochondral ossification. The participating osteoblasts originate from proliferation and differentiation of osteoprogenitors in multipotential mesenchymal cells of the periosteum and marrow stroma cells. Loss of NF1 function with subsequent Ras deregulation in NF1 may lead to altered osteoblastic/osteoprogenitor differentiation and proliferation, impaired bony callus formation, and overgrowth of cellular tissue due to preferred fibroblast/myofibroblast differentiation of multipotential mesenchymal cells in stroma and periosteal cells. Further studies on prospectively obtained PA tissue will help confirm these findings and support future treatment protocols that could be focused on local diminution of aberrant Ras signaling.
Acknowledgments
We thank the study participants, research coordinators, and laboratory assistants (Heather Wintch, Jeanne Siebert, Susan Geyer, Meredith Winn, Diane Hartford, Bronte Clifford, and Terri S. Nielsen-Rogers), for their help in this study. We thank the members of the Genomics Core Facility at the University of Utah for their assistance, Dr. Lor Randall for his assistance and referral of study subject, and Dr. Cheryl Coffin for valuable insight in clinical pathology. We thank Tom Callens, from the Medical Genomics Lab at the University of Alabama at Birmingham, for help with the NF1 mutational analysis. This research was performed with support in part from the Shriners Research Foundation, National Center for Research Resources Public Health Services research grant M01-RR00064, National Institute of Neurological Disorders and Stroke research grant 1 K23 NS052500-01, the Children’s Health Research Center at the University of Utah, and the Clinical Genetics Research Program supported by the Primary Children’s Research Foundation. The ideas for this research originated in part from discussion at a symposium for bone-related issues in NF1 that was supported by Shriners Hospital for Children Intermountain and the National Neurofibromatosis Foundation, now known as the “Children’s Tumor Foundation.”
Web Resources
The URLs for data presented herein are as follows:
- Genomics Core Facility, http://www.cores.utah.edu/genomics/ (for the Utah Index Set at the University of Utah)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.gov/Omim/ (for NF1) [PubMed]
- UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway (for the May 2004 Assembly)
References
- 1.Crawford AH, Schorry EK (1999) Neurofibromatosis in children: the role of the orthopaedist. J Am Acad Orthop Surg 7:217–230 [DOI] [PubMed] [Google Scholar]
- 2.McElhannon FM Jr (1975) Congenital pseudoarthrosis of the tibia. South Med J 68:824–827 [DOI] [PubMed] [Google Scholar]
- 3.Riccardi VM (1981) Von Reckinghausen neurofibromatosis. N Engl J Med 305:1617–1626 [DOI] [PubMed] [Google Scholar]
- 4.Stevenson DA, Birch PH, Friedman JM, Viskochil DH, Balestrazzi P, Boni S, Buske A, Korf BR, Niimura M, Pivnick EK, Schorry EK, Short MP, Tenconi R, Tonsgard JH, Carey JC (1999) Descriptive analysis of tibial pseudarthrosis in patients with neurofibromatosis 1. Am J Med Genet 84:413–419 [DOI] [PubMed] [Google Scholar]
- 5.Paulson TG, Galipeau PC, Reid BJ (1999) Loss of heterozygosity analysis using whole genome amplification, cell sorting, and fluorescence-based PCR. Genome Res 9:482–491 [PMC free article] [PubMed] [Google Scholar]
- 6.López Correa C, Brems H, Lázaro C, Estivill X, Clementi M, Mason S, Rutkowski JL, Marynen P, Legius E (1999) Molecular studies in 20 submicroscopic neurofibromatosis type 1 gene deletions. Hum Mutat 14:387–393 [DOI] [PubMed] [Google Scholar]
- 7.Xu G, Nelson, L, O’Connell P, White R (1991) An Alu polymorphism intragenic to the neurofibromatosis type 1 gene. Nucleic Acids Research 19:3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazaro C, Gaona A, Xu G, Weiss R, Estivill X (1993) A highly informative CA/GT repeat polymorphism in intron 38 of the human neurofibromatosis type 1 (NF1) gene. Hum Genet 92:429–430 10.1007/BF01247353 [DOI] [PubMed] [Google Scholar]
- 9.Sawada S, Florell S, Purandare S, Ota M, Stephens K, Viskochil D (1996) Identification of NF1 mutations in both alleles of a dermal neurofibroma. Nat Genet 14:110–112 10.1038/ng0996-110 [DOI] [PubMed] [Google Scholar]
- 10.Rasmussen SA, Overman J, Thomson SA, Colman SD, Abernathy CR, Trimpert RE, Moose R, Virdi G, Roux K, Bauer M, Rojiani AM, Maria BL, Muir D, Wallace MR (2000) Chromosome 17 loss-of-heterozygosity studies in benign and malignant tumors in neurofibromatosis type 1. Genes Chromosomes Cancer 28:425–431 [DOI] [PubMed] [Google Scholar]
- 11.Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, Speleman F, De Paepe A (2000) Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat 15:541–555 [DOI] [PubMed] [Google Scholar]
- 12.Briner J, Yunis E (1973) Ultrastructure of congenital pseudarthrosis of the tibia. Arch Pathol 95:97–99 [PubMed] [Google Scholar]
- 13.Boyd HB (1982) Pathology and natural history of congenital pseudarthrosis of the tibia. Clin Orthop 166:5–13 [PubMed] [Google Scholar]
- 14.Blauth M, Marms D, Schmidt D, Blauth W (1984) Light- and electron-microscopic studies in congenital pseudarthrosis. Arch Orthop Trauma Surg 103:269–277 10.1007/BF00387333 [DOI] [PubMed] [Google Scholar]
- 15.Hefti F, Bollini G, Dungl P, Fixsen J, Grill F, Ippolito E, Romanus B, Tudisco C, Wientroub S (2000) Congenital pseudarthrosis of the tibia: history, etiology, classification, and epidemiologic data. J Pediatr Orthop 9:11–15 [DOI] [PubMed] [Google Scholar]
- 16.Ippolito E, Corsi A, Grill F, Wientroub S, Bianco P (2000) Pathology of bone lesions associated with congenital pseudarthrosis of the leg. J Ped Ortho Part B 9:3–10 [DOI] [PubMed] [Google Scholar]
- 17.Yu X, Chen S, Potter OL, Murthy SM, Li J, Pulcini JM, Ohashi N, Winata T, Everett ET, Ingram D, Clapp WD, Hock JM (2005) Neurofibromin and its inactivation of Ras are prerequisites for osteoblast functioning. Bone 36:793–802 10.1016/j.bone.2005.01.022 [DOI] [PubMed] [Google Scholar]
- 18.Kuorilehto T, Nissinen M, Koivunen J, Benson MD, Peltonen J (2004) NF1 tumor suppressor protein and mRNA in skeletal tissues of developing and adult normal mouse and NF1-deficient embryos. J Bone Miner Res 19:983–989 [DOI] [PubMed] [Google Scholar]
- 19.Kuorilehto T, Ekholm E, Nissinen M, Hietaniemi K, Hiltunen A, Paavolainen P, Penttinen R, Peltonene J (2006) NF1 gene expression in mouse fracture healing and in experimental rat pseudarthrosis. J Histochem Cytochem 54:363–370 10.1369/jhc.5A6784.2005 [DOI] [PubMed] [Google Scholar]