

Abstract
We determined the ventilatory, cardiovascular and cerebral tissue oxygen response to two protocols of normobaric, isocapnic, intermittent hypoxia. Subjects (n = 18, male) were randomly assigned to short-duration intermittent hypoxia (SDIH, 12% O2 separated by 5 min of normoxia for 1 h) or long-duration intermittent hypoxia (LDIH, 30 min of 12% O2). Both groups had 10 exposures over a 12 day period. The hypoxic ventilatory response (HVR) was measured before each daily intermittent hypoxia exposure on days 1, 3, 5, 8, 10 and 12. The HVR was measured again 3 and 5 days after the end of intermittent hypoxia. During all procedures, ventilation, blood pressure, heart rate, arterial oxyhaemoglobin saturation and cerebral tissue oxygen saturation were measured. The HVR increased throughout intermittent hypoxia exposure regardless of protocol, and returned to baseline by day 17 (day 1, 0.84 ± 0.50; day 12, 1.20 ± 1.01; day 17, 0.95 ± 0.58 l min−1%SaO2−1; P < 0.01). The change in systolic blood pressure sensitivity (r =+0.68; P < 0.05) and the change in diastolic blood pressure sensitivity (r =+0.73; P < 0.05) were related to the change in HVR, while the change in heart rate sensitivity was not (r =+0.32; NS). The change in cerebral tissue oxygen saturation sensitivity to hypoxia was less on day 12, and returned to baseline by day 17 (day 1, −0.51 ± 0.13; day 12, −0.64 ± 0.18; day 17, −0.51 ± 0.13; P < 0.001). Acute exposure to SDIH increased mean arterial pressure (+5 mmHg; P < 0.01), but LDIH did not (P > 0.05). SDIH and LDIH had similar effects on the ventilatory and cardiovascular response to acute progressive hypoxia and hindered cerebral oxygenation. Our findings indicate that the vascular processes required to control blood flow and oxygen supply to cerebral tissue in a healthy human are hindered following exposure to 12 days of isocapnic intermittent hypoxia.
Exposure to intermittent hypoxia has several unique effects on human and animal physiology. In response to acute isocapnic hypoxia, there are increases in ventilation, blood pressure and muscle sympathetic nerve activity (Xie et al. 2001), and cerebral blood flow (Ainslie & Poulin, 2004). Daily exposure to repeated bouts of hypoxia increases not only hypoxic chemosensitivity (Garcia et al. 2000a, b; Katayama et al. 2001a, b, 2002), but also the blood pressure (Katayama et al. 2001b) and sympathetic responses to hypoxia (Greenberg et al. 1999). Evidence from rodent studies suggests that the pattern of the intermittent hypoxic stimulus may affect hypoxic chemosensitivity differently. Peng & Prabhakar (2004) exposed rats to either short-duration intermittent hypoxia (SDIH, 15 s of 5% O2 at 5 min intervals, 8 h day−1) or long-duration hypobaric intermittent hypoxia (LDIH, 4 h day−1, 0.4 atm (40.53 kPa)) for 10 days. Their results indicate that exposure to SDIH enhances hypoxic chemosensitivity, but exposure to LDIH does not. The results from the latter study may be confounded because the hypoxic duration and intensity were not the same for both groups of animals. Exposure to SDIH may also affect cerebral vascular responsiveness. In another rodent study, exposure to SDIH (1 min of 10% O2 at 4 min intervals, 12 h day−1) for 14 days severely blunted the vasodilator response to both hypoxia and acetylcholine in the isolated middle cerebral artery and gracilis muscle artery (Phillips et al. 2004). Altered peripheral vascular responsiveness has also been observed in sleep apnoea patients (Kato et al. 2000). Altered cerebral and peripheral vascular responsiveness may impair blood flow regulation, and could compromise tissue perfusion and O2 delivery during episodes of hypoxia.
In response to progressive isocapnic hypoxia, cerebral blood flow velocity (Vovk et al. 2002), heart rate (HR) and arterial blood pressure increase (Yasuma & Hayano, 2000). Few studies have examined the cardiovascular response to repeated bouts of intermittent hypoxia in humans. Katayama et al. (2001b) showed increased systolic and diastolic blood pressure responses to progressive isocapnic hypoxia following 7 days of hypobaric LDIH (1 h day−1, 4500 m). Similar results were found in men at high altitude (5050 m) for 24 days (Insalaco et al. 1996). The increases in the systolic and diastolic blood pressure responses were linearly related to the increase in hypoxic chemosensitivity (Katayama et al. 2001b).
Based on the above brief summary, the primary purpose of this study was to compare normobaric isocapnic exposure to SDIH and LDIH, and to follow changes in human ventilatory, cardiovascular and cerebral tissue oxygen responses to hypoxia over a 12 day period, and again over a 5 day period after the intermittent hypoxic intervention had ended. We hypothesized that intermittent hypoxia would augment the ventilatory and cardiovascular response to acute isocapnic progressive hypoxia and would attenuate the cerebral response. The hypercapnic ventilatory response was not expected to be affected by intermittent hypoxic exposure. It was further hypothesized that those subjects exposed to SDIH would have a greater increase in cardiovascular and ventilatory responses, and a greater depression of the cerebral response, when compared with those exposed to LDIH.
Methods
All experimental procedures and protocols were approved by the Clinical Screening Committee for Research of the University of British Columbia, and they conformed to the Declaration of Helsinki. All subjects provided written informed consent prior to participating in the investigation.
Subjects
Eighteen active, healthy male human volunteers were randomly assigned to one of two intermittent hypoxia groups (SDIH or LDIH). Subjects were similar for age (25.7 ± 4.3 years), height (176.1 ± 13.9 cm) and mass (81.4 ± 9.0 kg). All subjects performed three forced vital capacity (FVC) manoeuvres using a calibrated spirometer (Spirolab II; Medical International Research, Via del Maggiolino, Roma, Italy). FVC and forced expiratory volume in 1 s (FEV1.0) were assessed in accordance with standardized procedures (American Thoracic Society, 1995). All participants had a normal FVC (5.0 ± 0.7 l) and FEV1.0 (4.2 ± 0.5 l). Subjects had normal cardiopulmonary function, and were excluded from participation if they had been diagnosed with asthma, sleep apnoea, had a history of smoking or if they were hypertensive (systolic >140 mmHg; diastolic >90 mmHg). Subjects were life-long residents of sea-level, and had not sojourned to high altitude (>3000 m) in the year prior to testing. None of the subjects participated in breath-hold diving or trained/competed as endurance athletes, as this has been shown to affect ventilatory responses to hypoxia and hypercapnia (Byrne-Quinn et al. 1971; Ferretti, 2001).
Experimental protocol
The experimental protocol is displayed in Fig. 1. Subjects were exposed to a total of 10 episodes of 30 min isocapnic intermittent hypoxia (FIO2 12%) throughout a 12 day period. The SDIH group (n = 9) was exposed to a 5 min hypoxia/5 min normoxia cycle for 1 h, while the LDIH group (n = 9) was exposed to 30 min of sustained hypoxia. Following the 12 day intermittent hypoxia intervention, subjects returned 3 and 5 days later to determine the time course of recovery for the ventilatory, cardiovascular and cerebral responses to hypoxia and hypercapnia. Each experimental day began with a minimum of 10 min of rest to ensure stable resting/baseline measures. The hyperoxic hypercapnic ventilatory response (HCVR) was determined following rest (eupnoea) on days 1, 12, 15 and 17. The isocapnic hypoxic ventilatory response (HVR) was determined immediately before intermittent hypoxic exposure on days 1, 3, 5, 8, 10, 12, 15 and 17. The HCVR preceded the HVR test by a minimum of 5 min or until cardiovascular and ventilatory parameters had returned to eupnoeic levels.
Figure 1. Schematic diagram illustrating the experimental protocol.

Each day of measurement is indicated along the top. HCVR, hypercapnic ventilatory response; HVR, hypoxic ventilatory response; SDIH, short-duration intermittent hypoxia, i.e. 5 min 12% O2/5 min room air cycled six times; LDIH, long-duration intermittent hypoxia, i.e. 30 min 12% O2.
Measurements and procedures
All data were acquired using an analog-to-digital converter (Powerlab/16SP ML 795; ADInstruments, Colorado Springs, CO, USA) interfaced with a computer. HR variability data were sampled at 1000 Hz; during all other procedures data were sampled at 200 Hz and stored for subsequent analysis. Commercially available software was used to analyse ventilatory and near-infrared spectroscopy variables (Chart V5.02; ADInstruments) and cardiovascular variables (Beatscope V1.1; FMS, Arhem, The Netherlands).
HCVR
HCVR was assessed by a modified rebreathing technique (Read, 1967). Subjects were asked to maximally expire prior to rebreathing from a bag containing 6% CO2 and 94% O2. Rebreathing continued until the end-tidal partial pressure of CO2 (PET,CO2) reached 60–65 mmHg or for a maximal duration of 5 min. Gas was sampled at the mouth and analysed using an infrared CO2 analyser (CD-3 A; AEI, Pittsburgh, PA, USA). Inspired minute ventilation 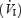 was plotted as a function of PET,CO2 and the linear regression relating these two variables was used to represent the HCVR (expressed as l min−1 mmHg−1).
was plotted as a function of PET,CO2 and the linear regression relating these two variables was used to represent the HCVR (expressed as l min−1 mmHg−1).
Isocapnic HVR
HVR was assessed using previously described methods (Weil et al. 1970; Guenette et al. 2004; Koehle et al. 2005). Subjects breathed room air from a mixing chamber and 100% N2 was gradually added to the inspiratory circuit to evoke a gradual drop in arterial oxyhaemoglobin saturation (SaO2) to 75% over an approximate 5 min period. SaO2 was measured at the finger using a pulse oximeter (3740; Ohmeda, Louisville, CO, USA). Isocapnia was maintained during the test by the manual addition of CO2 to the inspiratory circuit. Resting PET,CO2 was determined at the beginning of each day and maintained throughout experimentation. The FIO2 was determined by analysing gas sampled from the proximal side of the inspiratory valve (S-3 A; AEI). 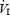 was plotted as a function of SaO2 and the slope of the linear regression was taken to represent the HVR (expressed as l min−1%SaO2−1). The relationship between
was plotted as a function of SaO2 and the slope of the linear regression was taken to represent the HVR (expressed as l min−1%SaO2−1). The relationship between 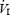 and SaO2 was considered acceptable when linearity (r = 0.7) was demonstrated.
and SaO2 was considered acceptable when linearity (r = 0.7) was demonstrated.
Cardiovascular parameters
Beat-by-beat HR, systolic (SBP) and diastolic (DBP) blood pressures were obtained throughout all procedures using finger pulse photoplethysmography (Finometer; FMS). Mean arterial pressure (MAP) was calculated directly from SBP and DBP. The photoplethysmograph was placed on the mid-phalanx of the middle digit of the left hand. Beat-by-beat blood pressure was calibrated against an automated blood pressure measurement (BPM-100; VSM Medtech Ltd, Vancouver, Canada) taken from the right arm at the level of the heart every 3 min. Cardiovascular sensitivity to hypoxia was determined as per previously published studies and was expressed as ΔSBP/ΔSaO2, ΔDBP/ΔSaO2 and ΔHR/ΔSaO2 (Insalaco et al. 1996; Katayama et al. 2001b). Similar analyses during hypercapnia were not undertaken because of the inability to demonstrate linearity (r < 0.7) of the cardiovascular variables and PET,CO2. All cardiovascular parameters were averaged over 10 s epochs. The relationship between each cardiovascular parameter (SBP, DBP and HR) and SaO2 was considered acceptable when linearity (r = 0.7) was demonstrated.
Near-infrared spectroscopy (NIRS)
Cerebral tissue oxygen saturation (SaO2), and changes in oxyhaemoglobin concentration (O2Hb), deoxyhaemoglobin concentration (HHb) and total haemoglobin concentration (cHb) were determined using NIRS at a sampling rate of 2 Hz (Hamamatsu Niro 300; Hamamatsu Phototonics, K.K., Japan). Optodes were placed in a black plastic holder and applied to the head with a bandage to shield the light and maintain optode separation. The path-length value was determined as the product of the source-detector probe spacing (in cm) multiplied by the differential path-length factor (DPF) for the brain (Madsen & Secher, 1999). A DPF of 5.92 was used for the brain as determined by Van Der Zee et al. 1992) where a 4 or 5 cm probe spacing would have a path length of 23.7 or 29.6 cm, respectively. The cerebral tissue response to hypoxia was expressed as ΔScO2/ΔSaO2, ΔO2Hb/ΔSaO2, ΔHHb/ΔSaO2 and ΔcHb/ΔSaO2.
HR variability
HR variability was measured to determine if resting shifts in autonomic function occurred. Subjects were monitored via an ECG (ML 132; ADInstruments), configured in the standard bipolar limb lead I. Analysis occurred off-line as previously described (Task Force, 1996). Measured R-R intervals were determined from the electrocardiogram, and the resulting tachogram was fast-Fourier transformed. The high-frequency, low-frequency and very low-frequency bands were defined as 0.15–0.4, 0.04–0.15 and <0.04 Hz, respectively.
Statistical analysis
All data are expressed as means ±s.d. unless otherwise indicated. Statistical software (Statistica v.6.1, Statsoft, Inc., Tulsa, OK, USA) was applied to detect differences between groups (SDIH and LDIH) and between days (8 days) using a repeated measures analysis of variance procedure. When significant F ratios were detected, Tukey's post hoc analysis was applied to determine where the differences lay. Pearson product moment correlations were implemented to determine relationships between selected dependent variables. The level of significance was set at P < 0.05 for all statistical comparisons.
Results
Basal ventilatory, cardiovascular and cerebral tissue oxygenation variables
All subjects completed 10 intermittent hypoxic exposures over the 12 day period. One subject did not complete the final day 17 ventilatory response testing. Mean ventilatory, cardiovascular, and cerebral tissue O2 variables measured during eupnoea are displayed in Table 1. The mean coefficient of variation for each variable across 8 days of resting measures is also displayed in Table 1. There were no differences between SDIH and LDIH groups for any resting ventilatory or cardiovascular variable. Slight increases in resting breathing frequency (+2–3 breaths min−1) were detected on days 8 and 10 (P < 0.05), but Vt and 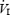 were not different (P > 0.05). Furthermore, PET,CO2 was not different on any day of measurement. Basal blood pressure and HR did not differ throughout experimentation. In addition, HR variability did not change over the course of the intermittent hypoxic exposures (data not shown). Mean ScO2 during eupnoea was ∼70%, and was consistent for all subjects across all days.
were not different (P > 0.05). Furthermore, PET,CO2 was not different on any day of measurement. Basal blood pressure and HR did not differ throughout experimentation. In addition, HR variability did not change over the course of the intermittent hypoxic exposures (data not shown). Mean ScO2 during eupnoea was ∼70%, and was consistent for all subjects across all days.
Table 1.
Basal ventilatory, cardiovascular, cerebral tissue oxygen and heart rate variability variables during eupnoea
| Day 1 | Day 3 | Day 5 | Day 8 | Day 10 | Day 12 | Day 15 | Day 17 | CV (%) | |
|---|---|---|---|---|---|---|---|---|---|
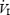 (l min−1) (l min−1) |
11.1 ± 2.4 | 11.8 ± 2.1 | 11.9 ± 2.3 | 12.4 ± 2.4 | 12.1 ± 2.6 | 11.7 ± 2.2 | 11.7 ± 1.9 | 11.8 ± 1.9 | 9.7 ± 0.5 |
| Fb (breaths min−1) | 16.8 ± 3.8 | 18.1 ± 3.6 | 17.8 ± 4.0 | 18.4 ± 3.2* | 18.7 ± 3.4* | 17.7 ± 2.7 | 17.6 ± 3.1 | 17.9 ± 3.0 | 8.6 ± 4.5 |
| Vt (l) | 0.69 ± 0.12 | 0.67 ± 0.11 | 0.69 ± 0.10 | 0.69 ± 0.11 | 0.67 ± 0.13 | 0.68 ± 0.12 | 0.69 ± 0.07 | 0.68 ± 0.10 | 10.6 ± 0.7 |
| PET,CO2 (mmHg) | 43.6 ± 3.8 | 43.2 ± 3.0 | 43.6 ± 3.6 | 43.0 ± 3.3 | 42.4 ± 2.6 | 43.6 ± 3.3 | 43.6 ± 3.1 | 43.1 ± 3.0 | 3.2 ± 1.2 |
| SBP (mmHg) | 124 ± 9 | 120 ± 11 | 121 ± 9 | 121 ± 10 | 118 ± 9 | 122 ± 7 | 123 ± 11 | 121 ± 12 | 5.4 ± 2.0 |
| DBP (mmHg) | 71 ± 7 | 70 ± 7 | 71 ± 6 | 71 ± 8 | 70 ± 8 | 70 ± 6 | 70 ± 9 | 70 ± 9 | 6.5 ± 1.9 |
| MAP (mmHg) | 89 ± 7 | 87 ± 8 | 87 ± 6 | 88 ± 9 | 86 ± 8 | 88 ± 6 | 88 ± 9 | 87 ± 10 | 5.7 ± 1.8 |
| HR (beats min−1) | 66 ± 11 | 66 ± 12 | 68 ± 13 | 67 ± 13 | 71 ± 11 | 70 ± 12 | 68 ± 13 | 68 ± 11 | 8.9 ± 3.2 |
| ScO2(%) | 70 ± 5 | 68 ± 7 | 69 ± 4 | 69 ± 5 | 69 ± 6 | 70 ± 6 | 70 ± 5 | 69 ± 4 | 4.7 ± 2.1 |
Values are means ±s.d.
Significantly different from day 1 (P < 0.05). 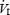 , inspired minute ventilation; Fb, breathing frequency; Vt, tidal volume; PET,CO2 end-tidal partial pressure of CO2; SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure; ScO2, cerebral tissue O2 saturation; CV, coefficient of variation.
, inspired minute ventilation; Fb, breathing frequency; Vt, tidal volume; PET,CO2 end-tidal partial pressure of CO2; SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure; ScO2, cerebral tissue O2 saturation; CV, coefficient of variation.
Effects of intermittent hypoxia on the HCVR
The HCVR was not different between SDIH and LDIH, and did not change systematically throughout exposure to either protocol of intermittent hypoxia. The mean ±s.d. values for all subjects on days 1, 12, 15 and 17 were 2.49 ± 1.49, 1.74 ± 2.37, 2.35 ± 1.88 and 2.12 ± 1.15 l min−1 mmHg−1, respectively.
Effects of intermittent hypoxia on the HVR
Displayed in Fig. 2 is an example of the HVR for one subject on day 1 prior to intermittent hypoxic exposure, and again on day 12 on the last day of the intermittent hypoxic intervention. Following 12 days of intermittent hypoxia, the HVR increased by nearly 78% for this particular subject. Group mean HVR data for SDIH and LDIH are displayed in Fig. 3. There were significant increases in HVR after 12 days of intermittent hypoxic exposure regardless of the type of intermittent hypoxic exposure (P < 0.01). This increase in HVR was at a maximum by day 12 and subsequently returned to baseline by 5 days after the intermittent hypoxic intervention had ended. The increase in HVR at day 12 was primarily mediated by a greater Vt response to hypoxia (day 1, ΔVt= 0.74 ± 0.38 l; day 12, ΔVt= 0.92 ± 0.41 l; P < 0.05). Not all subjects displayed a peak in HVR on day 12. Several subjects peaked before day 12 (n = 4), and others peaked on days 15 and 17 (n = 4). PET,CO2 was maintained within 1–2 mmHg for all HVR trials and hypoxic exposures.
Figure 2. The relationship between ventilation and arterial oxyhemoglobin saturation.

The relationship between ventilation and arterial oxyhemoglobin saturation (i.e. the HVR) for one individual subject on (A) day 1 and (B) on day 12. 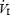 , inspired minute ventilation; SaO2, arterial oxyhaemoglobin saturation.
, inspired minute ventilation; SaO2, arterial oxyhaemoglobin saturation.
Figure 3. The HVR measured before the daily exposure to intermittent hypoxia for subjects in SDIH and LDIH.

•, SDIH;Ο, LDIH. The box indicates the period of the intermittent hypoxic intervention. Data are means ± s.e.m.*Significantly different from days 1 and 3 (P < 0.01).
Effects of intermittent hypoxia on cardiovascular sensitivity to hypoxia
Cardiovascular sensitivity to hypoxia was assessed during the HVR and is displayed in Table 2 as the ΔSBP/ΔSaO2, the ΔDBP/ΔSaO2 and the ΔHR/ΔSaO2 for all trials. SDP, DBP and HR were linearly (r = 0.7) related to the change in SaO2. There was no difference in cardiovascular sensitivity to hypoxia between SDIH and LDIH subjects. The ΔSBP/ΔSaO2 and the ΔDBP/ΔSaO2 seemed to increase after 12 days of intermittent hypoxic exposure, and returned to baseline by 3–5 days after the end of the intermittent hypoxic intervention; however, this increase was not statistically significant because of large interindividual variability. The ΔHR/ΔSaO2 did not change throughout LDIH or SDIH. The change in ΔSBP/ΔSaO2 (δΔSBP/ΔSaO2; r =+0.68) and the ΔDBP/ΔSaO2 (δΔSBP/ΔSaO2; r =+0.73) between days 1 and 12 was significantly correlated (P < 0.05) to the change in HVR (δHVR), while the change in ΔHR/ΔSaO2 (δΔHR/ΔSaO2; r =+0.32; P > 0.05) between days 1 and 12 was not significantly correlated to the change in HVR (δHVR).
Table 2.
Effects of short and long duration intermittent hypoxia on systolic (ΔSBP/ΔSaO2) and diastolic (ΔDBP/ΔSaO2) blood pressure sensitivity to hypoxia, and heart rate (ΔHR/ΔSaO2) sensitivity to hypoxia
| Day 1 | Day 3 | Day 5 | Day 8 | Day 10 | Day 12 | Day 15 | Day 17 | ||
|---|---|---|---|---|---|---|---|---|---|
| ΔSBP/ΔSaO2 | SDIH | 0.78 ± 0.57 | 0.86 ± 0.87 | 0.94 ± 0.73 | 1.14 ± 0.70 | 0.99 ± 0.50 | 1.20 ± 1.22 | 0.98 ± 0.99 | 1.01 ± 0.67 |
| (mmHg %SaO2−1) | LDIH | 0.71 ± 0.30 | 0.68 ± 0.52 | 0.58 ± 0.51 | 0.70 ± 0.31 | 0.68 ± 0.34 | 0.88 ± 0.69 | 0.50 ± 0.74 | 0.68 ± 0.56 |
| ΔDBP/ΔSaO2 | SDIH | 0.37 ± 0.22 | 0.42 ± 0.50 | 0.48 ± 0.45 | 0.58 ± 0.42 | 0.45 ± 0.35 | 0.56 ± 0.65 | 0.45 ± 0.42 | 0.45 ± 0.34 |
| (mmHg %SaO2−1) | LDIH | 0.28 ± 0.17 | 0.28 ± 0.22 | 0.21 ± 0.25 | 0.32 ± 0.24 | 0.26 ± 0.18 | 0.40 ± 0.30 | 0.20 ± 0.35 | 0.29 ± 0.22 |
| ΔHR/ΔSaO2 | SDIH | 1.16 ± 0.43 | 1.08 ± 0.60 | 1.17 ± 0.49 | 1.25 ± 0.57 | 1.26 ± 0.54 | 1.19 ± 0.69 | 1.29 ± 0.83 | 1.19 ± 0.68 |
| (beats min−1%SaO2−1) | LDIH | 1.05 ± 0.29 | 0.96 ± 0.29 | 1.12 ± 0.43 | 0.94 ± 0.47 | 0.89 ± 0.29 | 1.06 ± 0.38 | 1.03 ± 0.47 | 0.90 ± 0.37 |
Values are means ±s.d. SDIH, short-duration intermittent hypoxia, LDIH, long-duration intermittent hypoxia.
Effects of intermittent hypoxia on the cerebral tissue O2 response to hypoxia
As SaO2 decreased during the HVR so did ScO2 and O2Hb, while both HHb and cHb increased. The relationships between these variables and SaO2 were linear (r≥ 0.9). There was no difference between the LDIH and SDIH groups for the following variables: ΔScO2/ΔSaO2, ΔO2Hb/ΔSaO2, ΔHHb/ΔSaO2 and ΔcHb/ΔSaO2. Figure 4A displays the mean ΔScO2/ΔSaO2 during the HVR for SDIH and LDIH subjects throughout the 12 day intermittent hypoxic intervention and following the end of intermittent hypoxia. The ΔScO2/ΔSaO2 that occurred during the HVR was significantly less on day 12 and subsequently returned to baseline by day 17 (P < 0.001). This response was driven by a much greater reduction in the ΔScO2/ΔSaO2 of the SDIH group. The change in ΔScO2/ΔSaO2 corresponds to a reduction in ScO2 (at equal levels of SaO2, i.e. iso-SaO2= 83 ± 3%) of −8.4 ± 2.8% on day 1, −10.0 ± 2.7% on day 12, and −8.3 ± 2.3% on day 17. Displayed in Fig. 4B and C is the absolute change in ScO2 to iso-SaO2 for all subjects in the SDIH and LDIH group on days 1 and 12. A greater reduction in ScO2 on day 12 was consistent for eight of nine subjects in SDIH, and five of nine subjects in LDIH.
Figure 4. Effects of intermittent hypoxia on cerebral tissue oxygenation.

A, the effects of intermittent hypoxia on the ratio of the fall in cerebral tissue oxygen saturation to the fall in arterial oxyhemoglobin saturation (ΔScO2/ΔSaO2) during progressive hypoxia for both SDIH and LDIH subjects (cerebral tissue oxygen saturation sensitivity). The box indicates the period of the intermittent hypoxia intervention. Data points are means ± s.e.m.*Significantly different from day 1 (P > 0.001). B, the absolute change in ScO2 on days 1 and 12 at iso-SaO2 (83 ± 3%) for each subject in SDIH. All but one subject showed a greater reduction in ScO2 at day 12. C, the absolute change in ScO2 on days 1 and 12 at iso-SaO2 (83 ± 3%) for each subject in LDIH. Five of nine subjects show a greater reduction in ScO2 at day 12. The large open circle data points represent the group mean (for both B and C).
The ΔO2Hb/ΔSaO2 and the ΔcHb/ΔSaO2 during the HVR were not different between SDIH and LDIH, and did not change systematically throughout the intermittent hypoxic intervention. However, the ΔHHb/ΔSaO2 during the HVR became progressively greater throughout the intermittent hypoxic intervention and was significantly different (P < 0.05) on day 12 (0.44 ± 0.12 μm%SaO2−1) compared with days 1 (0.34 ± 0.21 μm%SaO2−1) and 3 (0.34 ± 0.09 μm%SaO2−1). This change in deoxyhaemoglobin sensitivity had returned to baseline levels 5 days following the end of the intermittent hypoxic intervention (day 17, 0.38 ± 0.10 μm%SaO2−1).
Ventilatory and cardiovascular function during daily intermittent hypoxic exposures
During each daily intermittent hypoxic exposure, ventilation, blood pressure, HR, ScO2 and SaO2 were averaged over a 3 min period at the end of the first 5 min of hypoxia (Ho) and at the end of the last 5 min of hypoxia (Hi). Ventilation was not different between SDIH and LDIH during Ho or Hi on any of the 12 day intermittent hypoxic interventions. In addition, 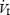 was not significantly different on day 12 when compared with day 1. MAP was not different between SDIH and LDIH at Ho, but during Hi, MAP was significantly greater than Ho for SDIH (+5 mmHg; P < 0.01) and not for LDIH (+2 mmHg; P > 0.05). The increase in MAP with SDIH did not become greater throughout the 12 day intermittent hypoxic intervention. Since the change in MAP did not change systematically throughout the 12 day intervention, the data for each day were combined and are displayed in Fig. 5. Although HR was not different between groups at Ho or Hi, it did increase slightly, but significantly, throughout exposure (+1 beat min−1; P < 0.01).
was not significantly different on day 12 when compared with day 1. MAP was not different between SDIH and LDIH at Ho, but during Hi, MAP was significantly greater than Ho for SDIH (+5 mmHg; P < 0.01) and not for LDIH (+2 mmHg; P > 0.05). The increase in MAP with SDIH did not become greater throughout the 12 day intermittent hypoxic intervention. Since the change in MAP did not change systematically throughout the 12 day intervention, the data for each day were combined and are displayed in Fig. 5. Although HR was not different between groups at Ho or Hi, it did increase slightly, but significantly, throughout exposure (+1 beat min−1; P < 0.01).
Figure 5. Group mean MAP values.

The group mean MAP averaged over the first 5 min of hypoxia (Ho) and the last 5 min of hypoxia (Hi) during the daily intermittent hypoxic exposure. *SDIH is significantly different from the first 5 min of hypoxia (P < 0.05).
Discussion
To our knowledge, this is the first study to compare ventilatory, cardiovascular, and cerebral tissue O2 responses following exposure to SDIH and LDIH in humans. We observed that during progressive isocapnic hypoxia, the reduction in ScO2 for any given SaO2 was greater following 12 days of exposure to SDIH and LDIH. We attribute the reduction in cerebral tissue oxygenation to cerebral vascular dysregulation following exposure to intermittent hypoxia. Exposure to two protocols of isocapnic intermittent hypoxia reversibly enhanced the hypoxic ventilatory response, and had no effect on the hypercapnic ventilatory response. We also found that the SBP and DBP responses to hypoxia were linearly related to the change in HVR, while the HR response to hypoxia was not.
Effects of intermittent hypoxia on cerebral tissue O2
During acute exposure to progressive hypoxia, ScO2 decreases. This occurs due to an increase in HHb and cHB, while O2Hb decreases (Madsen & Secher, 1999). The change in each variable is linearly related to the change in SaO2. In the cerebral circulation, vascular autoregulation is responsible for the vasodilator response to hypoxia, which aids in increasing cerebral blood flow and subsequently cerebral tissue oxygenation. Several lines of evidence suggest that exposure to intermittent hypoxia affects vascular function negatively (Earley & Walker, 2002; Gonzales & Walker, 2002; Jernigan & Resta, 2002; Phillips et al. 2004); for example, exposure to chronic intermittent hypoxia markedly attenuates the acute vasodilator responses to hypoxia in isolated vessels (Phillips et al. 2004). Phillips et al. (2004) exposed rats to chronic intermittent hypoxia (FIO2= 10% for 1 min at 4 min intervals 12 h day−1) for 14 days. Following 14 days of intermittent hypoxia, the middle cerebral arteries were isolated and placed in a tissue bath, pressurized to 90 mmHg, and vessel diameters were measured before and after exposure to acetylcholine and acute reductions of PO2. Dilation of the middle cerebral artery induced by acetylcholine was greatly attenuated while dilation induced by acute reductions in PO2 was virtually abolished in animals exposed to chronic intermittent hypoxia. This suggests that vascular regulation is altered following intermittent hypoxia and may affect the ability to regulate tissue perfusion and oxygenate cerebral tissue during episodes of hypoxia. In the current human study, the ability to oxygenate the cerebral tissue was determined during acute exposure to progressive hypoxia using NIRS. The ΔScO2/ΔSaO2 became significantly less following exposure to both SDIH and LDIH (day 1, −0.51 ± 0.13; day 12, −0.64 ± 0.18). This change in ScO2 sensitivity was mediated by an increase in the ΔHHb/ΔSaO2 (day 1, 0.34 ± 0.21; day 12, 0.44 ± 0.12; P < 0.001). These changes in the ability to oxygenate the brain were reversible and, following the end of exposure to intermittent hypoxia, had returned to baseline (see Fig. 4A). There were no differences in the ΔO2Hb/ΔSaO2 or the ΔcHb/ΔSaO2. In addition, the change in ScO2 sensitivity seen throughout intermittent hypoxia cannot be explained by shifts in resting ScO2, as it did not change and the coefficient of variation (CV) was relatively low (4.7%). Taken together, the results indicate that there was a greater reduction in ScO2 at equal levels of arterial oxyhaemoglobin during progressive hypoxia following 12 days of both SDIH and LDIH. This finding was a consistent observation where eight of nine subjects in the SDIH group, and five of nine subjects in the LDIH group demonstrated a greater reduction in cerebral tissue O2 saturation (Fig. 4B and C). To more fully characterize the cerebral response to hypoxia we determined the change in ScO2 for each subject at a SaO2 value that was the same on days 1 and 12 (iso-SaO2). On day 1 the subjects demonstrated a −8.3% change in ScO2, and on day 12 the change was −10.3% when SaO2 was progressively reduced to 83 ± 3% (P < 0.05). We do not know the specific mechanism leading to the altered oxygenation of cerebral tissue. However, the results seem to suggest cerebral vascular dysregulation in the human subject following exposure to SDIH and LDIH, and support the findings seen in the rat (Phillips et al. 2004). In response to hypoxia, the small vessel dilation and vascular smooth muscle membrane hyperpolarization that occurs in these vessels appears to be primarily due to the release of nitric oxide from the vascular endothelium which acts to open vascular smooth muscle membrane Ca2+-activated K+ channels (Frisbee et al. 2002). Previous reports suggest that exposure to intermittent hypoxia associated with recurrent apnoeas in sleep apnoea patients elevates the generation of reactive oxygen species which depletes vascular nitric oxide formation (Schulz et al. 2000; Dyugovskaya et al. 2002; Steiner et al. 2002). We cautiously suggest that exposure to SDIH and LDIH may generate reactive oxygen species, deplete vascular nitric oxide formation, and blunt the vasodilatory response to hypoxia in the cerebral circulation. Blunted vasodilatory responses in the cerebral circulation could impair blood flow regulation, tissue perfusion, and cerebral tissue oxygenation.
Ventilatory effects of intermittent hypoxia
The isocapnic hypoxic ventilatory response was significantly increased following 10 daily episodes of intermittent hypoxia spaced over a 12 day period. This increase in HVR occurred regardless of the intermittent hypoxic protocol employed in the current study (i.e. SDIH or LDIH). Following the end of the intermittent hypoxic protocol, the HVR returned to baseline within 5 days. The change in HVR we observed was primarily attributable to an augmentation of the tidal volume response to hypoxia. The tidal volume response to hypoxia increased by ∼5% at iso-SaO2 levels. Other studies involving human subjects have reported similar results (Garcia et al. 2000a, b; Katayama et al. 2001a, b). A 62% increase in HVR was seen in subjects following 7 days of 1 h daily exposure to hypobaric hypoxia (432 mmHg) (Katayama et al. 2001b). In the present study a 37% increase in HVR was seen in subjects exposed to 10 30 min episodes of a similar level of isocapnic hypoxia spaced over 12 days. Several human studies have used SDIH methods similar to ours and have reported increases in HVR (Serebrovskaya et al. 1999; Bernardi et al. 2001). In contrast to the results of our current study, Peng & Prabhakar (2004) showed, in rodents, that exposure to LDIH does not enhance carotid body hypoxic chemosensitivity, while SDIH does. It may be that a species difference is the cause of the discrepancy between studies. We have shown that human subjects exposed to both 30 min of sustained hypoxia or to six 5 min bouts of hypoxia per day for 12 days, sustained significant increases in HVR. While the hypoxic intensity used in our human study was much less than that used in the rodent study, it is reasonable to expect that a larger hypoxic stimulus may elicit even greater increases in HVR.
In four subjects we observed further increases in the HVR following the end of intermittent hypoxia on days 15 and 17. A possible explanation for this may be related to changes in protein synthesis and gene expression that require a longer time domain before effects are manifested. For example, during intermittent hypoxia, episodic spinal serotonin receptor activation may initiate a cell-signalling cascade that increases spinal protein synthesis and leads to enhanced glutamatergic synaptic currents in phrenic motoneurones (Mitchell et al. 2001). The above process has been shown to play a role in long-term facilitation that occurs immediately following a single session of intermittent hypoxia and may be unrelated to the increase in HVR that we observed. However, a similar process may underlie the continued increase in HVR even after the termination of intermittent hypoxia.
The HCVR was unaltered by exposure to both SDIH and LDIH. This finding is similar to other studies which have measured the HCVR using the rebreathing method and the single breath CO2 response test (Katayama et al. 1998, 1999, 2002).
Cardiovascular effects of intermittent hypoxia
There were no changes in any resting cardiovascular variable throughout the 12 days of intermittent hypoxia or the 5 days following the end of intermittent hypoxia. In addition, we did not detect any changes in the high- and low-frequency spectral components of HR variability, indicating that baseline autonomic control of the heart did not change throughout the experimental period and that all subjects were in a comparable autonomic state prior to each hypoxic trial. Very few studies have studied the cardiovascular effects of intermittent hypoxia. Katayama et al. (2001b) found a 48% increase in the ΔSBP/ΔSaO2 and a 127% increase in the ΔDBP/ΔSaO2. They also found significant relationships between the change in SBP sensitivity (δΔSBP/ΔSaO2; r =+0.66) and the change in DBP sensitivity (δΔDBP/ΔSaO2; r =+0.62) with the change in HVR (δHVR). The results from the present study are comparable. We observed a nonsignificant increase in ΔSBP/ΔSaO2 (42%) and ΔDBP/ΔSaO2 (50%). We also showed significant relationships between the δΔSBP/ΔSaO2 (r =+0.68; P < 0.05) and the δΔDBP/ΔSaO2 (r =+0.73; P < 0.05) with δHVR. Also, in agreement with Katayama et al. (2001b), we found no change in the ΔHR/ΔSaO2. The results from Insalaco et al. (1996) also demonstrate comparable findings in humans at 5050 m for 24 days. Our results coupled with those of others suggest that the blood pressure response to hypoxic stimuli increases during exposure to short-term intermittent hypoxia. More research is required to understand the reasons for the absence of change in the HR response to hypoxia. As pointed out by Katayama et al. (2001a), it is likely to be a complex interaction between lung inflation receptors, baroreceptors and chemoreceptors.
A significant increase in MAP (+∼5 mmHg; P < 0.05) was found throughout the daily exposure to SDIH but not LDIH (Fig. 5). The magnitude of the change in MAP did not increase over the 12 days of SDIH. HR was similar between both groups throughout exposure to either LDIH or SDIH, and only small increases in HR occurred during the daily exposure (+∼1 beat min−1; P < 0.01). This has not been previously documented. It is not understood why SDIH would result in a rise in MAP while LDIH would not. It is possible that repeated states of deoxygenation and reoxygenation are a more important stimulus than sustained hypoxaemia for increasing blood pressure. Furthermore, this phenomenon may relate to the secondary hypertension that is present in patients with obstructive sleep apnoea (Morgan & Joyner, 2002). Although under different experimental conditions, Fletcher et al. (1992) demonstrated, using rodents, that increases in resting, normoxic daytime blood pressure following exposure to 35 days of SDIH were similar to those associated with sleep apnoea. This increase in resting daytime blood pressure was dependent upon intact carotid chemoreceptors. Sympathetic responsiveness to hypoxia and hypercapnia has also been shown to increase following 30 days of SDIH (Fletcher et al. 1992; Greenberg et al. 1999). Or perhaps more simply the rise in MAP can be explained by the absence of a hypoxia mediated local vasodilatory response (Doherty & Liang, 1984; Blauw et al. 1995). In the LDIH-conditioned individuals systemic hypoxia probably mediated local hypoxic vasodilatation, thus preventing the rise in blood pressure seen in the SDIH-conditioned subjects.
Critique of methods
Measurement of HVR can vary greatly from day-to-day (Sahn et al. 1977; Beidleman et al. 1999). The CV for the HVR has been reported to range from 26 to 76% (Zhang & Robbins, 2000; Fahlman et al. 2002). Using identical methods described in this study, repeated HVR measurements were performed in our laboratory over five consecutive days, and the mean individual CV was 27 ± 4% (Koehle et al. 2005). This is identical to the findings of Zhang & Robbins (2000) who found a CV of 26%.
SaO2 was measured by pulse oximetry and not determined from arterial blood gases. Changes in arterial pH and body temperature affect the haemoglobin–oxygen dissociation curve. Pulse oximetry fails to account for this. During this study, however, subjects were at rest and as a result, changes in temperature were unlikely during the experiment, and changes in pH were minimized by maintaining isocapnia.
NIRS provides a unique and detailed measurement of cerebral oxygenation. It can be argued that NIRS determines local venous O2 saturation rather than tissue O2 saturation. This is because nearly 70% of the blood contained within the cerebral circulation is within the venules (Madsen & Secher, 1999). However, ScO2 measured by NIRS is still greater than venous oxygen saturation measured directly from the internal jugular vein (Madsen & Secher, 1999). This suggests that the blood contained in the capillaries and arteries, although small, is still a significant part of the measurement of ScO2. In addition, O2Hb measured by NIRS correlates well with changes in cerebral blood flow velocity measured using transcranial Doppler (r =+0.61) (Smielewski et al. 1995). In this latter study, jugular occlusion was not used, and the vascular reactivity was assessed using the cerebral blood flow velocity response to hypercapnia.
Summary
In summary, this is the first study to demonstrate that the vascular processes required to control blood flow and O2 supply to cerebral tissue in a healthy human are hindered following exposure to 12 days of isocapnic intermittent hypoxia. This is in line with evidence from animal studies that suggest cerebral artery vascular dysregulation occurs following intermittent hypoxia. The results from this study indicate that exposure to 12 days of isocapnic intermittent hypoxia will transiently increase human hypoxic chemosensitivity. The increase in hypoxic chemosensitivity is short-lived as the HVR returns to baseline within 3–5 days after intermittent hypoxia has ended. Exposure to isocapnic intermittent hypoxia does not alter hypercapnic chemosensitivity, probably because respiratory alkalosis is necessary to evoke changes in hypercapnic chemosensitivity. We hypothesized that increases in the blood pressure response to hypoxia would be greater with the increased hypoxic chemosensitivity and as in other studies (Insalaco et al. 1996; Katayama et al. 2001b). Our hypothesis was supported and there is evidence to suggest that the increased blood pressure response probably occurred through a carotid-body-associated increase in muscle sympathetic nerve activity. The HR response was unaltered by intermittent hypoxia. This is the first study to demonstrate a rise in MAP that occurs throughout the daily exposure to SDIH. This rise in MAP did not occur during exposure to LDIH.
Acknowledgments
We thank Mr George Foster for his technical assistance, and our subjects for their enthusiastic participation. This study was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Foundation for Innovation. A. W. S. was supported by a scholar award from the Michael Smith Foundation for Health Research (British Columbia, Canada) and a New Investigator award from the Canadian Institutes of Health Research.
References
- Ainslie PN, Poulin MJ, American Thoracic Society Ventilatory, cerebrovascular, and cardiovascular interactions in acute hypoxia: regulation by carbon dioxide. J Appl Physiol. 2004;97:149–159. doi: 10.1152/japplphysiol.01385.2003. [DOI] [PubMed] [Google Scholar]
- American Thoracic Society. Standardization of spirometry, 1994 update. Am J Respir Crit Care Med. 1995;152:1107–1136. doi: 10.1164/ajrccm.152.3.7663792. [DOI] [PubMed] [Google Scholar]
- Beidleman BA, Rock PB, Muza SR, Fulco CS, Forte VA, Jr, Cymerman A. Exercise VE and physical performance at altitude are not affected by menstrual cycle phase. J Appl Physiol. 1999;86:1519–1526. doi: 10.1152/jappl.1999.86.5.1519. [DOI] [PubMed] [Google Scholar]
- Bernardi L, Passino C, Serebrovskaya Z, Serebrovskaya TV, Appenzeller O. Respiratory and cardiovascular adaptations to progressive hypoxia. Eur Heart J. 2001;22:879–886. doi: 10.1053/euhj.2000.2466. [DOI] [PubMed] [Google Scholar]
- Blauw GJ, Westendorp RG, Simons M, Chang PC, Frolich M, Meinders AE. β-Adrenergic receptors contribute to hypoxaemia induced vasodilation in man. Br J Clin Pharmacol. 1995;40:453–458. [PMC free article] [PubMed] [Google Scholar]
- Byrne-Quinn E, Weil JV, Sodal IE, Filley GF, Grover RF. Ventilatory control in the athlete. J Appl Physiol. 1971;30:91–98. doi: 10.1152/jappl.1971.30.1.91. [DOI] [PubMed] [Google Scholar]
- Doherty JU, Liang CS. Arterial hypoxemia in awake dogs. Role of the sympathetic nervous system in mediating the systemic hemodynamic and regional blood flow responses. J Lab Clin Med. 1984;104:665–677. [PubMed] [Google Scholar]
- Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med. 2002;165:934–939. doi: 10.1164/ajrccm.165.7.2104126. [DOI] [PubMed] [Google Scholar]
- Earley S, Walker BR. Endothelium-dependent blunting of myogenic responsiveness after chronic hypoxia. Am J Physiol Heart Circ Physiol. 2002;283:H2202–2209. doi: 10.1152/ajpheart.00125.2002. [DOI] [PubMed] [Google Scholar]
- Fahlman A, Jackson S, Terblanche J, Fisher JA, Vesely A, Sasano H, Myburgh KH. A simple breathing circuit to maintain isocapnia during measurements of the hypoxic ventilatory response. Respir Physiol Neurobiol. 2002;133:259–270. doi: 10.1016/s1569-9048(02)00166-0. [DOI] [PubMed] [Google Scholar]
- Ferretti G. Extreme human breath-hold diving. Eur J Appl Physiol. 2001;84:254–271. doi: 10.1007/s004210000377. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Behm R, Miller CC, III, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol. 1992;72:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- Frisbee JC, Maier KG, Falck JR, Roman RJ, Lombard JH. Integration of hypoxic dilation signaling pathways for skeletal muscle resistance arteries. Am J Physiol Regul Integr Comp Physiol. 2002;283:R309–319. doi: 10.1152/ajpregu.00741.2001. [DOI] [PubMed] [Google Scholar]
- Garcia N, Hopkins SR, Powell FL. Intermittent vs continuous hypoxia: effects on ventilation and erythropoiesis in humans. Wilderness Environ Med. 2000a;11:172–179. doi: 10.1580/1080-6032(2000)011[0172:ivcheo]2.3.co;2. [DOI] [PubMed] [Google Scholar]
- Garcia N, Hopkins SR, Powell FL. Effects of intermittent hypoxia on the isocapnic hypoxic ventilatory response and erythropoiesis in humans. Respir Physiol. 2000b;123:39–49. doi: 10.1016/s0034-5687(00)00145-6. [DOI] [PubMed] [Google Scholar]
- Gonzales RJ, Walker BR. Role of CO in attenuated vasoconstrictor reactivity of mesenteric resistance arteries after chronic hypoxia. Am J Physiol Heart Circ Physiol. 2002;282:H30–37. doi: 10.1152/ajpheart.2002.282.1.H30. [DOI] [PubMed] [Google Scholar]
- Greenberg HE, Sica A, Batson D, Scharf SM. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol. 1999;86:298–305. doi: 10.1152/jappl.1999.86.1.298. [DOI] [PubMed] [Google Scholar]
- Guenette JA, Diep TT, Koehle MS, Foster GE, Richards JC, Sheel AW. Acute hypoxic ventilatory response and exercise-induced arterial hypoxemia in men and women. Respir Physiol Neurobiol. 2004;143:37–48. doi: 10.1016/j.resp.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Insalaco G, Romano S, Salvaggio A, Braghiroli A, Lanfranchi P, Patruno V, Donner CF, Bonsignore G. Cardiovascular and ventilatory response to isocapnic hypoxia at sea level and at 5050 m. J Appl Physiol. 1996;80:1724–1730. doi: 10.1152/jappl.1996.80.5.1724. [DOI] [PubMed] [Google Scholar]
- Jernigan NL, Resta TC. Chronic hypoxia attenuates cGMP-dependent pulmonary vasodilation. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1366–1375. doi: 10.1152/ajplung.00273.2001. [DOI] [PubMed] [Google Scholar]
- Katayama K, Sato Y, Ishida K, Mori S, Miyamura M. The effects of intermittent exposure to hypoxia during endurance exercise training on the ventilatory responses to hypoxia and hypercapnia in humans. Eur J Appl Physiol. 1998;78:189–194. doi: 10.1007/s004210050406. [DOI] [PubMed] [Google Scholar]
- Katayama K, Sato Y, Morotome Y, Shima N, Ishida K, Mori S, Miyamura M. Ventilatory chemosensitive adaptations to intermittent hypoxic exposure with endurance training and detraining. J Appl Physiol. 1999;86:1805–1811. doi: 10.1152/jappl.1999.86.6.1805. [DOI] [PubMed] [Google Scholar]
- Katayama K, Sato Y, Morotome Y, Shima N, Ishida K, Mori S, Miyamura M. Intermittent hypoxia increases ventilation and SaO2 during hypoxic exercise and hypoxic chemosensitivity. J Appl Physiol. 2001a;90:1431–1440. doi: 10.1152/jappl.2001.90.4.1431. [DOI] [PubMed] [Google Scholar]
- Katayama K, Sato Y, Shima N, Qiu JC, Ishida K, Mori S, Miyamura M. Enhanced chemosensitivity after intermittent hypoxic exposure does not affect exercise ventilation at sea level. Eur J Appl Physiol. 2002;87:187–191. doi: 10.1007/s00421-002-0594-4. [DOI] [PubMed] [Google Scholar]
- Katayama K, Shima N, Sato Y, Qiu JC, Ishida K, Mori S, Miyamura M. Effect of intermittent hypoxia on cardiovascular adaptations and response to progressive hypoxia in humans. High Alt Med Biol. 2001b;2:501–508. doi: 10.1089/152702901753397063. [DOI] [PubMed] [Google Scholar]
- Kato M, Roberts-Thomson P, Phillips BG, Haynes WG, Winnicki M, Accurso V, Somers VK. Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation. 2000;102:2607–2610. doi: 10.1161/01.cir.102.21.2607. [DOI] [PubMed] [Google Scholar]
- Koehle MS, Foster GE, McKenzie DC, Sheel AW. Repeated measurement of hypoxic ventilatory response as an intermittent hypoxic stimulus. Respir Physiol Neurobiol. 2005;145:33–39. doi: 10.1016/j.resp.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Madsen PL, Secher NH. Near-infrared oximetry of the brain. Prog Neurobiol. 1999;58:541–560. doi: 10.1016/s0301-0082(98)00093-8. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Invited review: Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Morgan BJ, Joyner MJ. Sleep apnea: a new ‘risk factor’ for cardiovascular disease? Exerc Sport Sci Rev. 2002;30:145–146. doi: 10.1097/00003677-200210000-00001. [DOI] [PubMed] [Google Scholar]
- Peng Y-J, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. [DOI] [PubMed] [Google Scholar]
- Phillips SA, Olson EB, Morgan BJ, Lombard JH. Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and skeletal muscle resistance arteries. Am J Physiol Heart Circ Physiol. 2004;286:H388–393. doi: 10.1152/ajpheart.00683.2003. [DOI] [PubMed] [Google Scholar]
- Read DJ. A clinical method for assessing the ventilatory response to carbon dioxide. Australas Ann Med. 1967;16:20–32. doi: 10.1111/imj.1967.16.1.20. [DOI] [PubMed] [Google Scholar]
- Sahn SA, Zwillich CW, Dick N, McCullough RE, Lakshminarayan S, Weil JV. Variability of ventilatory responses to hypoxia and hypercapnia. J Appl Physiol. 1977;43:1019–1025. doi: 10.1152/jappl.1977.43.6.1019. [DOI] [PubMed] [Google Scholar]
- Schulz R, Mahmoudi S, Hattar K, Sibelius U, Olschewski H, Mayer K, Seeger W, Grimminger F. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapy. Am J Respir Crit Care Med. 2000;162:566–570. doi: 10.1164/ajrccm.162.2.9908091. [DOI] [PubMed] [Google Scholar]
- Serebrovskaya TV, Karaban IN, Kolesnikova EE, Mishunina TM, Kuzminskaya LA, Serbrovsky AN, Swanson RJ. Human hypoxic ventilatory response with blood dopamine content under intermittent hypoxic training. Can J Physiol Pharmacol. 1999;77:967–973. [PubMed] [Google Scholar]
- Smielewski P, Kirkpatrick P, Minhas P, Pickard JD, Czosnyka M. Can cerebrovascular reactivity be measured with near-infrared spectroscopy? Stroke. 1995;26:2285–2292. doi: 10.1161/01.str.26.12.2285. [DOI] [PubMed] [Google Scholar]
- Steiner DR, Gonzalez NC, Wood JG. Interaction between reactive oxygen species and nitric oxide in the microvascular response to systemic hypoxia. J Appl Physiol. 2002;93:1411–1418. doi: 10.1152/japplphysiol.00251.2002. [DOI] [PubMed] [Google Scholar]
- Task Force of the European Society of Cardiology. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Heart rate variability: standards of measurement, physiological interpretation and clinical use. Circulation. 1996;93:1043–1065. [PubMed] [Google Scholar]
- Van Der Zee P, Cope M, Arridge SR, Essenpreis M, Potter LA, Edwards AD, et al. Experimentally measured optical pathlengths for the adult head, calf and forearm and the head of the newborn infant as a function of inter optode spacing. Adv Exp Med Biol. 1992;316:143–153. doi: 10.1007/978-1-4615-3404-4_17. [DOI] [PubMed] [Google Scholar]
- Vovk A, Cunningham DA, Kowalchuk JM, Paterson DH, Duffin J. Cerebral blood flow responses to changes in oxygen and carbon dioxide in humans. Can J Physiol Pharmacol. 2002;80:819–827. doi: 10.1139/y02-105. [DOI] [PubMed] [Google Scholar]
- Weil JV, Byrne-Quinn E, Sodal IE, Friesen WO, Underhill B, Filley GF, Grover RF. Hypoxic ventilatory drive in normal man. J Clin Invest. 1970;49:1061–1072. doi: 10.1172/JCI106322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Puleo DS, Morgan BJ. Exposure to hypoxia produces long-lasting sympathetic activation in humans. J Appl Physiol. 2001;91:1555–1562. doi: 10.1152/jappl.2001.91.4.1555. [DOI] [PubMed] [Google Scholar]
- Yasuma F, Hayano JI. Impact of acute hypoxia on heart rate and blood pressure variability in conscious dogs. Am J Physiol Heart Circ Physiol. 2000;279:H2344–2349. doi: 10.1152/ajpheart.2000.279.5.H2344. [DOI] [PubMed] [Google Scholar]
- Zhang S, Robbins PA. Methodological and physiological variability within the ventilatory response to hypoxia in humans. J Appl Physiol. 2000;88:1924–1932. doi: 10.1152/jappl.2000.88.5.1924. [DOI] [PubMed] [Google Scholar]