

Abstract
OBJECTIVES
The purpose of this study was to compare the effect of intravenous flecainide and ajmaline with respect to their ability to induce or accentuate the typical ECG pattern of Brugada syndrome.
BACKGROUND
Brugada syndrome is associated with a high incidence of sudden cardiac death. The typical ECG pattern of ST-segment elevation in the right precordial leads often is concealed, but it can be unmasked with sodium channel blockers such as flecainide and ajmaline. Little is known about the relative effectiveness of these provocative agents in unmasking Brugada syndrome.
METHODS
Intravenous pharmacologic challenge with flecainide and ajmaline was performed. Whole-cell patch clamp techniques were used to assess the relative potency of ajmaline and flecainide to inhibit the transient outward current (Ito).
RESULTS
A coved-type ST-segment elevation in the right precordial leads was induced or enhanced in 22 of 22 patients following ajmaline administration. Among the 22 patients, only 15 patients showed positive response to flecainide, resulting in a positive concordance of 68%. Both drugs produced equivalent changes in QRS and PQ intervals, suggesting similar effects on sodium channel current. Whole-cell patch clamp experiments revealed a reduction of the total charge provided by Ito with an IC50 of 216 and 15.2 μM for ajmaline and flecainide, respectively.
CONCLUSIONS
Our data demonstrate disparate response of Brugada patients to flecainide and ajmaline, with a failure of flecainide in 7 of 22 cases (32%). Greater inhibition of Ito by flecainide may render it less effective. These observations have important implication for identification of patients at risk for sudden death.
Keywords: Brugada syndrome, Arrhythmia, Electrocardiogram, Ajmaline, Flecainide
Introduction
Brugada syndrome is characterized by an ECG pattern of right bundle branch block and ST-segment elevation in the right precordial leads (V1–V3).1 Intensive screening among patients with aborted sudden death or syncope has increased the number of patients since the first report by Brugada et al.2 Pharmacologic challenge with intravenous administration of sodium channel blockers has been suggested to unmask the ECG pattern in patients with Brugada syndrome.3,4 A variety of drugs, such as flecainide, ajmaline, procainamide, and pilsicainide, reportedly provoke typical ST-segment elevation.5-9 However, comparative studies for ajmaline and flecainide are lacking. Therefore, in the present study the effects of intravenous flecainide and ajmaline were studied with respect to their ability to unmask the typical Brugada ECG and their effects on the 12-lead surface ECG. In addition, the effect of ajmaline and flecainide on the Ito current in canine ventricular epicardial cells was measured in in vitro experiments to determine the mechanisms for possible differences in the response to the two sodium channel blockers in patients diagnosed with Brugada syndrome.
Patients and methods
A total of 22 consecutive, unrelated patients with Brugada syndrome diagnosed between August 2000 and March 2003 were prospectively enrolled into the study. All patients gave informed consent to clinical investigation. Diagnostic work-up included full noninvasive and invasive testing, including echocardiography, exercise testing, pharmacologic challenge, right ventricular angiography, coronary angiography, and programmed stimulation. Up to three extra-stimuli at three different basic driving cycle lengths were applied in the right ventricular outflow tract and right ventricular apex. Basal abnormal ECG, defined as a coved-type pattern with at least 2-mm ST-segment elevation in leads V1 to V3, was present in 15 of 22 patients at least once during repetitive ECG recordings.
Intravenous administration of flecainide and ajmaline was routinely performed as part of the diagnostic work-up. Diagnosis of Brugada syndrome was based on the criteria of the 2002 Brugada syndrome consensus report.10 The drug challenge using intravenous ajmaline was positive in all 22 patients.
In addition to a positive ajmaline test, diagnosis of Brugada syndrome was based on inducibility of ventricular fibrillation (VF), positive basal ECG, and presyncope (n=3); syncope, negative basal ECG, and inducible ventricular tachycardia (VT)/VF (n = 1); syncope and positive basal ECG (n = 2); presyncope, inducible VT/VF, and positive family history of sudden cardiac death (SCD; n = 1); positive family history of SCD, positive basal ECG, and syncope (n = 2); only previous syncope (n = 2); positive basal ECG and inducible VT/VF (n = 5); positive family history of SCD, positive basal ECG, and inducible VT/VF (n = 3); positive family history for SCD (n = 1); palpitations, suspicious but nondiagnostic basal ECG, and inducible VT/VF (n = 2).
Drug administration
Pharmacologic challenge was performed only if baseline ECG was not positively defined as coved-type ECG with at least 2-mm ST-segment elevation in two leads among V1 through V3 immediately before drug challenge. All patients were connected to a 12-lead ECG machine (Mortara Instrument, Inc., Milwaukee, WI, USA). ECG was continuously recorded at a paper speed of 10 mm/s and at 1-minute intervals at paper speeds of 25 and 50 mm/s. After administration of the last dose, the ECG was recorded for another 10 minutes. Flecainide was given intravenously at a dosage of 2.0 mg/kg body weight (maximum 150 mg) over 10 minutes. Ajmaline was administered at a dosage of 1 mg/kg body weight over 10 minutes. After administration of the last dose, the ECG was recorded for another 10 minutes. ECG-related stopping criteria for the two drugs were 30% prolongation of QRS duration and occurrence of ventricular arrhythmias. The two tests were separated by a minimum of 72 hours. Testing with either drug was performed in random order.
ECG analysis
ECGs were analyzed by three physicians blinded to the drugs. QRS duration, intraventricular conduction delays, PQ interval, and QTc interval were analyzed at the maximum of ST-segment elevation during challenge or 10 minutes after infusion was started. The test was considered positive if a coved-type ECG pattern appeared in more than one right precordial lead with an ST-segment elevation ≥2 mm. ST-segment elevation was determined for leads V1 through V3. In addition, the maximal ST elevation in any lead was obtained. QTc interval was determined using the Bazett method (QTc = QT/√RR). Maximal QTc interval was obtained from among the 12 leads.
Cellular electrophysiology
Isolation of myocytes
Myocytes were isolated by enzymatic dissociation as previously described.11 Briefly, adult mongrel dogs of either sex were anesthetized with sodium pentobarbital 30 mg/kg IV. The hearts were quickly removed and placed in Tyrode's solution. A wedge consisting of the portion of the left ventricular free wall supplied by the left circumflex coronary artery was excised. The artery was cannulated and flushed with Ca2+-free “Krebs buffer” supplemented with 0.1% bovine serum albumin ([BSA] fraction V; Sigma-Aldrich Co., St. Louis, MO) and gassed with 95% O2/5%CO2 for 5 minutes at a rate of 12 ml/min. Perfusion then was switched to 75 ml Ca2+-free Krebs buffer containing 75 mg BSA and 37.5 mg collagenase (CLS 2, 171 U/mg; Worthington, Freehold, NJ, USA) for 15 to 20 minutes at 37°C (95% O2/5% CO2, with recirculation). Following perfusion, thin slices of ventricular tissues were dissected from the epicardium, minced, and incubated in fresh Krebs buffer containing 0.5 mg/ml collagenase, 3% BSA, and 0.3 mM CaCl2, and agitated with 95%O2/5% CO2. Incubation was repeated three to five times at 15-minute intervals with fresh enzyme solution. The supernatant from each digestion was filtered (220-μm mesh) and centrifuged (200–300 RPM, 2 minutes). Cells were stored in HEPES-buffered Tyrode's solution (see later) supplemented with 0.5 mM Ca2+ at room temperature for later use.
“Krebs buffer” used in the cell dissociation procedure contained the following (in mM): NaCl 118.5, KCl 2.8, NaHCO3 14.5, KH2PO4 1.2, MgSO4 1.2, and glucose 11.1. The composition of the Tyrode's solution was as follows (in mM): NaCl 132, KCl 4, CaCl2 2, MgSO4 1.2, HEPES 20, and glucose 11.1; pH adjusted to 7.35 with NaOH.
Myocytes were superfused with HEPES-buffered Tyrode's solution at a flow rate of 2 to 3 ml/min. Only relaxed, quiescent cells displaying clear cross-striations were used. All experiments were performed at 37° ± 1°C.
Recording of Ito currents
Ionic currents were recorded using an Axopatch-1D amplifier with a CV-4 1/100 head stage (Axon Instruments, Foster City, CA, USA). Suction pipettes made of borosilicate glass (Becton, Dickinson and Company, Parsippany, NJ, USA) were pulled on a Flaming-Brown type pipette puller (Sutter Instrument Co., Novato, CA, USA). Pipette tip resistance measured in Tyrode's solution was 1.5 to 4 MΩ when filled with pipette solution (passed through a 0.22-μm sterile filter; Millipore Corp., Bedford, MA, USA). Cell capacitance was calculated by integrating the area under the uncompensated capacitance transient produced by 5-mV hyperpolarizing and depolarizing steps from 0 mV and dividing this area by the voltage step.
Ito was elicited by 300-ms depolarization steps to test potentials ranging from −10 to +70 mV from a holding potential of −80 mV, every 10 seconds, in myocytes superfused with Tyrode's solution containing the following (in mM): NaCl 140, CaCl2 0.1, BaCl2 0.1, KCl 3, MgCl2 1, glucose 10, and HEPES 10; pH adjusted to 7.4 with NaOH. Pipette solution contained the following (in mM): NaCl 10, KCl 10, K aspartate 110, EGTA 10, MgCl2 1, HEPES 10, and MgATP 5; pH adjusted to 7.1 with KOH. CdCl2 (300 μM) was added to block ICa-L. Step depolarization was preceded by a prepulse to −45 mV for 10 ms to inactivate the sodium channels. The current elicited by a similar voltage protocol, but from a holding potential of 0 mV, was subtracted from the previous recording. The capacitance transient preceding the Ito deflection and leak current during the test pulse were thus eliminated. Ito was measured as the difference between the peak outward current and the current at the end of the test pulse. Because the current traces showed an acceleration of inactivation with ajmaline and flecainide, the actual amount of current available after the drug was reduced more than peak current. Accordingly, we quantified the change in total charge provided by the current by measuring the area under the current trace at each concentration of drug.
Statistical analysis
The SPSS software package (version 10; SPSS Inc., Chicago, IL, USA) was used for statistical analysis. Data are given as mean ± SD. Paired Student's t-test was used to compare measurements of PQ and QTc intervals, QRS duration, and mean maximal ST-segment elevation before and after administration of flecainide and ajmaline. Unpaired Student's t-test was used to compare measurements between ajmaline and flecainide. P < .05 was considered significant.
Results
A total of 22 patients diagnosed with Brugada syndrome underwent ajmaline and flecainide challenge. No ventricular tachyarrhythmias were observed during the pharmacologic challenge, except for isolated premature ventricular beats in three patients. No severe side effects occurred. One of 22 patients developed urticaria and flush during ajmaline infusion. At baseline immediately before drug testing, seven patients displayed a type I ECG in no more than one right precordial lead. The remaining patients had either a saddle-back ECG or an incomplete right bundle branch block without ST-segment elevations.
Drug challenge
Ajmaline
At baseline (n = 22), mean ST-segment elevation was 0.12 mV ± 0.09 in lead V1 and 0.18 mV ± 0.15 in lead V2. Mean maximal ST-segment elevation was 0.2 ± 0.08 mV. Ajmaline increased or induced ST-segment elevation in 22 of 22 patients to a mean of 0.22 mV (± 0.12) in lead V1, 0.39 mV (± 0.19) on V2, and 0.1 mV (± 0.14) in lead V3. Mean maximal ST-elevation was augmented to 0.43 ± 0.15 mV (Figure 1).
Figure 1.
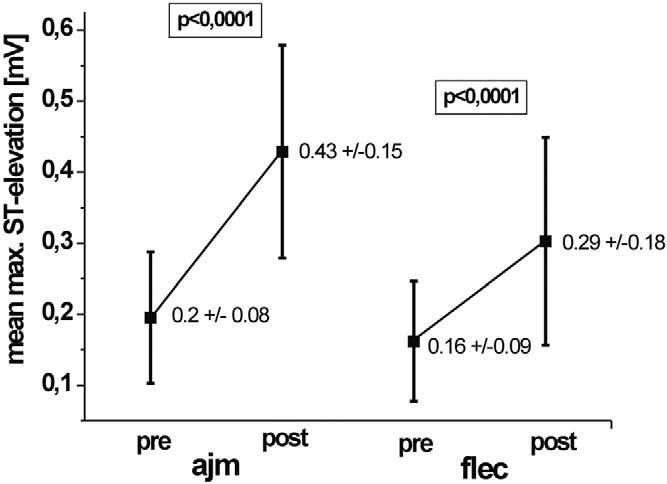
Change in maximal ST-segment elevation before (pre) and after (post) intravenous ajmaline (ajm) and flecainide (flec) administration.
Flecainide
Baseline ST-segment elevation before flecainide infusion was 0.09 ± 0.06 mV in lead V1, 0.13 ± 0.1 mV in lead V2, and 0.09 ± 0.06 in lead V3 for all 22 patients. Mean maximal elevation was 0.16 ± 0.09 mV (Figure 1). Flecainide challenge led to significant ST-segment elevations in 15 of 22 patients, resulting in a discordance of 32%. Figure 2 shows a typical example. No ST-segment elevations with a typical coved-type ECG could be induced at completion of drug infusion in seven patients (Figure 3). In these seven patients, mean ST-segment elevation during corresponding ajmaline challenge was 0.26 ± 0.15 mV in lead V1 and 0.3 ± 0.13 mV in lead V2. In the remaining 15 patients, flecainide challenge was positive with a mean amplitude in lead V1 of 0.19 ± 0.1, 0.31 ± 0.18 in lead V2, and 0.1 ± 0.13 mV in lead V3, resulting in a concordance of 68%. Mean maximal ST-segment elevation was 0.29 ± 0.18 mV.
Figure 2.
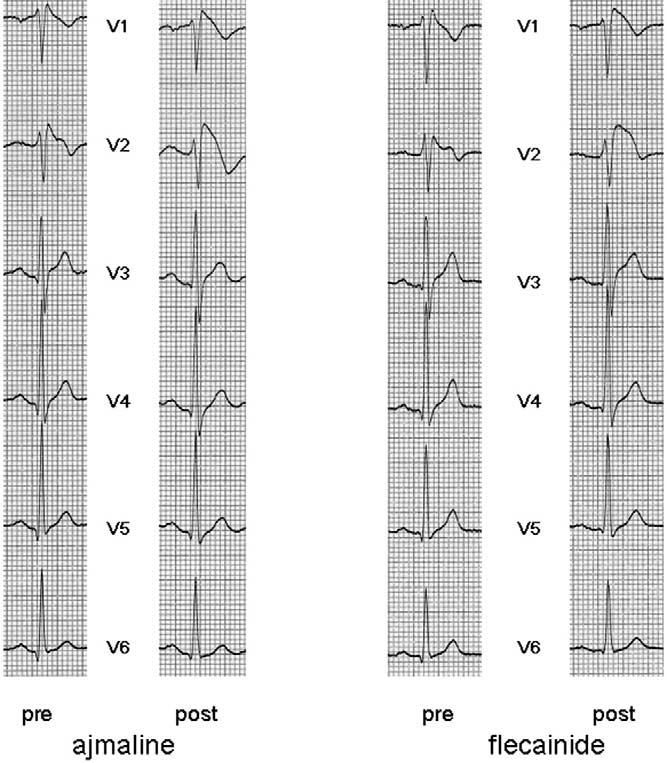
Left: Precordial leads before (pre) and after (post) ajmaline. Right: ECG before (pre) and after (post) flecainide. Induction of ECG changes after the two corresponding tests in the same patient is clearly visible.
Figure 3.

All precordial leads in three different patients before (pre) and after (post) ajmaline and flecainide. Ajmaline provoked or enhanced typical ECG changes diagnostic of Brugada syndrome in all patients. The corresponding flecainide challenge did not alter the ECG to diagnostic ST-segment changes in all patients.
QRS duration, PQ interval, and QTc interval after flecainide and ajmaline infusion
QRS duration was 98 ± 13 ms before and 109 ± 15 ms after ajmaline infusion and 95 ± 12 ms before and 104 ± 12 ms after flecainide infusion, respectively. PQ interval was 181 ± 28 ms before and 207 ± 39 ms after ajmaline. Flecainide prolonged PQ interval from baseline 178 ± 24 ms to 202 ± 31 ms. QTc interval increased from 401 ± 21 ms before to 432 ± 38 ms after ajmaline and from 402 ± 23 ms to 420 ± 25 ms after flecainide. No statistically significant difference between QRS duration, PQ interval, and QTc interval comparing flecainide versus ajmaline challenge was found.
Effects of ajmaline and flecainide on transient outward current Ito
The greater sensitivity to ajmaline may be attributable to differences in the effectiveness of the two drugs in blocking the sodium channel current INa at the doses used. This situation seems unlikely in view of their similar effects in prolonging QRS and PQ intervals. Alternatively, ajmaline and flecainide may exert a differential effect on Ito, a current whose density is critically important in the manifestation of Brugada syndrome. To test this hypothesis, we used whole-cell patch clamp techniques to measure the effect of ajmaline and flecainide on Ito in canine ventricular epicardial cells. Because the two agents produced a prominent acceleration of current decay, we considered it more appropriate to quantitate the effect of ajmaline and flecainide on the total charge provided by Ito because this parameter more accurately reflects the contribution of Ito to the early phases of the epicardial action potential. Ajmaline reduced the total charge provided by Ito with an IC50 of 216 μM, whereas flecainide reduced it with an IC50 of 15.2 μM. The greater inhibition of Ito by flecainide is expected to counter the sodium-blocking effects of the drug in unmasking the ECG characteristics of Brugada syndrome, thus rendering it less effective.
Discussion
In this prospective study, we investigated the response of the surface ECG to two different intravenously administered sodium channel blockers—ajmaline and flecainide. In 15 of 22 patients (68%) with Brugada syndrome, the study of intravenous drug challenge with flecainide and ajmaline revealed concordant results, whereas a discordant result was found in another seven patients (32%). In the seven patients, intravenous flecainide did not produce the typical ECG changes diagnostic of Brugada syndrome. ECG changes were provoked in these patients only after ajmaline infusion.
Because some patients with Brugada syndrome have only transient ECG changes with normal ECGs at baseline, pharmacologic challenge represents a substantial tool in the identification of patients with Brugada syndrome at the time of first evaluation, and the highest possible sensitivity of sodium channel blockers for pharmacologic challenge is warranted.12 Brugada et al2 reported a large series of 334 patients, of whom 100 patients (29.9%) could be diagnosed only after pharmacologic challenge. In a study reported by Priori et al,4 of 176 patients in whom repetitive baseline ECGs were recorded, only 90 patients had at least one positive baseline ECG. These findings underline the importance of pharmacologic challenge.
Comparison to previous reports
In the present series, the effect of intravenous ajmaline on QRS duration, PQ interval, and QTc interval were comparable to the findings of Rolf et al,6 except for QRS duration, for which the authors found a larger mean increase of 27 ms versus 11 ms in our study. Regarding the effect of flecainide on surface ECG, PQ interval, QRS duration, and QTc interval in our series after flecainide infusion were 202 ± 31 ms, 105 ± 13 ms, and 420 ± 26 ms, respectively. Shimizu et al9 described almost identical QTc intervals after flecainide infusion but a wider QRS complex after flecainide infusion, with a greater change to baseline. This difference may be explained by our stopping criteria, which allowed a maximum increase in QRS duration of 30%.9 Smits et al13 investigated the difference in baseline ECG and ECG recorded after sodium channel blocker administration in patients with and without SCN5A mutation, using mainly ajmaline or flecainide. However, they did not provide a subanalysis of the effect of the different drugs; therefore, our results cannot be compared to their findings for each sodium channel blocker.
As to the degree of induced ST-segment elevation in the 15 patients in whom flecainide challenge was positive, the mean amplitude of ST-segment elevation was 0.19 ± 0.1 mV in lead V1, 0.31 ± 0.19 mV in lead V2, and 0.1 ± 0.13 mV in lead V3. The absolute change of ST-segment elevation after flecainide in patients with a positive response was 0.2 mV in our patients.
This difference was greater for flecainide in the study by Shimizu et al.9 The difference may be explained by the fact that the patients reported by Shimizu et al revealed mean baseline ST-segment elevations of 0.39 mV, which already is diagnostic of Brugada syndrome, compared to a mean 0.16 ± 0.09 mV in our series. Of note, our series included patients with suspicion of Brugada syndrome and normal baseline ECG.
Priori et al12 reported six of 41 patients (14.5%) had a negative response to sodium channel blockers. However, they did not specify which drug was used in the patients. Three of the six patients (50%) with a negative drug response had been symptomatic. Given that symptomatic patients at first evaluation may have a negative ECG at baseline, a negative response to drug challenge may lead to underdiagnosis of Brugada syndrome. Of note, among our seven patients with a negative response to flecainide, three patients suffered previous syncope and four had inducible VT/VF at programmed ventricular stimulation. According to a published report by Brugada et al,14 the risk of VT/VF in a patient with a basal abnormal ECG, syncope, and inducible VT/VF is 27.2% over a mean follow-up of 24 ± 32 months. In our series, this risk value applies for two of seven patients with a negative flecainide and positive ajmaline challenge. Furthermore, patients without syncope and inducible VT/VF in their population had a 14% risk in the presence of a basal abnormal ECG and a 9.7% risk in case of an abnormal ECG only after pharmacologic challenge.
Patients with a negative response to flecainide more frequently presented with a normal basal ECG (5/7 vs 2/15). Thus, one could argue that patients with a presumably better prognosis because of a normal basal ECG tend to have a higher likelihood of a negative response to flecainide. This observation most likely reflects a less severe substrate in the drug-free state and should not be interpreted as a false-positive response to ajmaline.
Potential mechanisms of different response to flecainide and ajmaline
Our data point to differences in the effectiveness of ajmaline and flecainide in blocking Ito as the basis for their differential ability to unmask the Brugada syndrome. The ECG characteristics of Brugada syndrome and the arrhythmogenic substrate responsible for the development of extrasystoles and polymorphic VT in Brugada syndrome are believed to be secondary to amplification of heterogeneities in the early phases of the action potential of cells residing in different transmural layers of the right ventricular wall of the heart.
The presence of a transient outward current (Ito)-mediated spike-and-dome morphology, or notch, in ventricular epicardium, but not endocardium, creates a transmural voltage gradient responsible for the inscription of the ECG J-wave.15,16 Rebalancing of currents active at the end of phase 1 is believed to underlie accentuation of the action potential notch in right ventricular epicardium, which is responsible for the augmented J-wave and ST-segment elevation associated with Brugada syndrome.16 Accentuation of the right ventricular action potential notch under pathophysiologic conditions and/or following inhibition of INa with drugs such as ajmaline and flecainide leads to exaggeration of J-wave or ST-segment elevation. The arrhythmogenic substrate is thought to develop following a further shift in the balance of current leading to loss of the action potential dome at some epicardial sites but not others. This situation creates a transmural dispersion of repolarization and a vulnerable window, which when captured by a premature extrasystole can trigger a reentrant arrhythmia. Conduction of the action potential dome from sites at which it is maintained to sites at which it is lost causes local reexcitation via phase 2 reentry. This situation leads to development of a closely coupled extrasystole capable of capturing the vulnerable window across the ventricular wall, thus triggering circus movement reentry in the form of VT/VF.8,17,18 Studies suggest sex-related differences in Ito contribute to the 8- to 10-fold greater prevalence of the Brugada phenotype in males compared with females.19 The smaller Ito in females was in large part due to a more rapid decay of the current. A more prominent transient outward current (Ito) underlies the larger action potential notch and greater sensitivity of right ventricular epicardium of males to sodium channel block. The more prominent Ito causes the end of phase 1 of the right ventricular epicardial action potential to repolarize to more negative potentials in tissue and arterially perfused wedge preparations from males, facilitating loss of the action potential dome and the development of phase 2 reentry and polymorphic VT. A rebalancing of currents active at the end of phase 1 also underlies the unmasking of the syndrome in response to drugs. Vagotonic agents, IK-ATP activators, and hypokalemia achieve this by augmenting outward currents. Sodium channel blockers, beta-blockers, cocaine, antidepressants, and antihistamines such as terfenadine likely accomplish this by reducing inward currents.20 In the case of flecainide and ajmaline, INa inhibition reduces inward current, whereas Ito inhibition counters this action of the drugs by blocking outward current. Ajmaline and flecainide, in addition to reducing the peak amplitude of Ito, significantly accelerated the decay of the current, causing a marked reduction in the total charge contributing to phase 1 of the epicardial action potential and the charge available to oppose the reduction of INa. Our data suggest the lesser effectiveness of flecainide largely results from its more potent inhibition of Ito. Differences in INa inhibition at the doses used are difficult to completely rule out as a contributing factor. However, the clinical observation of a similar effect of the two drugs on QRS and PQ intervals suggests such differences less likely are a discriminating factor in the effectiveness of ajmaline and flecainide in unmasking Brugada syndrome. Another drug that we have not studied but that often is used in the United States for unmasking the Brugada syndrome is procainamide. Like disopyramide, procainamide is a class IA antiarrhythmic agent that displays a more rapid dissociation from the sodium channel and consequently a lower level of use-dependent sodium channel block. This characteristic of the drug is believed to underlie its lesser potency compared with class IC agents such as flecainide in unmasking the syndrome. In contrast to ajmaline and flecainide, procainamide produces no block of Ito at clinically relevant drug concentrations.9
Study limitations
This was not a randomized study, and neither of the tests was repeated. Therefore, day-to-day variations in response to both drugs cannot be excluded. However, the baseline ECG changes, including morphology and ST-segment elevation, were not different immediately before drug challenge with either drug. This study combines clinical and experimental findings in humans and canines. With respect to potential differences of the effect of flecainide and ajmaline in canines versus humans, several distinctions have been described for Ito in human versus canine ventricular myocytes, including slower current decay, more rapid recovery from inactivation, and a depolarizing shift of steady-state inactivation in human cells. Pharmacologic sensitivity of human Ito to flecainide reportedly is greater than that of canine Ito. The electrophysiologic and pharmacologic distinctions between humans and dogs do not appear to result from differential expression of the genes encoding for Ito but rather to differences in molecular structure and/or posttranslational modification of these subunits. No data on the differential effects of ajmaline on human versus canine Ito are available.21,22
Clinical implications
The observation of 32% of patients with discordant results using flecainide and ajmaline for intravenous drug challenge may further complicate correct phenotyping of patients with Brugada syndrome.
Footnotes
Supported by Grant HL47678 from the National Heart, Lung and Blood Institute to Dr. Antzelevitch and a grant from the American Heart Association to Dr. Antzelevitch.
References
- 1.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391–1396. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 2.Brugada J, Brugada R, Antzelevitch C, Towbin J, Nademanee K, Brugada P. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation. 2002;105:73–78. doi: 10.1161/hc0102.101354. [DOI] [PubMed] [Google Scholar]
- 3.Brugada J, Brugada P. Further characterization of the syndrome of right bundle branch block, ST segment elevation, and sudden cardiac death. J Cardiovasc Electrophysiol. 1997;8:325–331. doi: 10.1111/j.1540-8167.1997.tb00796.x. [DOI] [PubMed] [Google Scholar]
- 4.Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Giordano U, Bloise R, Giustetto C, De Nardis R, Grillo M, Ronchetti E, Faggiano G, Nastoli J. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105:1342–1347. doi: 10.1161/hc1102.105288. [DOI] [PubMed] [Google Scholar]
- 5.Brugada R, Brugada J, Antzelevitch C, Kirsch GE, Potenza D, Towbin JA, Brugada P. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101:510–515. doi: 10.1161/01.cir.101.5.510. [DOI] [PubMed] [Google Scholar]
- 6.Rolf S, Bruns HJ, Wichter T, Kirchhof P, Ribbing M, Wasmer K, Paul M, Breithardt G, Haverkamp W, Eckardt L. The ajmaline challenge in Brugada syndrome: diagnostic impact, safety, and recommended protocol. Eur Heart J. 2003;24:1104–1112. doi: 10.1016/s0195-668x(03)00195-7. [DOI] [PubMed] [Google Scholar]
- 7.Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol. 1996;27:1061–1070. doi: 10.1016/0735-1097(95)00613-3. [DOI] [PubMed] [Google Scholar]
- 8.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100:1660–1666. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 9.Shimizu W, Antzelevitch C, Suyama K, Kurita T, Taguchi A, Aihara N, Takaki H, Sunagawa K, Kamakura S. Effect of sodium channel blockers on ST segment, QRS duration, and corrected QT interval in patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2000;11:1320–1329. doi: 10.1046/j.1540-8167.2000.01320.x. [DOI] [PubMed] [Google Scholar]
- 10.Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, Corrado D, Hauer RN, Kass RS, Nademanee K, Priori SG, Towbin JA. Study Group on the Molecular Basis of Arrhythmias of the European Society of Cardiology. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation. 2002;106:2514–2519. doi: 10.1161/01.cir.0000034169.45752.4a. [DOI] [PubMed] [Google Scholar]
- 11.Thomas GP, Gerlach U, Antzelevitch C. HMR 1556, a potent and selective blocker of slowly activating delayed rectifier potassium current. J Cardiovasc Pharmacol. 2003;41:140–147. doi: 10.1097/00005344-200301000-00018. [DOI] [PubMed] [Google Scholar]
- 12.Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Brignole M, Giordano U, Giovannini T, Menozzi C, Bloise R, Crotti L, Terreni L, Schwartz PJ. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: a prospective evaluation of 52 families. Circulation. 2000;102:2509–2515. doi: 10.1161/01.cir.102.20.2509. [DOI] [PubMed] [Google Scholar]
- 13.Smits JP, Eckardt L, Probst V, Bezzina CR, Schott JJ, Remme CA, Haverkamp W, Breithardt G, Escande D, Schulze-Bahr E, LeMarec H, Wilde AA. Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J Am Coll Cardiol. 2002;40:350–356. doi: 10.1016/s0735-1097(02)01962-9. [DOI] [PubMed] [Google Scholar]
- 14.Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation. 2003;108:3092–3096. doi: 10.1161/01.CIR.0000104568.13957.4F. [DOI] [PubMed] [Google Scholar]
- 15.Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J-wave. Circulation. 1996;93:372–379. doi: 10.1161/01.cir.93.2.372. [DOI] [PubMed] [Google Scholar]
- 16.Antzelevitch C. The Brugada syndrome: ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol. 2001;12:268–272. doi: 10.1046/j.1540-8167.2001.00268.x. [DOI] [PubMed] [Google Scholar]
- 17.Antzelevitch C, Brugada P, Brugada J, Brugada R, Shimizu W, Gussak I, Perez Riera AR. Brugada syndrome: a decade of progress. Circ Res. 2002;91:1114–1118. doi: 10.1161/01.res.0000046046.53721.90. [DOI] [PubMed] [Google Scholar]
- 18.Kurita T, Shimizu W, Inagaki M, Suyama K, Taguchi A, Satomi K, Aihara N, Kamakura S, Kobayashi J, Kosakai Y. The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. J Am Coll Cardiol. 2002;40:330–334. doi: 10.1016/s0735-1097(02)01964-2. [DOI] [PubMed] [Google Scholar]
- 19.Di Diego JM, Cordeiro JM, Goodrow RJ, Fish JM, Zygmunt AC, Perez GJ, Scornik FS, Antzelevitch C. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation. 2002;106:2004–2011. doi: 10.1161/01.cir.0000032002.22105.7a. [DOI] [PubMed] [Google Scholar]
- 20.Antzelevitch C, Brugada P, Brugada J, Brugada R, Towbin JA, Nademanee K. Brugada syndrome: 1992–2002. A historical perspective. J Am Coll Cardiol. 2003;41:1665–1671. doi: 10.1016/s0735-1097(03)00310-3. [DOI] [PubMed] [Google Scholar]
- 21.Akar FG, Wu RC, Deschenes I, Armoundas AA, Piacentino V, 3rd, Houser SR, Tomaselli GF. Phenotypic differences in transient outward K+ current of human and canine ventricular myocytes: insights into molecular composition of ventricular Ito. Am J Physiol Heart Circ Physiol. 2004;286:H602–609. doi: 10.1152/ajpheart.00673.2003. [DOI] [PubMed] [Google Scholar]
- 22.Liu QY, Wang XL. The blocking effects of six antiarrhythmic drugs on transient outward current in rat ventricular myocytes. Yao Xue Xue Bao. 1997;32:183–187. [PubMed] [Google Scholar]