

Abstract
The small GTPase Rac cycles between the membrane and the cytosol as it is activated by nucleotide exchange factors (GEFs) and inactivated by GTPase-activating proteins (GAPs). Solubility in the cytosol is conferred by binding of Rac to guanine-nucleotide dissociation inhibitors (GDIs). To analyze the in vivo dynamics of Rac, we developed a photobleaching method to measure the dissociation rate constant (koff) of membrane-bound GFP-Rac. We find that koff is 0.048 s−1 for wtRac and ∼10-fold less (0.004 s−1) for G12VRac. Thus, the major route for dissociation is conversion of membrane-bound GTP-Rac to GDP-Rac; however, dissociation of GTP-Rac occurs at a detectable rate. Overexpression of the GEF Tiam1 unexpectedly decreased koff for wtRac, most likely by converting membrane-bound GDP-Rac back to GTP-Rac. Both overexpression and small hairpin RNA-mediated suppression of RhoGDI strongly affected the amount of membrane-bound Rac but surprisingly had only slight effects on koff. These results indicate that RhoGDI controls Rac function mainly through effects on activation and/or membrane association.
INTRODUCTION
Rho family small GTPases control many cellular functions, including cell polarity, migration, cell cycle progression, apoptosis, gene expression, and vesicular trafficking. Rho GTPases cycle between an active (GTP-bound) and an inactive (GDP-bound) state. They also cycle between the membrane, where active GTPases interact with downstream effectors, and the cytosol, where GTPases interact with GDP dissociation inhibitors (GDIs). Membrane targeting is predominantly mediated by carboxy-terminal sequences that include geranylgeranyl modification and a polybasic motif. Three classes of molecules regulate the activation and/or localization of Rho GTPases: Guanine nucleotide exchange factors (GEFs) activate by catalyzing the exchange of GDP for GTP; GTPase-activating proteins (GAPs) inactivate by accelerating the low intrinsic rate of GTP hydrolysis; and GDIs act as chaperone proteins that confer solubility in the cytoplasm and inhibit both activation by GEFs and inactivation by intrinsic and GAP-catalyzed GTP hydrolysis (Van Aelst and D'Souza-Schorey, 1997).
According to the currently accepted model for the activation/targeting cycle of Rho GTPases, inactive Rho GTPases are maintained in the cytosol of quiescent cells bound to RhoGDI. On cell stimulation, activation by GEFs occurs coincident with dissociation from RhoGDI and binding to the membrane. Activated, membrane-bound GTPases then interact with downstream effectors to initiate signaling. Inactivation and termination of signaling involves GAP-stimulated GTP hydrolysis, followed by RhoGDI-mediated extraction from the membrane. An important feature of this model is that activation and membrane targeting of Rho GTPases are coupled. Thus, the majority of inactive GTPases are complexed with RhoGDI in the cytosol, whereas the majority of the active GTPases are membrane-bound. According to some reports, RhoGDI has a higher affinity for GDP-bound GTPases (Olofsson, 1999). In this scheme, activation by GEFs precedes membrane targeting. However, Dbl-catalyzed nucleotide exchange is inhibited by RhoGDI in the absence of membranes (Yaku et al., 1994). Additionally, membrane-bound Rac is a better substrate for Tiam1 than soluble Rac (Robbe et al., 2003). These results suggest that partial or complete dissociation of the Rho GTPase/RhoGDI complex precedes and is required for activation by GEFs.
There is, however, evidence that contradicts this model. First, RhoGDI was reported to exhibit similar affinities for the GTP- and GDP-bound forms of Cdc42 and Rac (Chuang et al., 1993b; Nomanbhoy and Cerione, 1996). Second, a significant portion of GAP-insensitive, constitutively active G12V Rac exists in the cytosol bound to RhoGDI (Del Pozo et al., 2002). These observations suggest that GAPs are not necessary for the dissociation of Rho GTPases from the membrane. In the same context, integrins promote membrane targeting by regulating the availability of membrane binding sites so that, in nonadherent cells, active GTPases are present in the cytosol bound to RhoGDI (del Pozo et al., 2000, 2002, 2004). Therefore, activation and membrane targeting of Rho GTPases can be separable events.
Overall, the current view of Rho GTPase–membrane interactions is based on in vitro or indirect in vivo experiments; the dynamics of these interactions have not been analyzed in living cells. Moreover, because cycling between the cytosol and the membrane is in principle bidirectional, the role of various regulators in controlling each direction has been difficult to delineate. Here, we set out to measure the in vivo rates of Rac interaction with the plasma membrane and investigate the roles of GAPs, GEFs, and GDI in these dynamics. We developed a fluorescence loss in photobleaching (FLIP) protocol together with a mathematical model to analyze the experimental data. Our results define rate constants for dissociation of Rac from the membrane, reveal effects of GAP and GEF activity, and show that RhoGDI unexpectedly controls Rac–membrane targeting mainly by affecting rates of association.
MATERIALS AND METHODS
Cell Culture and Transfection
NIH3T3 cells were grown in DMEM supplemented with 10% fetal bovine serum, penicillin, and streptomycin (Invitrogen, Carlsbad, CA). For photobleaching experiments, cells were trypsinized and replated (105) in Delta T Dishes (Bioptechs, Butler, PA) coated with 2 μg/ml fibronectin. DNA plasmids were transfected with Effectene according to the manufacturer's instructions (Qiagen, San Diego, CA). For cotransfections, plasmids encoding GFP-Rac plus Tiam1 or mRFP-RhoGDI were used at molar ratios of 1:4, respectively. Cells were analyzed ∼24 h after transfection. For interfering RNA (RNAi) experiments, pSuper constructs encoding short hairpin RNA (shRNA; 40 μg) together with pcDNA3.1(+)-mRFP (4 μg) were electroporated using GenePulser Xcell (Bio-Rad Laboratories, Hercules, CA), and cells were processed 48 and 72 h after electroporation.
DNA Plasmids and Constructs
Plasmids encoding EGFP-tagged wtRac (wild-type Rac) and G12VRac have been described previously (Del Pozo et al., 2002). A plasmid encoding EGFP-PBR/C189SRac was obtained from A. L. Wilson-Delfosse (Case Western Reserve University, Cleveland, OH) and pcDNA3-HA-C1199Tiam1 was obtained from J. G. Collard (Amsterdam, The Netherlands). pRSETB-mRFP (gift of R. Y. Tsien, University of California, San Diego, CA), pEGFP-Q61LRac (gift of C. J. Der, University of North Carolina, Chapel Hill, NC), and pEFBOS-myc-RhoGDI were used as templates for PCR amplification of fragments that were subcloned in pcDNA3.1(+) to generate mRFP-Q61LRac and mRFP-RhoGDI (mRFP was N-terminal). pCMV5-Fyn-GFP (gift of L. G. Berthiaume, University of Alberta, Canada) was used to PCR-amplify the Fyn fragment that was subsequently subcloned into pEGFP-N3 (Clontech Laboratories, Palo Alto, CA). For RNAi experiments, duplexed oligos containing a sequence corresponding to nucleotides 97–116 in the coding region of mouse RhoGDI or control (mismatch) sequence were subcloned in pSuper (OligoEngine, Seattle, WA), according to the manufacturer's instructions.
Subcellular Fractionation
Cells were washed with cold phosphate-buffered saline, scraped, and homogenized with 500 μl of buffer containing 10 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 1 mM DTT, 0.25 M sucrose, and a cocktail of protease inhibitors (Sigma, St. Louis, MO). Nuclei and unbroken cells were removed by centrifugation at 1000 × g for 10 min at 4°C, and the postnuclear supernatant was centrifuged at 100,000 × g for 1 h at 4°C to separate the cytosolic and particulate fractions.
Immunoprecipitation and Western Blotting
Cytosolic fractions of cells transfected with EGFP-fusion constructs were immunoprecipitated with polyclonal anti-GFP for 4 h at 4°C (2 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA), and immune complexes were immobilized on protein A Sepharose CL-4B (Amersham Biosciences, Uppsala, Sweden). Immunoprecipitates or equivalent volumes of cytosol, membrane, and unfractionated material or aliquots of RIPA lysates containing equal amounts of protein were separated by SDS-PAGE, electrophoretically transferred to nitrocellulose (Bio-Rad Laboratories, Hercules, CA), and immunoblotted with the following primary antibodies: B-2 monoclonal anti-GFP (1/1000; Santa Cruz Biotechnology), monoclonal anti-RhoGDI (1/5000; clone 16; BD Transduction Laboratories, San Jose, CA), polyclonal anti-D4-GDI (1/1000; Spring Bioscience, Fremont, CA), AC-40 monoclonal anti-actin (1/1000; Sigma), and polyclonal anti-integrin β1 (gift of A. F. Horwitz, University of Virginia, Charlottesville, VA). Secondary antibodies were horseradish peroxidase–conjugated anti-mouse or anti-rabbit immunoglobulin, followed by ECL substrate (Amersham Biosciences). Densitometric analysis was performed with Image J software.
Confocal Microscopy and Photobleaching
Imaging and photobleaching were performed with a Zeiss LSM 510 Meta confocal microscope operated by LSM-FCS software (Carl Zeiss, Oberkochen, Germany). Temperature was controlled with a Delta T stage adapter (Bioptechs) and maintained at 37°C. Medium pH was controlled by addition of 25 mM HEPES buffer. Media were overlaid with mineral oil to prevent evaporation. EGFP and mRFP were excited with the 488-nm line of an Argon laser and the 543-nm line of a HeNe laser, respectively. The photobleaching protocol consisted of 24 bleaching events at 100% laser power, targeting the whole cell except for a narrow area (protrusive or quiescent) that was left unbleached. Images of the whole cell at 0.1% laser power were recorded after every bleaching event. A focal plane proximal to the basal membrane was chosen for both photobleaching and scanning at low laser power. The optical slice was 1.3 μm. Total pixel intensities in the unbleached and adjacent bleached areas were measured, background signal was subtracted, and intensities were expressed as a percentage of the intensity at time zero.
Mathematical Model
A simple compartmental model that assumed that cytosol diffusion over the width of the unbleached region is faster than binding and photobleaching was used to fit the experimental data. In this model, the concentration of fluorescent GFP-Rac in the unbleached membrane, c1, is affected by dissociation from and binding to the membrane:
 |
where c2 is the concentration of fluorescent GFP-Rac in the bleached cytosol (if diffusion is not sufficiently fast with respect to bleaching, c2 should refer to the unbleached portion of the cytosol, and the model would contain an additional fitting parameter; in most cases, these two versions of the model yielded close results); kon and koff stand for membrane association and dissociation rate constants, respectively. Taking into account that in all experiments the normalized intensities of the bleached area were fitted well to a two-exponential function:
where A, α, B, and β were constrained to be positive numbers and c2 in Equation (1) was substituted with Equation (2). By assuming that the system was at steady state before photobleaching, Equation (1) was solved analytically to yield the following expression for c1,norm:
where a = A/(A + B) and b = B/(A + B). One can interpret Equation (3) as the normalized intensity of the unbleached area if the signal comes solely from the membrane. To account for the cytosolic component of the signal in the unbleached region, Equation (3) should be extended to
where values of the parameter r range from 0 to 1, with 0 representing unbleached signal coming entirely from the cytosol and 1 representing signal solely from the membrane; see Supplementary Text for a full description of the model and derivation details. Equation (4) has been used for fitting the normalized intensities from the unbleached compartment to estimate koff and r. Fitting was performed by minimizing the mean squared error,
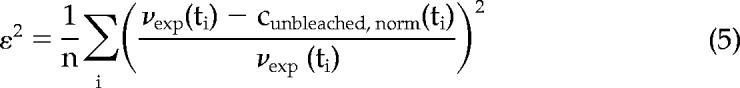 |
with the constraints: koff >; 0 and 0 <; r <; 1. In the expression for the mean squared error, νexp(ti) is the experimental value at time point ti, and n is the total number of time points in each experiment. Values of the square root of the mean squared error, ε, typically ranged from 0.01 to 0.1. See Supplementary Text for a detailed discussion of the reliability of this method.
Computer Simulations
The effect of membrane diffusion on fluorescence decay was modeled by solving a reaction-diffusion equation, 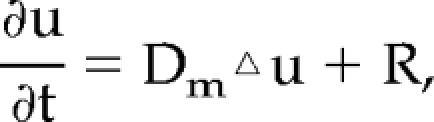 for the concentration of a fluorescent protein, u. In the equation, Dm is the membrane diffusion coefficient of the protein, ▵ is a two-dimensional Laplace operator,
for the concentration of a fluorescent protein, u. In the equation, Dm is the membrane diffusion coefficient of the protein, ▵ is a two-dimensional Laplace operator, 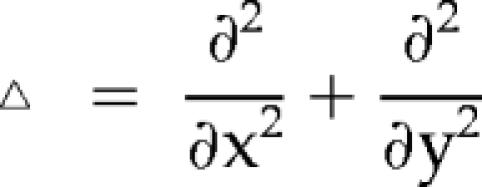 in Cartesian coordinates, and R is a reaction term describing the rate of bleaching: R = −Vbu(1 − m(x)), where Vb is a bleaching rate constant and
in Cartesian coordinates, and R is a reaction term describing the rate of bleaching: R = −Vbu(1 − m(x)), where Vb is a bleaching rate constant and
 |
The equation was solved numerically on a two-dimensional circular domain with a diameter of 55 μm, with zero flux boundary conditions and the uniform initial protein distribution, u(x, 0) = 1.
Computer simulations were performed using the Virtual Cell framework (Slepchenko et al., 2003). By taking into account axial symmetry, the computational domain was reduced in half to 56 × 28 μm and contained 50,880 grid points on a structured orthogonal mesh. Integration was performed with a 25-ms time step, and an average numerical error of the solution is estimated to be <;2.5%.
RESULTS
Methodology
To obtain consistent and robust lamellipodia, we replated NIH3T3 cells on fibronectin and allowed them to spread for 15–45 min. Under these conditions, Rac is activated and translocates to the membrane (Price et al., 1998; del Pozo et al., 2000). Large lamellipodial protrusions formed around most of the cell during this period (Figure 1A, and unpublished data). A photobleaching protocol was used in which 24 photobleaching events at 100% laser power targeted most of the cell apart from a protrusive area that was left unbleached (Figure 1A). After each bleach, the entire cell was imaged at low laser power (Figure 1B). Fluorescence measurements without photobleaching showed negligible signal decay due to scanning at low laser power (Figure 1G).
Figure 1.

Experimental method for determining Rac dissociation rate from the membrane. (A) Representative GFP-wtRac–expressing cells, either 30 min (left) or 12 h (right) after replating on fibronectin. The red lines delimit the photobleached areas. (B) Cell in A at the indicated times during the photobleaching protocol. (C) Normalized fluorescence in the unbleached region (protrusive or quiescent) of individual cells expressing GFP-wtRac and the unbleached and bleached region of cells expressing GFP-PBR/C189SRac, plotted as a function of time. Lines are the best-fit curves to Equation (4). (D) Average best-fit curves to Equation (4) for protrusive (n = 11) and quiescent (n = 5) unbleached regions of cells expressing GFP-wtRac. Values are means ± SD. (E and F) Apparent koff and r values for GFP-wtRac in protrusive (n = 11) and quiescent (n = 5) areas of cells. Values are means ± SD (∗p <; 0.05 by Student's t test). (G) Fluorescence intensity as a function of time (relative to time 0) in the absence of photobleaching. Values represent the mean of three independent experiments ± SD. (H) Apparent koff in protrusive areas of cells expressing low (n = 8), medium (n = 11), and high (n = 10) levels of GFP-wtRac. Levels of expression were monitored by the total fluorescence intensities across the cell and normalized to the lowest value. koff values are the means ± SD.
To estimate dissociation rate constants, a mathematical model was developed and used to fit the experimental data (for details see Materials and Methods). Briefly, the normalized intensities within the bleached area were fitted to a two-component exponential equation (Equation (2)). The model assumes that cytosol diffusion is fast relative to the time of bleaching and measurement, so that free cytoplasmic components equilibrate across the boundary. This assumption was validated by analysis of GFP and a Rac mutant that does not associate with membranes (Figure 1C). Thus, the parameters derived for the bleached area were applied to the three-component exponential function that was used to fit the data from the unbleached area (Equation (4)). In this equation, koff denotes the dissociation rate constant of Rac from the membrane.
Because fluorescence intensity in the unbleached area consisted of both membrane and cytosolic components, a parameter, r in Equation (4), was used as a qualitative indicator of the proportion of membrane-bound Rac within this zone. Values of r can range from 0 to 1, where 0 is 100% cytosolic and 1 is 100% membrane-bound. It should be noted that r is a nonlinear function of Rac membrane targeting compared with the fraction of membrane-bound Rac within the whole cell, as measured by cell fractionation. This difference can be attributed to the increased surface to volume ratio in protrusive areas and the higher density of Rac targeting sites within these regions. Because r depends on the thickness of protrusions, we analyzed z-stack profiles of cells under experimental conditions that yield distinct r values (GFP-wtRac, with and without RFP-RhoGDI and GFP-G12VRac; see below). No differences in the geometry of protrusions were detectable by this method (Supplementary Figure 3).
Figure 3.

Effect of Rac regulators on membrane targeting of GFP-Rac. (A) Postnuclear lysates from NIH3T3 cells expressing the indicated constructs were separated into membrane (M) and cytosolic (C) fractions. Equal percentages of cytosolic and membrane material were resolved by SDS-PAGE and immunoblotted for GFP (Rac), and integrin β1 and RhoGDI as membrane and cytosol markers, respectively. (B) Percentage of GFP-Rac in the membrane fraction; values are means ± SD (n = 3).
To assess the validity of this approach, we first examined a Rac mutant (PBR/C189SRac) that fails to target to the membrane because of lack of prenylation and charge neutralization of the positive residues (substitution by glutamine) in the polybasic region (Del Pozo et al., 2002; see Figure 3). As shown in Figure 1C, fluorescence decay in the unbleached region of cells expressing GFP-PBR/C189SRac was almost indistinguishable from the decay in the bleached area. Significantly, analysis of protrusive areas in these cells did not produce well-defined dissociation parameters. Next, the dynamics of GFP-wtRac in actively protruding areas of the cell during spreading were compared with concave, quiescent areas of stably adherent cells (Figure 1, A, C, and D). At protrusive regions, average koff = 0.067 s−1 with a 4% fitting error. In quiescent areas, koff was similar (0.065 s−1; 10% fitting error) (Figure 1E). However, quiescent regions produced nearly threefold lower values of the parameter r (Figure 1F). These results indicate that nonprotrusive areas of the cell have less membrane-bound Rac, though the Rac that is present dissociates at a similar rate.
To test whether overexpression of GFP-Rac affects dissociation kinetics, we analyzed Rac dissociation over a sevenfold range of GFP-wtRac expression. Within this range, the level of GFP-wtRac had no effect on the dissociation rate (Figure 1H). These results therefore suggest that under these conditions GFP-Rac is regulated normally. For the experiments described below, cells that correspond to medium expressors in Figure 1H were examined.
Contribution from Membrane Diffusion
In the mathematical model, we assumed that fluorescence decay in the unbleached area is brought about by the dissociation of fluorescent Rac from the membrane. However, two-dimensional diffusion within the plasma membrane should also contribute to this decay. To distinguish diffusion from dissociation, we took advantage of the fact that the rate of membrane diffusion is dependent on the size of the unbleached area, whereas the rate of dissociation is not. Therefore, we varied the width of the unbleached areas, keeping other variables constant. For these experiments, we examined constitutively active GFP-V12Rac, because it associates with the membrane more strongly (Del Pozo et al., 2002; see Figure 3) and therefore would be more sensitive to effects of lateral diffusion. Figure 2, A and B, shows that the rate of decay in the unbleached areas decreased as the width of the unbleached area increased. Thus, membrane diffusion is a significant factor, but above 6 μm effects were small.
Figure 2.

Estimation of Rac membrane diffusion rate constant. (A) Fluorescence intensity in the bleached and unbleached regions of individual cells expressing GFP-G12VRac. The width of the unbleached area was varied as indicated in the inset. Values are relative to t = 0. (B) Apparent koff values for GFP-G12VRac as a function of the width of the unbleached area. (C) Postnuclear lysates of NIH3T3 cells expressing GFP or Fyn-GFP were fractionated into membrane (Memb.) and cytosolic (Cyt.) fractions. Equal percentages of cytosolic, membrane, and total material were resolved by SDS-PAGE and immunoblotted for GFP, integrin β1, and RhoGDI. (D) Normalized fluorescence intensity in the unbleached region (8-μm width) of a representative cell expressing Fyn-GFP. Line is the best fit to a single exponential function. The average membrane diffusion rate constant (kdiff) was 0.017 s−1 (n = 13). (E) Computer simulations of the effect of membrane diffusion on fluorescence decay in the unbleached region. Cell geometry and the masked region (in red): cell diameter is 52 μm, mask width is 8.5 μm, and mask arc length is 42 μm (left). Fluorescence distribution at 22.5 s into bleaching simulated on the same domain (middle). A color scale with the equivalent numerical values is displayed (right). The simulated bleaching rate is 1 s−1, and the protein diffusion coefficient is 0.25 μm2/s. (F) Normalized decay of protein concentration (fluorescence) averaged over the unbleached region. The simulation parameters are as in E. The best fit to a single exponential function Ae−kdifft yields A = 0.95 and kdiff = 0.016 s−1. (G) Dependence of the simulated decay rate constant due to membrane diffusion (kdiff) on the width of the unbleached region. kdiff values were derived as in F. The arc length was held constant at 42 μm. The estimated relative numerical error in the presented data is 5%.
To correct for effects of membrane diffusion on signal decay in the unbleached area, we took advantage of the fact that Rac is anchored to the plasma membrane mainly through a lipid moiety; thus, its diffusion should be similar to other lipid-linked proteins. We therefore examined a GFP chimera carrying the myristoylation and palmitoylation sequences of Fyn at its N-terminus (Fyn-GFP). These acylation signals confer localization of Fyn and Fyn-GFP at cholesterol- and GM1-enriched regions of the plasma membrane (Alland et al., 1994; McCabe and Berthiaume, 1999, 2001) similarly to Rac (del Pozo et al., 2004). Fyn-GFP is present almost entirely in the particulate fraction of NIH3T3 cells, unlike GFP, which is entirely cytoplasmic (Figure 2C). Integrin β1 and RhoGDI were used as markers for the membrane and cytosol, respectively. To estimate the decay rate due to diffusion, cells expressing Fyn-GFP were subjected to a FLIP protocol using 8-μm unbleached areas, and data from the unbleached area were fitted to a single-component exponential function (Figure 2D). The decay rate in these experiments was 0.017 s−1.
Computer simulations of the effect of membrane diffusion have shown that this rate corresponds to a diffusion coefficient of 0.25 μm2/s, which fits well with diffusion of membrane proteins that ranges from 0.1 to 0.5 μm2/s (Cowan et al., 1997). Simulations were run on a two-dimensional domain within a circular cell that mimicked the geometry of the experimental setups (Figure 2E). In the model, a membrane-bound species was bleached with a rate of 1 s−1 everywhere except for the masked (red) region, and the signal decay due to diffusion in the unbleached region was measured. Results were insensitive to the rate of bleaching so long as it was sufficiently fast. Figure 2F shows the fluorescence decay of an unbleached region with the width of 8.5 μm and arc length of 42 μm. When fitted to a single exponential function, the decay rate constant was 0.016 s−1. As expected, the signal decay due to membrane diffusion of the protein was size-dependent, as illustrated in Figure 2G, where the decay rate constants were derived from unbleached regions with varying widths (the arc length was kept constant at 42 μm). Note that the simulated time dependence of the fluorescence decay in Figure 2F is similar to the experimental data in Figure 2D, and the rate constant is very close to 0.017 s−1, obtained in the experiments with 8-μm unbleached areas. Thus, this rate was used to correct (by subtraction) the dissociation rate constants of Rac, obtained under the same experimental conditions.
Role of GAP Activity
To what extent GTP-Rac on the membrane must interact with a GAP and convert to GDP-Rac before dissociation from the membrane is currently unclear. GTP-Rac can also be present in the cytoplasm bound to RhoGDI; thus, it must also dissociate from the membrane at some rate. To examine the role of GAP activity in membrane dissociation, we examined G12VRac, which lacks both intrinsic and GAP-stimulated GTPase activity. Comparison of the membrane dissociation kinetics of wt versus G12VRac would provide a measure of the role of GAPs in this process. NIH3T3 cells expressing similar amounts of GFP-Rac (wt or G12V) were analyzed as described above, where the unbleached area was 8 μm. Figure 4, A and B, shows that the fluorescence decay in the unbleached area was markedly slower for G12V than wt Rac. The apparent dissociation rate constant was about threefold lower (0.066 s−1 for wt vs. 0.02 s−1 for G12VRac). Importantly, r values were significantly higher for G12VRac (Figure 4D), which paralleled increased membrane targeting by biochemical fractionation (Figure 3) and higher activity in pulldown assays (Supplementary Figure 4). When the apparent rate constants were corrected for membrane diffusion by subtracting 0.017 s−1, dissociation of G12VRac was ∼10-fold slower than wt (0.004 and 0.048 s−1, respectively; Figure 4C). Thus, under these experimental conditions, conversion of GTP- to GDP-Rac through GAP activity is the major pathway for dissociation of Rac from the membrane.
Figure 4.

Role of GAPs and GEFs on Rac–membrane dissociation. (A) Fluorescence decay in the unbleached region of representative individual cells expressing GFP-wtRac, alone or coexpressed with active Tiam1, or GFP-G12VRac. Lines are the best-fit curves to Eq (4). The width of the unbleached region was 8 μm. (B) Best-fit curves to Eq (4) averaged from multiple cells as in A. Values are means ± SD (n = 11–13). (C–D) Average koff (after correcting for membrane diffusion) and r values in protrusive areas of cells expressing GFP-wtRac (n = 11), GFP-wtRac and active Tiam1 (n = 13) or GFP-G12VRac (n = 12). Error bars: SD (∗p <; 0.05, ∗∗p <; 0.01 by Student's t test). (E) koff for GFP-wtRac as a function of RFP-Q61LRac (filled circles) determined from total RFP intensity (arbitrary units). Open circle represents the mean koff ± SD of GFP-wtRac alone as in C. (F) Cytosolic fractions of cells transfected with GFP-wtRac, GFP-G12VRac, GFP-Q61LRac or empty vector were subjected to immunoprecipitation with anti-GFP and immune complexes were resolved by SDS-PAGE and blotted for GFP and RhoGDI. Equal loading was verified by immunoblotting the cytosolic fractions (Total) for RhoGDI. Data are representative of three independent experiments.
Role of GEFs
GEFs mediate activation and membrane targeting of Rho GTPases, but a role in membrane dissociation has not been suspected. To test this component of the standard paradigm, GFP-wtRac was coexpressed with constitutively active Tiam1 (C1199). Biochemical fractionation showed that Tiam1 increased Rac membrane targeting by about twofold (Figure 3). In addition, Tiam1 elevated the activity of GFP-wtRac (Supplementary Figure 4). Surprisingly, when analyzed by the FLIP protocol, the dissociation rate for GFP-wtRac was significantly slower in the presence of active Tiam1 (0.028 s−1 after correction for membrane diffusion, compared with 0.048 s−1 for wt; Figure 4, A–C). As expected, Tiam1 also increased r, consistent with increased membrane targeting (Figure 4D). Thus, GEF overexpression appears to promote membrane targeting of Rac to some extent by inhibition of membrane dissociation.
One mechanism by which Tiam1 could inhibit Rac–membrane dissociation is through activating Rac downstream signals. For example, the Rac effector PAK1 phosphorylates RhoGDI to attenuate its binding to Rac and increase Rac membrane targeting (DerMardirossian et al., 2004). To test this hypothesis, we coexpressed GFP-wtRac with constitutively active Q61L Rac fused to red fluorescent protein (mRFP-Q61LRac). We reasoned that Q61LRac should increase downstream Rac signaling through a different mechanism. The Q61L mutation has been shown to abolish binding of Cdc42 to RhoGDI (Michaelson et al., 2001); coimmunoprecipitation experiments show that GFP-Q61LRac is also unable to associate with RhoGDI (Figure 4F). Therefore, this mutant was used because it cannot compete with wtRac for RhoGDI. mRFP-Q61LRac resulted in extensive lamellipodia all over the cell periphery in all cells tested (unpublished data). However, mRFP-Q61LRac, even at high levels, did not alter the dissociation rate of GFP-wtRac (Figure 4E). Therefore, the effect of Tiam1 on Rac dissociation rate is not due to elevated Rac signaling.
RhoGDI Overexpression
Several studies have shown that RhoGDI can extract Rho GTPases from membranes in vitro (Olofsson, 1999), but the role of RhoGDI in the dynamics of membrane/Rho GTPase interactions in vivo has not been addressed directly. RhoGDI appears to be the major GDI family member in NIH 3T3 cells (RhoGDI2 protein level was 1–2% of RhoGDI, using bacterially expressed and purified GDI1/2 proteins as standards; unpublished data). Thus, we focused on RhoGDI1 for these studies. Endogenous RhoGDI was in about threefold molar excess over transfected GFP-Rac, as quantified against purified, recombinant Rac and RhoGDI proteins (unpublished data).
To test the role of RhoGDI in Rac–membrane dissociation in vivo, FLIP measurements were made in cells coexpressing GFP-Rac with different mRFP-RhoGDI levels. High levels of RhoGDI expression led to cell rounding and detachment from the substratum (unpublished data), consistent with its ability to potently inhibit Rho family GTPases. These cells could not be analyzed using the photobleaching protocol; thus, we examined low to moderate expressors. Higher levels of RhoGDI, as monitored by mRFP fluorescence intensity, correlated with decreased r values (Figure 5B), indicating a decrease in membrane-bound Rac, as expected. However, no significant change was observed in the dissociation rate or activation of Rac (Figure 5A and Supplementary Figure 4). To test the role of nucleotide in this process, we also examined G12VRac. We found that for G12VRac, coexpression of mRFP-RhoGDI had a smaller effect on r and no significant effect on koff (Figure 5, A and B). Thus, the ability of RhoGDI to inhibit membrane targeting of Rac is influenced by nucleotide loading. Biochemical fractionation (Figure 3) confirmed that mRFP-RhoGDI strongly inhibited membrane targeting of wt Rac (by 9-fold) and also inhibited G12VRac though less dramatically (3-fold). The results from fractionation are more drastic than photobleaching, most likely because high expressing, rounded cells were not excluded. Overall, the result that RhoGDI overexpression strongly affects membrane targeting but has little effect on membrane off rate suggests that the major effect must be to inhibit membrane on rate.
Figure 5.

Role of RhoGDI in Rac–membrane dissociation. (A and B) koff values (A) and r values (B) of GFP-wtRac (•) or GFP-G12VRac (○) with increasing RFP-RhoGDI as determined by total RFP intensity. ▴ and ▵, the mean koff and r values ± SD of GFP-wtRac and GFP-G12VRac alone, respectively, as in Figure 4, C and D. (C) Cells were electroporated with pSUPER RNAi constructs for RhoGDI or control sequences. Total lysates were analyzed by immunoblotting for RhoGDI and RhoGDI2. The antibody to RhoGDI2 cross-reacts with RhoGDI, but RhoGDI2 runs at a lower position on the gel (arrow). Equal loading was verified by immunoblotting with anti-actin. Data are representative of at least three independent experiments. (D) Average best-fit curves to Equation (4) from cells expressing GFP-wtRac in the presence (n = 8) or absence (n = 11) of RhoGDI RNAi. Values are means ± SD. (E and F) koff (E) and r (F) values in protrusive areas of cells expressing GFP-wtRac alone (n = 11) or GFP-wtRac in the presence of RhoGDI RNAi (n = 8). Values are means ± SD (∗p <; 0.05 by Student's t test).
RhoGDI Knockdown
To further investigate the role of RhoGDI in Rac–membrane dissociation, we carried out shRNA-mediated knockdown of RhoGDI. Electroporation of a vector encoding RNAi for mouse RhoGDI resulted in 95% decrease in RhoGDI levels at 72 h after electroporation, whereas RhoGDI2 expression was unchanged (Figure 5C). Given the threefold molar excess of endogenous RhoGDI over transfected GFP-Rac in control experiments, RhoGDI levels should become a limiting factor on RNAi. Down-regulation of RhoGDI increased membrane targeting of wtRac, as indicated by both higher r values in FLIP measurements and by biochemical fractionation, where the membrane fraction increased by about twofold (Figures 5E and 3, respectively). However, RhoGDI knockdown decreased the rate of dissociation of wtRac from the membrane by only ∼20% (Figure 5, D and E). No significant change in Rac activity was observed (Supplementary Figure 4). These results confirm the data from RhoGDI overexpression that RhoGDI levels must affect mainly the association of Rac with the membrane.
DISCUSSION
Rho GTPases undergo a cycle of activation/inactivation and a cycle of exchange between membranes and cytosol, but how these two regulatory cycles are coupled and how GEFs, GAPs, and GDIs affect each step are not well understood. We therefore developed a photobleaching protocol to measure in vivo dissociation rates of Rac and investigated the relative role of GAPs, GEFs, and RhoGDI in this process. We found that the membrane off rate for wtRac is 0.048 s−1, corresponding to a half-time of ∼14 s. This rate is about what might be expected for a regulator of membrane protrusions that are mobile on this time scale (Giannone et al., 2004). These experiments focused on protrusive areas of cells spreading on fibronectin. Because signaling at lamellipodia of spreading cells resembles in many respects the leading edge of migrating cells, our findings are likely to be relevant to Rac dynamics during cell migration. These dissociation rates are much faster, however, than persistence times for migrating cells (i.e., tens of minutes; Gu et al., 1999), indicating that the pathways that determine local Rac activation and deactivation during migration must operate on slower time scales.
Second, the data also show that GAP-dependent conversion to GDP-Rac is the rate-limiting step in the dissociation reaction of Rac under the experimental conditions used here. koff for GTPase-defective G12VRac was decreased by an order of magnitude. These results are in accordance with several in vitro studies showing that RhoGDI more efficiently extracts the GDP-bound form of Rho GTPases from the membrane (Olofsson, 1999). This difference may be because GTP-Rac binds less well to GDI, or because, in vivo, GTP-Rac interacts with effector proteins that limit its access to GDI. The latter is based on both structural studies showing that effector interaction sites on Rho GTPases overlap with GDI-binding sites (Abdul-Manan et al., 1999; Mott et al., 1999; Hoffman et al., 2000; Scheffzek et al., 2000; Grizot et al., 2001) and direct measurements (Del Pozo et al., 2002). Although dissociation of G12VRac at membrane protrusions is substantially slower, the rate is not negligible (half-time 173 s), consistent with the substantial amount of cytosolic G12VRac complexed with RhoGDI (Figure 4F and Del Pozo et al., 2002). It is also possible that active Rac could exchange faster between membrane and cytosol in other cellular compartments, such as the endomembranes of the endoplasmic reticulum (ER) and Golgi (Choy et al., 1999; Michaelson et al., 2001) or internalized micropinosomes (Schlunck et al., 2004).
GEFs mediate activation of Rac and translocation to the membrane (Bokoch et al., 1994). Indeed, membrane partitioning and activation of wtRac were elevated by active Tiam1 in our system. However, our results showed that expression of Tiam1 unexpectedly slowed the dissociation of Rac from the membrane. Analysis of GFP-wtRac coexpressed with Q61L-Rac showed no effect on dissociation rate, ruling out the possibility that the effect of Tiam1 was due to stimulation of downstream Rac signaling. The most likely mechanism for this effect is that Tiam1 competes with the GAP-RhoGDI pathway by converting GDP-Rac at the membrane to GTP-Rac before its dissociation.
In our system, RhoGDI overexpression reduced and RhoGDI down-regulation increased membrane targeting of Rac without affecting Rac activity. This finding is consistent with Rac activation being separable from membrane targeting (Del Pozo et al., 2002). Significantly, changing RhoGDI levels over this range had only slight effects on the dissociation of Rac from the membrane. These results therefore lead to the conclusion that, from a kinetic perspective, the major effect of RhoGDI is on the rate of association of Rac with the membrane. This result would appear to disagree with in vitro studies showing that RhoGDI can extract RhoGTPases from membranes (Olofsson, 1999). It may be that these in vitro systems do not accurately recapitulate conditions in vivo. Alternatively, it may be that, although equimolar RhoGDI is required for dissociation from the membrane, it is normally present in some excess and in this concentration range has effects mainly on association. It may also be that different membrane compartments in vivo have differential sensitivity to the GTPase-dissociating function of RhoGDI. For example, impairment of RhoGDI binding results in perinuclear but not plasma membrane accumulation of Cdc42 (Lin et al., 2003), pointing to distinct Cdc42 dynamics in these compartments, despite the otherwise uniform distribution of RhoGDI. Finally, the data suggest that, because there was still a pool of cytoplasmic Rac in cells subjected to nearly complete knockdown of RhoGDI, there must be other GDI activities present in 3T3 cells. RhoGDI2 expression appears to be too low to account for much of this activity, and RhoGDI3 is reported to have distinct GTPase specificity (Brunet et al., 2002), but several other proteins have been reported to show GDI-like activity (Anastasiadis et al., 2000; Degani et al., 2002). Sorting out these issues will be an interesting direction for future work.
Though contrary to paradigm, the concept that RhoGDI controls Rac–membrane association is consistent with several recent studies. GDI displacement factors (GDFs), including proteins and lipids, have been proposed to partially dissociate the Rho GTPase/GDI complex at a step before membrane targeting (Bourmeyster et al., 1992; Chuang et al., 1993a; Takahashi et al., 1997; Faure et al., 1999; Kim et al., 2002; Yamashita and Tohyama, 2003). Among them, members of the ezrin/radixin/moesin (ERM) family of proteins and the neurotrophin receptor (p75) were also shown to interact with RhoGDI. We speculate that, to the extent that GDFs can interact with free RhoGDI as well as the GDI-Rac complex, free RhoGDI could antagonize the function of GDFs during GTPase membrane targeting. Additionally, phosphorylation of RhoGDI by PAK was found to stimulate Rac activation and membrane targeting, which could be antagonized by high RhoGDI levels. Taken together, these data suggest that the activation and membrane targeting of Rac is more complex than previously thought and may involve concerted events in which GEFs, the Rac-GDI complex and perhaps other factors participate.
Supplementary Material
ACKNOWLEDGMENTS
We thank L. G. Berthiaume, R. Y. Tsien, J. G. Collard, C. J. Der, and A. L. Wilson-Delfosse for providing DNA plasmids. We also thank I. Novak and L. M. Loew for helpful discussions of modeling aspects. This work was supported by National Institutes of Health (NIH) Grants R01 GM47214 and U54 GM64346 to M.A.S. B.M.S. acknowledges support of NIH through Grants P41 RR13186 and U54 GM64346.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E06-01-0005) on April 5, 2006.
 The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
REFERENCES
- Abdul-Manan N., Aghazadeh B., Liu G. A., Majumdar A., Ouerfelli O., Siminovitch K. A., Rosen M. K. Structure of Cdc42 in complex with the GTPase-binding domain of the 'Wiskott-Aldrich syndrome' protein. Nature. 1999;399:379–383. doi: 10.1038/20726. [DOI] [PubMed] [Google Scholar]
- Alland L., Peseckis S. M., Atherton R. E., Berthiaume L., Resh M. D. Dual myristylation and palmitylation of Src family member p59fyn affects subcellular localization. J. Biol. Chem. 1994;269:16701–16705. [PubMed] [Google Scholar]
- Anastasiadis P. Z., Moon S. Y., Thoreson M. A., Mariner D. J., Crawford H. C., Zheng Y., Reynolds A. B. Inhibition of RhoA by p120 catenin. Nat. Cell Biol. 2000;2:637–644. doi: 10.1038/35023588. [DOI] [PubMed] [Google Scholar]
- Bokoch G. M., Bohl B. P., Chuang T. H. Guanine nucleotide exchange regulates membrane translocation of Rac/Rho GTP-binding proteins. J. Biol. Chem. 1994;269:31674–31679. [PubMed] [Google Scholar]
- Bourmeyster N., Stasia M. J., Garin J., Gagnon J., Boquet P., Vignais P. V. Copurification of rho protein and the rho-GDP dissociation inhibitor from bovine neutrophil cytosol. Effect of phosphoinositides on rho ADP-ribosylation by the C3 exoenzyme of Clostridium botulinum. Biochemistry. 1992;31:12863–12869. doi: 10.1021/bi00166a022. [DOI] [PubMed] [Google Scholar]
- Brunet N., Morin A., Olofsson B. RhoGDI-3 regulates RhoG and targets this protein to the Golgi complex through its unique N-terminal domain. Traffic. 2002;3:342–357. doi: 10.1034/j.1600-0854.2002.30504.x. [DOI] [PubMed] [Google Scholar]
- Choy E., Chiu V. K., Silletti J., Feoktistov M., Morimoto T., Michaelson D., Ivanov I. E., Philips M. R. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 1999;98:69–80. doi: 10.1016/S0092-8674(00)80607-8. [DOI] [PubMed] [Google Scholar]
- Chuang T. H., Bohl B. P., Bokoch G. M. Biologically active lipids are regulators of RacGDI complexation. J. Biol. Chem. 1993a;268:26206–26211. [PubMed] [Google Scholar]
- Chuang T. H., Xu X., Knaus U. G., Hart M. J., Bokoch G. M. GDP dissociation inhibitor prevents intrinsic and GTPase activating protein-stimulated GTP hydrolysis by the Rac GTP-binding protein. J. Biol. Chem. 1993b;268:775–778. [PubMed] [Google Scholar]
- Cowan A. E., Nakhimovsky L., Myles D. G., Koppel D. E. Barriers to diffusion of plasma membrane proteins form early during guinea pig spermiogenesis. Biophys. J. 1997;73:507–516. doi: 10.1016/S0006-3495(97)78089-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degani S., Balzac F., Brancaccio M., Guazzone S., Retta S. F., Silengo L., Eva A., Tarone G. The integrin cytoplasmic domain-associated protein ICAP-1 binds and regulates Rho family GTPases during cell spreading. J. Cell Biol. 2002;156:377–387. doi: 10.1083/jcb.200108030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pozo M. A., Alderson N. B., Kiosses W. B., Chiang H. H., Anderson R. G., Schwartz M. A. Integrins regulate Rac targeting by internalization of membrane domains. Science. 2004;303:839–842. doi: 10.1126/science.1092571. [DOI] [PubMed] [Google Scholar]
- Del Pozo M. A., Kiosses W. B., Alderson N. B., Meller N., Hahn K. M., Schwartz M. A. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat. Cell Biol. 2002;4:232–239. doi: 10.1038/ncb759. [DOI] [PubMed] [Google Scholar]
- del Pozo M. A., Price L. S., Alderson N. B., Ren X. D., Schwartz M. A. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J. 2000;19:2008–2014. doi: 10.1093/emboj/19.9.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DerMardirossian C., Schnelzer A., Bokoch G. M. Phosphorylation of RhoGDI by Pak1 mediates dissociation of Rac GTPase. Mol. Cell. 2004;15:117–127. doi: 10.1016/j.molcel.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Faure J., Vignais P. V., Dagher M. C. Phosphoinositide-dependent activation of Rho A involves partial opening of the RhoA/Rho-GDI complex. Eur. J. Biochem. 1999;262:879–889. doi: 10.1046/j.1432-1327.1999.00458.x. [DOI] [PubMed] [Google Scholar]
- Giannone G., Dubin-Thaler B. J., Dobereiner H. G., Kieffer N., Bresnick A. R., Sheetz M. P. Periodic lamellipodial contractions correlate with rearward actin waves. Cell. 2004;116:431–443. doi: 10.1016/s0092-8674(04)00058-3. [DOI] [PubMed] [Google Scholar]
- Grizot S., Faure J., Fieschi F., Vignais P. V., Dagher M. C., Pebay-Peyroula E. Crystal structure of the Rac1-RhoGDI complex involved in nadph oxidase activation. Biochemistry. 2001;40:10007–10013. doi: 10.1021/bi010288k. [DOI] [PubMed] [Google Scholar]
- Gu J., Tamura M., Pankov R., Danen E. H., Takino T., Matsumoto K., Yamada K. M. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J. Cell Biol. 1999;146:389–403. doi: 10.1083/jcb.146.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman G. R., Nassar N., Cerione R. A. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell. 2000;100:345–356. doi: 10.1016/s0092-8674(00)80670-4. [DOI] [PubMed] [Google Scholar]
- Kim O., Yang J., Qiu Y. Selective activation of small GTPase RhoA by tyrosine kinase Etk through its pleckstrin homology domain. J. Biol. Chem. 2002;277:30066–30071. doi: 10.1074/jbc.M201713200. [DOI] [PubMed] [Google Scholar]
- Lin Q., Fuji R. N., Yang W., Cerione R. A. RhoGDI is required for Cdc42-mediated cellular transformation. Curr. Biol. 2003;13:1469–1479. doi: 10.1016/s0960-9822(03)00613-4. [DOI] [PubMed] [Google Scholar]
- McCabe J. B., Berthiaume L. G. Functional roles for fatty acylated amino-terminal domains in subcellular localization. Mol. Biol. Cell. 1999;10:3771–3786. doi: 10.1091/mbc.10.11.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe J. B., Berthiaume L. G. N-terminal protein acylation confers localization to cholesterol, sphingolipid-enriched membranes but not to lipid rafts/caveolae. Mol. Biol. Cell. 2001;12:3601–3617. doi: 10.1091/mbc.12.11.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson D., Silletti J., Murphy G., D'Eustachio P., Rush M., Philips M. R. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 2001;152:111–126. doi: 10.1083/jcb.152.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott H. R., Owen D., Nietlispach D., Lowe P. N., Manser E., Lim L., Laue E. D. Structure of the small G protein Cdc42 bound to the GTPase-binding domain of ACK. Nature. 1999;399:384–388. doi: 10.1038/20732. [DOI] [PubMed] [Google Scholar]
- Nomanbhoy T. K., Cerione R. Characterization of the interaction between RhoGDI and Cdc42Hs using fluorescence spectroscopy. J. Biol. Chem. 1996;271:10004–10009. doi: 10.1074/jbc.271.17.10004. [DOI] [PubMed] [Google Scholar]
- Olofsson B. Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell Signal. 1999;11:545–554. doi: 10.1016/s0898-6568(98)00063-1. [DOI] [PubMed] [Google Scholar]
- Price L. S., Leng J., Schwartz M. A., Bokoch G. M. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell. 1998;9:1863–1871. doi: 10.1091/mbc.9.7.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe K., Otto-Bruc A., Chardin P., Antonny B. Dissociation of GDP dissociation inhibitor and membrane translocation are required for efficient activation of Rac by the Dbl homology-pleckstrin homology region of Tiam. J. Biol. Chem. 2003;278:4756–4762. doi: 10.1074/jbc.M210412200. [DOI] [PubMed] [Google Scholar]
- Scheffzek K., Stephan I., Jensen O. N., Illenberger D., Gierschik P. The Rac-RhoGDI complex and the structural basis for the regulation of Rho proteins by RhoGDI. Nat. Struct. Biol. 2000;7:122–126. doi: 10.1038/72392. [DOI] [PubMed] [Google Scholar]
- Schlunck G., Damke H., Kiosses W. B., Rusk N., Symons M. H., Waterman-Storer C. M., Schmid S. L., Schwartz M. A. Modulation of Rac localization and function by dynamin. Mol. Biol. Cell. 2004;15:256–267. doi: 10.1091/mbc.E03-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepchenko B. M., Schaff J. C., Macara I., Loew L. M. Quantitative cell biology with the Virtual Cell. Trends Cell Biol. 2003;13:570–576. doi: 10.1016/j.tcb.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Sasaki T., Mammoto A., Takaishi K., Kameyama T., Tsukita S., Takai Y. Direct interaction of the Rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the Rho small G protein. J. Biol. Chem. 1997;272:23371–23375. doi: 10.1074/jbc.272.37.23371. [DOI] [PubMed] [Google Scholar]
- Van Aelst L., D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Yaku H., Sasaki T., Takai Y. The Dbl oncogene product as a GDP/GTP exchange protein for the Rho family: its properties in comparison with those of Smg GDS. Biochem. Biophys. Res. Commun. 1994;198:811–817. doi: 10.1006/bbrc.1994.1116. [DOI] [PubMed] [Google Scholar]
- Yamashita T., Tohyama M. The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat. Neurosci. 2003;6:461–467. doi: 10.1038/nn1045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.