

Abstract
Many oxides tend to form different structures (polymorphs) for small particles. High temperature oxide melt solution calorimetry has been used to measure the enthalpy as a function of polymorphism and surface area for oxides of Al, Ti, and Zr. The results confirm crossovers in polymorph stability at the nanoscale. The energies of internal and external surfaces of zeolitic silicas with open framework structures are an order of magnitude smaller than those of oxides of normal density.
Introduction
Nanoscale phenomena are ubiquitous and important to geoscience. The realization is growing that many outcomes and rates of geochemical processes are governed by phenomena at the nanometer scale, often in or at the surface of small particles. Surface interactions probably exert a disproportionate influence on the structure, chemistry, and movement of natural and anthropogenic nanomaterials in the Earth. In many cases, traditional distinctions between solutes, colloids and solids become vague. Nanoparticles, with sizes in the 1–100 nm range, lie in the realm between dissolved molecular clusters and crystalline macroscopic solids. Their properties are different from those of either.
Fine grained oxides precipitated from aqueous solution often crystallize in structures different from those of coarsely crystalline materials: γ-alumina instead of corundum, anatase and brookite instead of rutile, maghemite instead of hematite, and a host of complex hydrous iron oxyhydroxides. It has been suspected that differences in surface energy stabilize, as nanoparticles, polymorphs that are metastable in the bulk.[1,2] Oxide melt solution calorimetry has recently been used to measure enthalpies of phase transformation and surface enthalpies of different oxide polymorphs.[3] Calorimetric measurements are generally confirming this hypothesis of energy crossover between polymorphs at the nanoscale. This review summarizes recent work in the UC Davis Thermochemistry Facility on the energetics of nanophase oxides. For a broader overview of nanoparticles and Earth processes, the recent review volume on nanoparticles in the environment[4] should be consulted.
Energetics of various oxides
Aluminum oxides and oxyhydroxides
Corundum, α-Al2O3, is the thermodynamically stable phase of coarsely crystalline aluminum oxide at standard temperature and pressure conditions, but syntheses of nanocrystalline A12O3 usually result in γ-Al2O3. Based on earlier molecular dynamics simulations and their own thermochemical data, McHale et al.[5] predicted that γ-Al2O3 should become the energetically stable polymorph for specific surface areas exceeding ~125 m2 g-1. The thermodynamic stability of γ-Al2O3 should be even greater than implied by this energy. Due to the presence of tetrahedral and octahedral sites in its spinel-type structure, and the fairly random distribution of A13+ and vacancies over these sites, γ-Al2O3 has a greater entropy than α-Al2O3. The entropy change of the α-Al2O3 to γ-Al2O3 transition, ΔSα→γ, is about +5.7 J K-1 mol-1.[6] Therefore, at room temperature, γ-Al2O3 could be thermodynamically stable with respect to α-Al2O3 at specific surface areas > 100 m2 g-1, and at 800 K (a temperature typical of oxyhydroxide decomposition) γ-Al2O3 might become thermodynamically stable at specific surface areas greater than only 75 m2 g-1.
McHale et al.[5] measured the enthalpies of drop solution in molten lead borate of several nanosized α- and γ-alumina samples. However, the surfaces of the A12O3 were modified by adsorbed H2O which could not be completely removed without severe coarsening. The surface energies of the hydrated polymorphs appeared nearly equal, indicating that the heat of chemisorption of H2O is directly proportional to the surface energy of the anhydrous phase. Consequently, McHale et al.[5] could not determine the anhydrous surface energies without accurate knowledge of the heats of chemisorption of H2O. These measurements were made on two samples each of α- and γ-Al2O3 with a Calvert type microcalorimeter operating near room temperature.[6] The differential heat of H2O adsorption on γ-Al2O3 decreases logarithmically with increasing coverage (Freundlich behavior). In contrast, the differential heat of H2O adsorption on α-Al2O3 does not show regular logarithmic decay, and decreases far less rapidly with increasing coverage. This indicates a greater number of high energy sites on α-Al2O3 per unit surface area, which are relaxed by the most strongly chemisorbed hydroxyls. This observation is strong evidence that the surface energy of α-Al2O3 is higher than that of γ-Al2O3.
A quantitative analysis of the heat of adsorption data enables the separation of hydration enthalpies and surface enthalpies for the two alumina polymorphs.[6] The resulting variation of enthalpy of the anhydrous material with surface area is shown in Fig. 1. The enthalpy (and free energy) crossover postulated above is clearly demonstrated. Table 1 lists the surface and transformation energies. Calorimetric studies of water adsorption on alumina[5,6] suggest that the higher energy surfaces have the strongest affinity for water, and that α-alumina has more strongly bonded H2O than γ-alumina.
Figure 1.
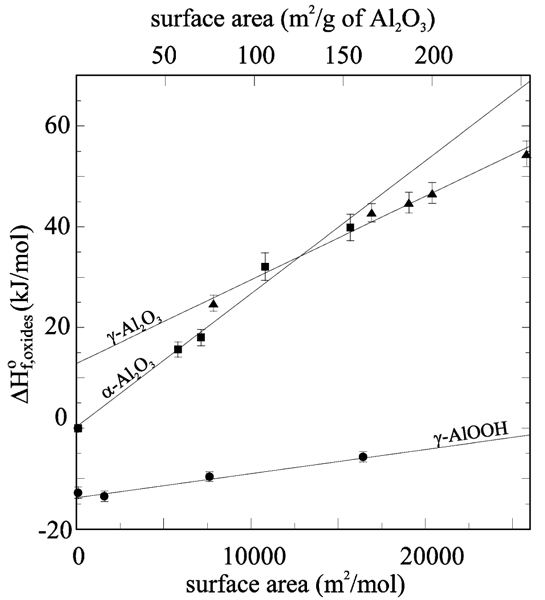
Enthalpy as a function of surface area for aluminum oxides and oxyhydroxides.[7]
Table 1.
Surface energies and transformation energies relative to bulk stable polymorph for several oxides
| Oxide | Surface energy/J m-2 | Transformation energya/kJ mol-1 |
| α-Al2O3 | 2.6 ± 0.2 | 0 |
| γ-A1203 | 1.7 ± 0.1 | 13.4 ± 2.0 |
| A1OOH (boehmite) | 0.5 ± 0.1 | -17 ± 1 |
| TiO2 (rutile) | 2.2 ± 0.2 | 0 |
| TiO2 (brookite) | 1.0 ± 0.2 | 0.7 ± 0.4 |
| TiO2 (anatase) | 0.4 ± 0.1 | 2.6 ± 0.4 |
| ZrO2 (monoclinic) | 6.5 ± 0.2 | 0 |
| ZrO2 (tetragonal) | 2.1 ± 0.05 | 9.5 ± 0.4 |
| ZrO2 (amorphous) | 0.5 ± 0.05 | 34 ± 4 |
| Zeolitic silicas | 0.09 ± 0.01 | 7–15 |
a Enthalpy relative to stable bulk anhydrous polymorph plus H2O (liquid) if applicable.
Polymorphs of aluminum oxyhydroxide (boehmite, diaspore) and aluminum hydroxide (gibbsite, bayerite, nordstrandite) are common fine-grained constituents of soils, unconsolidated sediments, and sedimentary rocks. Their occurrence in nature is controlled by their thermodynamic properties and the kinetics involved in transformations. A recent study of the surface enthalpy of boehmite[7] was prompted by an apparent contradiction between common occurrence of boehmite and gibbsite and the thermodynamic stability of diaspore. Furthermore, fine-grained boehmite is also an important industrial product used because of its relative ease of synthesis and its ability to retain high surface area.
The results of calorimetric studies are shown in Table 1 and Fig. 1. The data clearly show that surface enthalpy is lowest for boehmite, intermediate for γ-alumina, and highest for corundum. In a first approximation, the surface energy of an ionic solid will be proportional to the charges of the ions within the structure. Thus, replacing the oxygen anions with hydroxyls will lower the surface energy, in agreement with experiment. Furthermore, the hydroxyls offer a termination at the crystal surface that is compatible with an aqueous environment.
Titania
Energy crossovers have also been confirmed for the rutile, anatase, and brookite forms of titanium dioxide.[8] Relative to bulk rutile, bulk brookite is 0.71 ± 0.38 kJ mol-1 and bulk anatase is 2.61 ± 0.41 kJ mol-1 higher in enthalpy. The surface enthalpies of rutile, brookite, and anatase are 2.2 ± 0.2 J m-2, 1.0 ± 0.2 J m-2 and 0.4 ± 0.1 J m-2, respectively. The closely balanced energetics (see Table 1) directly confirm the crossover in stability of nanophase TiO2 polymorphs inferred by Zhang and Banfield.[2] The heavy lines in Fig. 2 show the energetically stable phases as a function of surface area. Depending on the competition between coarsening and phase transformation kinetics; the transition anatase-to-brookite or the transition brookite-to-anatase can lower the enthalpy and free energy of the system. Thus the observation of both transitions in heated samples is not a violation of thermodynamic principles. The final stable phase upon grain growth is always rutile.
Figure 2.
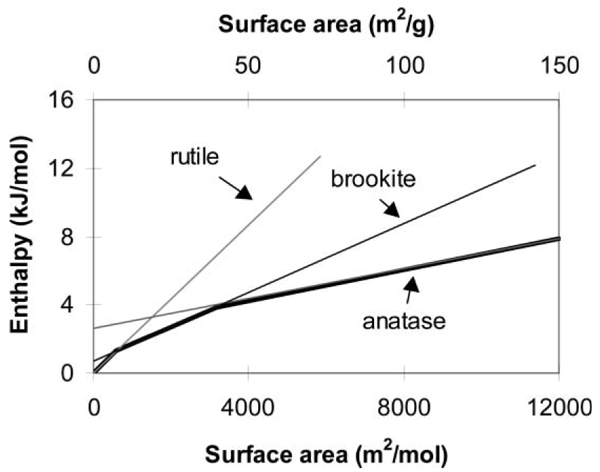
Enthalpy relative to bulk rutile for rutile, anatase, and brookite of various surface area. Figure modified from Ranade et al.[8] The slope of each line gives the surface energy, see Table 1.
Zirconia
Fig. 3 shows the energetics, as measured by oxide melt solution calorimetry, of monoclinic, tetragonal, and amorphous zirconia.[9] Once more, clear enthalpy crossovers are seen, with surface energy decreasing in the order monoclinic, tetragonal, amorphous. These crossovers correspond well to observation that monoclinic zirconia, the stable bulk polymorph, is readily synthesized with particle size greater than 50 nm, tetragonal zirconia is synthesized with intermediate particle size, and the amorphous material exists for particles smaller than about 4–8 nm. Calorimetric data, Rietveld refinements of crystal structures, and Raman spectra all indicate that nanophase crystalline zirconia is tetragonal rather than cubic.
Figure 3.
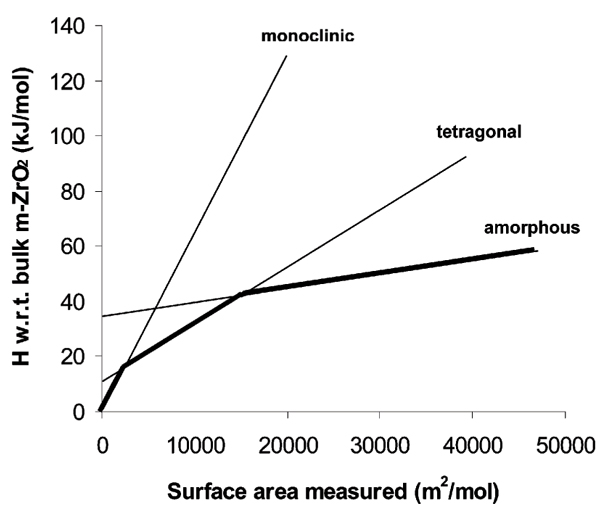
Enthalpy relative to bulk monoclinic zirconia (baddeleyite) for monoclinic, tetragonal, and amorphous ZrO2 of various surface areas (in preparation). The slope of each line gives the surface energy, see Table 1.
Iron and manganese oxides and oxyhydroxides
These are characterized by extensive polymorphism. Measurements of the enthalpies of several iron oxides and oxyhydroxides[10,11] complement earlier work on MnO2 phases.[11,12] Our focus thus far has been on polymorphism rather than on control of surface area; the latter studies are just beginning. What is noteworthy is that many polymorphs are only 5–10 kJ mol-1 higher in enthalpy than the stable well-crystallized forms, opening the door to crossovers in stability.
Silica
Silica shows extensive polymorphism. Phases having a reasonable degree of P–T stability are quartz, tridymite, cristobalite, coesite, stishovite, and melt. Phases which are probably always metastable, but can be synthesized from aqueous solutions, with or without inorganic or organic structure directing agents, include amorphous silica, moganite, clathrasils, and zeolitic and mesoporous materials. These very low density metastable materials have been studied extensively by oxide melt solution calorimetry. [13-16] They all lie in a narrow band or energies about 7–15 kJ mol-1 above quartz and overlapping amorphous silica.
In general, the less dense silicas have higher energies. Moloy et al.[17] have shown that the metastability of the silica polymorph (ΔHtr for the hypothetical transition quartz → less dense silica phase) correlates linearly with the internal surface area calculated by molecular modeling (see Fig. 4). The internal surface energy calculated from the slope of the line relating enthalpy to internal surface area[16] is 0.09 ± 0.01 J m-2, which is more similar to the external surface energy of amorphous silica than to that of quartz, the latter being significantly higher (see ref. 17 for discussion). Melanophlogite, a clathrasil with large but not interconnected pores falls on the same trend as the zeolitic silicas.[16]
Figure 4.
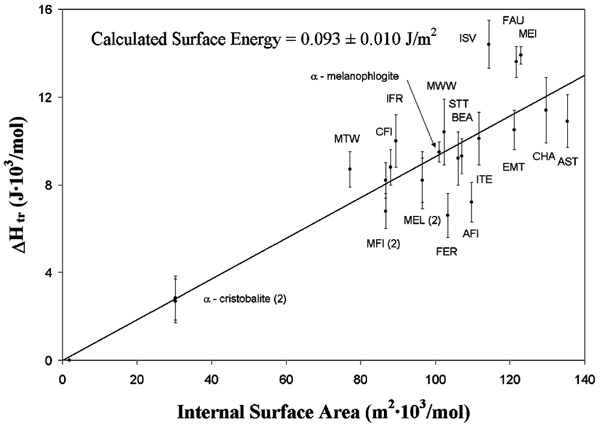
Correlation between enthalpy of less dense silica phases relative to quartz and calculated internal surface area.[15,16] Symbols are International Zeolite Association (IZA) codes for various zeolite frameworks.
One of these zeolitic silicas, silicalite-1, has been synthesized as nanocrystals with different sizes (40, 95 and 180 nm) and the organic structure directing agent removed by calcination. X-ray diffraction, SEM, nitrogen adsorption, FT IR and thermogravimetric analysis were used for characterization. Nitrogen adsorption of the zeolite containing the organic template gave the external surface area, while that of the calcined sample gave the total surface area. The internal surface area is then the total surface area minus the external surface area. High-temperature solution calorimetry was used to determine the enthalpies (ΔHtran) relative to quartz at 298 K. The ΔHtran values are 8.2 ± 0.4 kJ mol-1 (40 nm), 8.3 ± 0.3 kJ mol-1 (95 nm) and 8.0 ± 0.5 kJ mol-1 (180 nm).[18] These values are essentially the same as ΔHtran for microsized MFI crystals, 8.0 ± 0.8 kJ mol -1.[15] Thus, the enthalpy does not depend on the particle size, that is, the external surface area, at least for particles ≥ 40 nm. The ratio of internal surface area to external surface area is 4.1 (40 nm); 10.6 (95 nm), and 18.1 (180 nm), indicating the internal surface area dominates the external and total surface area even for nanosized zeolites. From the viewpoint of energetics, the large internal surface area of zeolite requires a small surface energy to make the system energetically accessible. If the surface enthalpy calculated by Moloy et al.[17] reflects the energetics of both internal and external surfaces, the external surface energy of silica zeolites can be also estimated to be 0.09 ± 0.01 J m-2.
Discussion
Table 1 presents surface energies for various oxides. Compared to that for zeolites and amorphous silica, and even quartz (see ref. 17 for discussion), the surface energy is considerably larger for oxides which do not form framework structures with low density. For 40 nm α-Al2O3 (which is unstable relative to 40 nm γ-Al2O3) the excess enthalpy relative to bulk is as large as 10 kJ mol-1. Even for 40 nm brookite (the stable polymorph at the nanometer scale, the excess enthalpy is 1.2 kJ mol-1. For 40 nm silicalite-1, the surface area of 95 m2 g-1 yields an excess enthalpy relative to bulk of only 0.53 kJ mol-1, which is within the experimental error of ΔHtran. This indicates that the effect of particle size on energetics is too small to measure accurately by oxide melt solution calorimetry for silica zeolites, at least for ≥ 40 nm particles.
These observations lead to several general inferences. (1) The existence of a large number of polymorphs with microporous structures requires them to be similar in energy and not too much higher than dense frameworks. If the energy of internal and external surfaces is similar, this implies little dependence of energy on particle size for microporous materials in general. Because of the small surface energy, crossovers in stability of microporous materials as a function of particle size are unlikely, in contrast to the behavior of dense polymorphs. The thermodynamic driving force for particle coarsening of microporous materials is very small. This may explain why it is often difficult to grow large crystals of porous materials. If similar trends hold for other types of microporous materials, for example manganese oxides, it can be predicted that the enthalpy of formation of a nanosized microporous MnO2 polymorph will be similar to that of micrometer sized MnO2 having the same structure. Many natural manganese oxide minerals are nanosized and do not coarsen readily, consistent with the arguments above.
The data presented above have several geochemical implications. If hydrous phases like oxyhydroxides generally have smaller surface energies than their anhydrous counterparts, then small particle size will favor the hydrated forms. This has been discussed[7] for the dehydration of boehmite to corundum plus water. It is concluded that the P–T stability field of the hydrous assemblage can be extended significantly in nanophase materials, and that dehydration and coarsening often occur simultaneously. Thus, the seemingly "metastable" retention of boehmite and other oxyhydroxides may in fact be "stable" under the additional constraint of retaining small particle size.
The extensive polymorphism of open framework micro-porous structures like zeolites and manganese oxides may be linked to their small internal and external surface energies as discussed above. Their tendency to remain fine-grained, and to produce intergrowths of different structures at the unit cell scale (as seen by TEM) may reflect not just the small energy differences between polymorphs, but small surface and interfacial energies as well.
The regime explored here shows a linear dependence between enthalpy and surface area, i.e., a constant surface energy (or enthalpy). As particles get even smaller, one may expect this linearity to break down. The transition from crystalline nanoparticle to amorphous nanoparticle to cluster needs further exploration of both structure and energetics. The current calorimetric data suggest that the amorphous phase generally has a smaller surface energy than the crystalline polymorphs. Is this a general feature?
Nanoparticles in the aqueous environment have hydrated surfaces. The enthalpy of hydration has been explored for alumina[5,6] but not for other oxides. Further work by adsorption and immersion calorimetry is planned. The enthalpy of adsorption of other inorganic ions and organic molecules on nanoparticles can also be studied by calorimetry. Such adsorption is expected to be sensitive to polymorphism and, for particles below 10 nm or so in size, new phenomena, reflecting the breakdown of the assumption of constant surface energies, may come into play.[19]
Note
† Presented at the ACS Division of Geochemistry Symposium 'The Impact of Nanoparticle Growth and Transformation Processes on Contaminant Geochemical Cycling', New Orleans, March 2003.
Acknowledgments
Acknowledgements
This work, performed over the past seven years, received funding from a series of NSF and DOE grants.
References
- Garvie RC. The occurrence of metastable tetragonal zirconia as a crystallite size effect. J Phys Chem. 1965;69:1238–1243. [Google Scholar]
- Zhang H, Banfield JF. Thermodynamic analysis of phase stability of nanocrystalline titania. J Mater Chem. 1998;8:2073–2076. doi: 10.1039/a802619j. [DOI] [Google Scholar]
- Navrotsky A. Nanoparticles and the Environment: Thermochemistry of Nanomaterials. Rev Mineral Geochem. 2001;44:73–103. [Google Scholar]
- Banfield JF, Navrotsky A, editor. Rev Mineral Geochem. Vol. 44. 2001. Nanoparticles and the Environment; p. 397. [Google Scholar]
- McHale JM, Navrotsky A, Perrotta AJ. Effects of Increased Surface Area and Chemisorbed H2O on the Relative Stability of Nanocrystalline γ-Al2O3 and α-Al2O3. J Phys Chem B. 1997;101:603–613. doi: 10.1021/jp9627584. [DOI] [Google Scholar]
- McHale JM, Auroux A, Perrotta AJ, Navrotsky A. Surface Energies and Thermodynamic Phase Stability in Nanocrystalline Aluminas. Science. 1997;277:788–791. doi: 10.1126/science.277.5327.788. [DOI] [Google Scholar]
- Majzlan J, Navrotsky A, Casey WH. Surface Enthalpy of Boehmite. Clays Clay Miner. 2000;48:699–707. [Google Scholar]
- Ranade MR, Navrotsky A, Zhang HZ, Banfield JF, Elder SH, Zaban A, Borse PH, Kulkarni SK, Doran GS, Whitfield HJ. Energetics of Nanocrystalline TiO2. Proc Natl Acad Sci. 2002;99:6476–6481. doi: 10.1073/pnas.251534898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher MW, Ushakov SV, Navrotsky A, Woodfield BF, Li G, Boerio-Goates J, Tissue BM. Energy Crossovers in Nanocrystalline Zirconia. J Am Ceram Soc. 2003. submitted.
- Laberty C, Navrotsky A. Energetics of Stable and Metastable Low-Temperature Iron Oxides and Oxyhydroxides. Geochim Cosmochim Acta. 1998;62:2905–2913. doi: 10.1016/S0016-7037(98)00208-7. [DOI] [Google Scholar]
- Majzlan J, Grevel KD, Navrotsky A. Thermodynamics of Iron Oxides: Part II. Enthalpies of Formation and Relative Stability of Goethite (α-FeOOH), Lepidocrocite (γ-FeOOH), and Maghemite (γ-Fe2O3) Am Mineral. 2003;88:855–859. [Google Scholar]
- Fritsch S, Post JE, Navrotsky A. Energetics of Low-Temperature Polymorphs of Manganese Dioxide and Oxyhydroxide. Geochim Cosmochim Acta. 1997;61:2613–2616. doi: 10.1016/S0016-7037(97)00121-X. [DOI] [Google Scholar]
- Petrovic I, Navrotsky A, Davis ME, Zones SI. Thermochemical Study of the Stability of Frameworks in High Silica Zeolites. Chem Mater. 1993;5:1805–1813. doi: 10.1021/cm00036a019. [DOI] [Google Scholar]
- Petrovic I, Navrotsky A, Chen C-Y, Davis ME. State of the Art 1994: High Temperature Calorimetry of MCM-41. In: Weitkamp J, Karge HG, Pfeifer H, Holderich W, editor. Zeolites Relat Microporous Mater: Proc Int Zeolite Conf 10th 1994 Parts A-C. 1994. pp. 677–684. [Google Scholar]
- Piccione PM, Laberty C, Yang S, Camblor MA, Navrotsky A, Davis ME. Thermochemistry of Pure-Silica Zeolites. J Phys Chem B. 2000;104:10001–10011. doi: 10.1021/jp002148a. [DOI] [Google Scholar]
- Navrotsky A, Xu H, Moloy EC, Welch MD. Thermochemistry of Guest-free Melanophiogite. Am Mineral. 2003;88:1212–1614. [Google Scholar]
- Moloy EC, Davila LP, Shackelford JF, Navrotsky A. High-Silica Zeolites: A Relationship Between Energetics and Internal Surface Area. Micropor Mesopor Mater. 2002;54:1–13. doi: 10.1016/S1387-1811(02)00328-1. [DOI] [Google Scholar]
- Li QH, Yang SY, Navrotsky A. Energetics of a Nanophase Zeolite Independent of Particle Size. Micropor Mesopor Mater. 2003;65:137–143. doi: 10.1016/S1387-1811(03)00480-3. [DOI] [Google Scholar]
- Zhang H, Penn RL, Hamers RJ, Banfield JF. Enhanced adsorption of molecules on surfaces of nanocrystalline particles. J Phys Chem B. 1999;103:4656–4662. doi: 10.1021/jp984574q. [DOI] [Google Scholar]