

Abstract
Major-histocompatibility-complex (MHC) proteins are used to display, on the surface of a cell, peptides derived from foreign material — such as a virus — that is infecting that cell. Cytotoxic T lymphocytes then recognize and kill the infected cell. The HIV-1 Nef protein downregulates the cell-surface expression of class I MHC proteins, and probably thereby promotes immune evasion by HIV-1. In the presence of Nef, class I MHC molecules are relocalized from the cell surface to the trans-Golgi network (TGN) through as-yet-unknown mechanisms. Here we show that Nef-induced downregulation of MHC-I expression and MHC-I targeting to the TGN require the binding of Nef to PACS-1, a molecule that controls the TGN localization of the cellular protein furin. This interaction is dependent on Nef's cluster of acidic amino acids. A chimaeric integral membrane protein containing Nef as its cytoplasmic domain localizes to the TGN after internalization, in an acidic-cluster- and PACS-1-dependent manner. These results support a model in which Nef relocalizes MHC-I by acting as a connector between MHC-I's cytoplasmic tail and the PACS-1-dependent protein-sorting pathway.
The Nef protein of HIV-1 downregulates the cell-surface expression of MHC-I proteins1. It thereby decreases the recognition and killing of HIV-infected cells by virus-specific cytotoxic T lymphocytes (CTLs), thus promoting immune evasion2. In the presence of Nef, MHC-I is internalized from the cell surface by endocytosis and relocalized to the TGN1,3, through mechanisms that have yet to be elucidated. Mutagenesis studies have pointed to the importance of specific regions of Nef in its ability to regulate MHC-1. These include Nef's myristoylation signal, which is essential for its membrane association, an amino-terminal-proximal α-helix and a centrally located SH3 (Src-homology-domain 3)-binding proline-based repeat, and a highly conserved cluster of acidic residues in the N-terminal third of the viral protein (EEEE65)3. The acidic cluster of Nef is highly similar to the PACS-1-binding, TGN-retrieval motif of furin and the mannose-6-phosphate receptor (MPR)4. PACS-1 (phosphofurin acidic cluster sorting protein-1) is the first identified member of a new family of coat proteins — proteins that enable the formation of transport vesicles from membranes such as the plasma membrane or those surrounding organelles. PACS-1 controls the endosome-to-Golgi trafficking of furin and the MPR by connecting the acidic-cluster-containing cytoplasmic domain of these molecules with the adaptor-protein complex-1 (AP-1) of endosomal clathrin-coated membrane pits (clathrin is another coat protein that enables the formation first of membrane ‘pits’, or invaginations, and then vesicles)4.
The parallels that exist between the retrieval of furin to the TGN and the Nef-mediated targeting of MHC-I to this compartment, in particular the dependence of both phenomena on acidic clusters in the transported proteins, led us to ask whether PACS-1 might participate in Nef-induced MHC-I downmodulation. Our results reveal that PACS-1 is indeed the downstream partner of the viral protein in this process.
Results
PACS-1 is required for Nef-induced MHC-I downregulation and relocalization. The possible importance of PACS-1 in Nef-induced MHC-I downregulation was revealed in A7 melanoma cells rendered deficient for PACS-1 by an antisense strategy4. In these cells (AS19), levels of PACS-1 are reduced roughly eightfold (Fig. 1a), and the localization of furin and the MPR to the TGN is selectively disrupted, whereas γ-adaptin (the γ-chain of adaptor complexes) and TGN46 are still properly targeted to this compartment4. Although HIV-1 Nef efficiently reduced levels of cell-surface MHC-I in the parental cells, the receptor was markedly less affected by the viral protein in the PACS-1-antisense derivatives. Confirming Nef's dependence on PACS-1, we found that the effects of Nef were restored in AS19 cells transduced with a lentiviral vector expressing PACS-1b (Fig. 1b) (although PACS-1b is a subspecies of PACS-1 of low abundance that corresponds to just the first 542 residues of PACS-1a, both PACS-1a and PACS-1b function in rescue experiments). Nef downmodulated the cell-surface expression of transfected CD4 equally well in control and antisense cells (Fig. 1c), consistent with the previously observed acidic-cluster independence of this other effect of Nef3,5.
Figure 1.

PACS-1 is required for efficient MHC-I downregulation by Nef. a, PACS-1a levels in lysates from C6 cells (control cells) and A7 cells rendered deficient in PACS-1b by an antisense strategy (antisense cells) were analysed by western blotting, using α-tubulin as a control. Mr (K), relative molecular mass (in thousands). b, Dose responsiveness of MHC-I downregulation in C6 (control) cells, antisense (AS) cells, and AS cells transfected with a lentiviral vector expressing PACS-1b; all of these cells were transfected with a bicistronic Nef–IRES–GFP vector. Steady-state surface levels of MHC-I were expressed as a function of GFP (green fluorescent protein) intensity. The results shown are means ± s.e.m. Inset, western blot analysis of exogenous PACS-1b protein in AS (−) and PACS-1b-complemented AS (+) cells. c, Steady-state surface levels of CD4, evaluated by flow cytometry in cells transfected with CD4 with (+Nef) or without (+ Empty vector) Nef–IRES–GFP. Results are representative of three independent experiments.
We then studied the putative role of PACS-1 in the Nef-induced relocalization of MHC-I to the TGN. Control and PACS-1-antisense cells, with or without Nef, were incubated with the MHC-I-specific antibody W6/32, which recognizes peptide-loaded MHC-I complexes, at 37 °C for 4 h to allow trafficking along the endocytic pathway, and studied by confocal microscopy. In control cells in the absence of Nef (or in the presence of acidic-cluster-mutated Nef; data not shown), MHC-I was found predominantly at the plasma membrane, with some faint staining of intracellular vesicles being found (Fig. 2a). In the presence of the viral protein, MHC-I underwent a dramatic relocalization to a paranuclear compartment also positive for TGN46 (Fig. 2b). In contrast, in PACS-1-antisense cells, MHC-I was retained in the periphery of the cells and failed to co-localize with this TGN marker (Fig. 2c). When PACS-1 was re-introduced into these cells, the Nef-induced MHC-I phenotype was restituted (Fig. 2d). Acidic-cluster-dependent binding of HIV-1 Nef to PACS-1. These results showed that PACS-1 is necessary for the relocalization of MHC-I to the trans-Golgi network in the presence of Nef. Revealing the molecular basis of this observation, Nef and PACS-1 could be co-immunoprecipitated in cells overexpressing the two proteins, in an acidic-cluster-dependent manner (Fig. 3a). This interaction was probably direct, because a wild-type, but not an acidic-cluster-mutated, glutathione-S-transferase (GST)–Nef fusion protein could capture in vitro recombinant PACS-1fbr, which corresponds to residues 116–257 of PACS-1 and contains its cargo-binding site (Fig. 3b).
Figure 2.
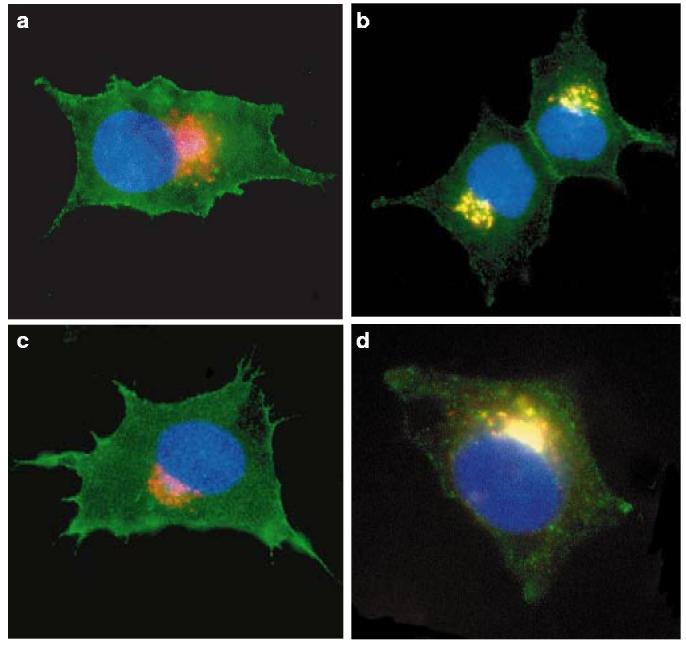
Nef targets MHC-I from the plasma membrane to the trans-Golgi network in a PACS-1-dependent manner. Transfected cells were incubated with the MHC-I-specific antibody W6/32 at 37 °C for 4 h to allow trafficking along the endocytic pathway. After fixation, cells were co-stained with a monoclonal antibody against TGN46 (a TGN marker) and the appropriate secondary antibodies. Representative cells are shown. Green, W6/32; red, TGN46; blue, CyanFP tagged with a nuclear-localization signal (a marker of transfected cells). a, Control cells transfected with an empty vector and CyanFP (ratio 10:1). b, Control cells transfected with Nef and CyanFP (ratio 10:1). MHC-I molecules are dramatically redistributed to the TGN area. c, PACS-1-antisense cells transfected with Nef and CyanFP (ratio 10:1). In these cells, the viral protein is unable to target MHC-I to the TGN. d, PACS-1-antisense cells transduced with a PACS-1-expressing lentivector and transfected with Nef and CyanFP (ratio 10:1). Targeting of MHC-I to the TGN is restored. Original magnification ×630.
Figure 3.
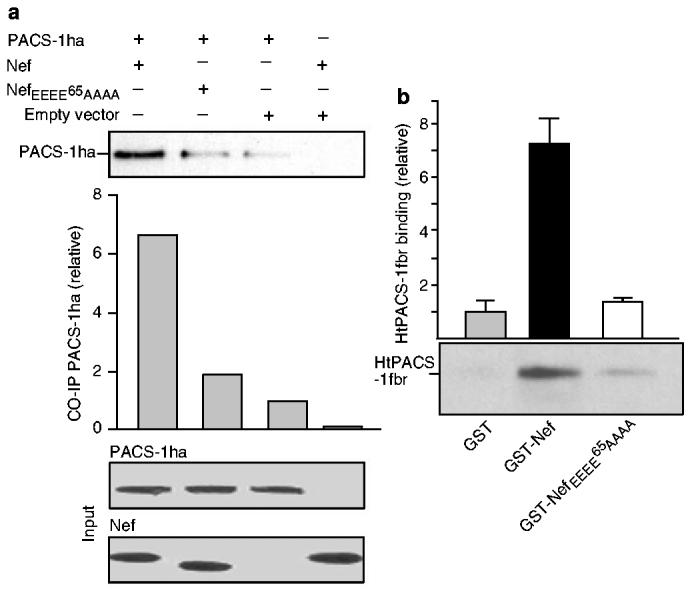
PACS-1 binds to Nef in an acidic-cluster-dependent manner. a, Top, acidic-cluster-dependent co-immunoprecipitation of PACS-1 and Nef (wild-type or acidic-cluster-mutated) in expressing cells. Centre, the relative amount of co-immunoprecipitated (CO-IP) PACS-1ha. Bottom, levels of Nef and PACS-1ha in the input before co-immunoprecipitation. b, HtPACS-1fbr interacts with GST–Nef, but only weakly with GST–Nef EEEE65AAAA and with GST. Proteins bound to GST or conjugates were analysed by western blotting with antiserum 678 (anti-PACS-1fbr). GST–Nef maximally bound 17% of input HtPACS-1fbr. Assays were performed in triplicate. Error bars represent s.d.Values on the ordinate in a, b are arbitrary units, the reference (empty vector in a, GST in b) being assigned a value of 1.
Nef as a connector with the PACS-1-mediated TGN targeting pathway. Nef also downmodulates the cell-surface protein CD4, an effect that preserves the infectivity of HIV-1 virions by preventing any interference between CD4 and release of viral particles or incorporation of the viral envelope into virions6,7. In the presence of Nef, CD4 is rapidly endocytosed from the cell surface and targeted for degradation in lysosomes8-10. This two-step process reflects the ability of Nef to connect the cytoplasmic tail of CD4 first with the adaptor-protein complex-2 (AP-2) of clathrin-coated pits at the plasma membrane, and then with COP-I coatomers (a different set of coat proteins) in early endosomes9,11-13. By analogy, we postulated that Nef serves as a viral connector between MHC-I and PACS-1, thereby directing MHC-I to a TGN retrieval pathway. To test this model, we created chimaeric integral membrane proteins that comprised the extracellular and transmembrane domains of CD4 fused to either wild-type Nef (44Nef) or acidic-cluster-mutated Nef (44NefEEEE65AAAA; in this mutant, the four glutamate residues that end at position 65 were converted to alanine). The Nef sequences thus became the cytoplasmic domains of the chimaera, allowing us to investigate the sorting motifs within the viral connector itself9,11,14. In transfected CD4-negative 293T cells, the wild-type and acidic-cluster-mutated Nef proteins were both rapidly internalized and did not recycle to the cell surface (data not shown). This confirms the previous demonstration that Nef contains determinants, distinct from the EEEE acidic stretch, that control Nef's accelerated internalization from the plasma membrane and sorting from early to late endosomes9,11,15-17.
We then studied the subcellular localization of the endocytosed 44Nef and 44NefEEEE65AAAA proteins. Transiently transfected CD4-negative melanoma cells (C6 cells) were surface-labelled with a CD4-specific antibody, incubated for 2 h at 37°C to allow transit through the endocytic compartment, and examined by confocal microscopy (Fig. 4). Whereas 44Nef localized in a γ-adaptin-positive paranuclear compartment likely to be the TGN (Fig. 4a–c), 44NefEEEE65AAAA accumulated in multiple vesicles dispersed throughout the periphery of the cell (Fig. 4d–f).
Figure 4.

The N-terminal acidic cluster of Nef mediates the TGN targeting of an internalized Nef-containing chimaeric integral-membrane protein. Cells expressing the indicated CD4–Nef chimaeras were labelled with a CD4-specific antibody for 2 h at 37 °C to allow trafficking along the endocytic pathway. a–c, Intracellular distribution of wild-type 44Nef. d–f, Intracellular distribution of 44NefEEEE65AAAA. g, Composite images of two representative cells, integrating fluorescence and intravesicular pH scaled in pseudocolours. The cell on the left expressed wild-type 44Nef, and that on the right expressed 44Nef EEEE65AAAA. In the left-hand cell, vesicular structures staining with anti-CD4 antibody exhibit the green–yellow colouration indicative of a pH around 6.1. In contrast, the blue colour of CD4-positive vesicles generated in the presence of 44Nef EEEE65AAAA corresponds to a pH of ∼5.4. The pseudocolour pH scale is at the left. h, Left, mean pH of CD4-positive vesicles; the numbers at the top of each bar indicate the mean vesicular pH ± s.d. and the number (n) of vesicles included in each analysis; error bars correspond to 1 s.d. from the mean. Right, the percentage of vesicles (indicated at the top of each bar) with a pH lower than 5.85, the pH of late endosomes/lysosomes. Original magnification ×630.
We analysed the pH of the compartments in which the two molecules resided by using a pH-sensitive antibody-coupled probe and fluorescence microscopy, as described9,18. Similar analyses have revealed a pH gradient along the endocytic pathway, with the pH being 6.5–6.0 in early/recycling endosomes, 5.9–5.0 in late endosomes, and 5.5–5.0 in lysosomes. In the TGN, the pH is around 6.0 (refs 18, 19). The compartment in which internalized 44Nef accumulated had a pH of 6.1, compatible with it being the TGN. In contrast, the 44NefEEEE65AAAA-containing vesicles had an average pH of 5.4, and thus probably represented a population of late endosomes/lysosomes (Fig. 4g, h).
Finally, we studied the fate of internalized 44Nef molecules in PACS-1-antisense cells (Fig. 5). In these cells, the chimaera was found in a dispersed pattern reminiscent of that observed for 44NefEEEE65AAAA in PACS-1-positive cells, and failed to co-localize with γ-adaptin in the TGN (Fig. 5d–f), in marked contrast with its distribution in control cells (Figs 4a–c, 5a–c). Most of the 44NefEEEE65AAAA-containing vesicles had a pH below 5.85, with an average of 5.78, consistent with these vesicles being part of the late-endosomal compartment (Fig. 5g).
Figure 5.

The TGN targeting of a CD4–Nef chimaera requires PACS-1. 44Nef-expressing cells were labelled with a CD4-specific antibody for 2 h at 37 °C to allow internalization. a–c, Intracellular distribution of CD4–44Nef in control cells. d–f, Intracellular distribution of CD4–44Nef in PACS-1-antisense cells. g, Left, mean pH of CD4-positive vesicles. The numbers at the top of bars indicate the mean vesicular pH ± s.d. and the number of vesicles (in parentheses) included in each analysis; error bars correspond to 1 s.d. from the mean. Right, the percentage of vesicles with an acidic pH (indicated at the top of each bar) compatible with late endosomes/lysosomes. Numbers in parentheses indicate the number of vesicles included in each analysis. Original magnification ×630.
Discussion
Our results support a model in which the Nef protein of HIV-1 downregulates cell-surface expression of MHC-I by connecting the cytoplasmic tail of the immune receptor, in the endosomes, with the PACS-1-based TGN retrieval pathway. An implication of our results is that Nef must physically recruit MHC-I, either directly or indirectly. In this respect, other determinants of HIV-1 Nef shown to be essential for MHC-I downregulation include an N-terminal α-helix and a proline-rich repeat located just downstream of the EEEE acidic cluster3,20. It is interesting that the targeting of a 44Nef–CD4 chimaera to the TGN was not prevented by mutating either one of these two motifs (data not shown), indicating that they are probably involved not in binding of PACS-1 but rather in some other step necessary for MHC-I modulation. Both the N-terminal helix and the proline-rich repeat participate in the binding of Nef to protein kinases, and the proline repeat constitutes the docking site for SH3-containing Src-family tyrosine kinases21,22. It is possible that one such Nef-interacting protein acts as a bridge between Nef and the MHC-I cytoplasmic tail.
Mutating the acidic-cluster motif of Nef abrogates the subcellular relocalization of MHC-I (ref. 3). We also found that acidic-cluster-deficient Nef is unable to accelerate MHC-I internalization (data not shown). Of note, the rates of MHC-I endocytosis induced by wild-type Nef are well below those measured for CD4, indicating that the surface removal of the two molecules is affected through distinct mechanisms. It is also interesting that MHC-I is similarly distributed in control cells and in nef-expressing PACS-1-antisense cells (Fig. 2). Thus Nef needs first to bind to PACS-1 to act on MHC-I. The residual Nef-induced MHC-I downregulation that is detected in PACS-1-antisense cells expressing high levels of the viral protein would then reflect the remainder of some PACS-1 in these cells, rather than a PACS-1-independent phenomenon.
The binding sites of CD4 and PACS-1 on HIV-1 Nef largely overlap, as CD4 interacts with a region of Nef that is centred around residues 57–59 (ref. 23). The recruitment of these two molecules thus appears to be mutually exclusive. This probably explains why Nef does not target CD4 to the TGN after triggering its endocytosis. It could be, however, that Nef is retrieved to the Golgi after delivering CD4 to late endosomes/lysosomes.
The dileucine-motif-mediated interaction of HIV-1 Nef with adaptor complexes controls the acceleration of CD4 endocytosis15-17, yet it plays no part in the modulation of MHC-I (refs 17, 20). However, adaptor-protein-containing clathrin-coated pits still participate in this latter process: PACS-1 is a sorting protein that connects membrane proteins containing acidic clusters with AP-1 to allow the membrane proteins to be retrieved from the endosome to the Golgi in clathrin-coated vesicles4. Our results show that the formation of a complex between a cellular connector, PACS-1, and a viral connector, Nef, increases the range of sorting pathways that a given protein, in this case MHC-I, can follow.
Several human viruses, including herpes simplex virus, cytomegalovirus (CMV) and adenovirus, escape surveillance by CTLs by downregulating the cell-surface expression of MHC-I. These viruses all encode factors that interfere with MHC-I assembly and transport along the exocytic pathway24,25. Thus, the mechanism of MHC-I downmodulation described here, whereby Nef redirects the trafficking of class I molecules after they are internalized from the cell surface, is unprecedented. Equally intriguing is the fact that Nef redirects MHC-I to the TGN, instead of simply targeting the receptor to lysosomes, as it does for CD4. These results strongly suggest that the Golgi targeting of MHC-I is advantageous for HIV-1, for reasons that are still obscure. An understanding of the molecular bases of this process should enable the design of anti-Nef drugs that will expose HIV-infected cells to the attacks of the immune system, and might in the long term facilitate viral eradication.
Methods
DNA constructions.
We used previously described CMV-promoter-based mammalian expression plasmids for CD4 and HIV-1 Nef8,14, prokaryotic vectors for production of GST–Nef fusion proteins11 or for ht-PACS-1fbr4, and the bicistronic pCDNef-NA7 IRES–GFP plasmid3. pECFP (Clontech) expresses CyanFP with a nuclear-localization signal.
Antibodies.
The mouse monoclonal antibody against γ-adaptin was from Sigma (antibody 100/3), and conjugated monoclonal antibodies against CD4 and MHC-I were from DAKO. The rabbit antiserum 7704 against PACS-1 has been described previously4. The anti-tubulin antibody (N356) was from Amersham. The sheep anti-human-TGN46 antibody was from Serotec.
Cell lines and transfections.
293T cells were grown in DMEM supplemented with 10% fetal calf serum (FCS). CD4-negative A7 melanoma cells expressing or not expressing antisense PACS-1 RNA have been described previously4. The antisense cells were complemented by transduction with a lentiviral vector expressing rat PACS-1b from the EF-1α promoter. Transfections were done using calcium phosphate8.
Protein analyses.
Equivalent amounts of postnuclear lysate (normalized with Dc Protein Assay, BioRad) from control (C6) and antisense (AS19) melanoma cells were resolved by SDS–PAGE and analysed by western blot using antiserum 7704 (anti-PACS-1) and monoclonal antibody N356 (anti-α-tubulin) followed by chemiluminescent detection. Signals were quantified using NIH Image software.
Binding assays.
Replicate plates of C12 cells were infected with vaccinia vectors expressing PACS-1ha (influenza virus haemagglutinin-A-tagged PACS-1) together with wild-type or 44NefEEEE65AAAA or an empty vector. After preclearing with protein-A–Sepharose beads, extracts were incubated with a Nef-specific antiserum (1:200 dilution) overnight at 4°C. Immune complexes were isolated using protein-A–Sepharose, and washed with 50 mM TrisCl pH 7.5, 200 mM NaCl, 1% NP40 and 1% deoxycholate (DOC). Coimmunoprecipitated PACS-1ha proteins were detected by western blotting with the monoclonal antibody HA.11 (1:1,000 dilution). For in vitro binding assays, htPACS-1fbr (5 μg) was combined with 10 μg of GST, GST–44Nef or GST–44NefEEEE65AAAA in binding buffer (50 mM Tris pH 7.5, 200 mM NaCl, 2 mM MgCl2 and 1% NP40) and incubated for 1 h at room temperature. Protein complexes were recovered with glutathione–agarose beads and analysed by western blotting with antiserum 678 (anti-PACS-1fbr). Detection was by enhanced chemiluminescence, and quantification by NIH Image software.
Flow cytometry.
Immunofluorescence microscopy.
Cells were grown on glass coverslips and incubated with fluorescein isothiocyanate (FITC)-conjugated CD4 or with anti-MHC-I antibody for various times at 37°C. The cells were then fixed with a 10-min incubation in methanol at −20°C or in 2% paraformaldehyde for 20 min at room temperature, and then permeabilized with 0.1% NP40 and washed several times with PBS containing 3% BSA. The anti γ-adaptin and anti-TGN46 antibodies were used at a dilution of 1:100. Secondary antibodies were from Jackson and used at a 1:200 dilution. Samples were analysed on a LSM 410 invert laser scan confocal microscope (Zeiss) or on a Zeiss Axiovert microscope equipped with a cooled charge-coupled-device (CCD) camera.
pH measurements in live cells.
Intracellular pH was measured by ratio fluorescence imaging of an internalized pH-sensitive antibody, as described9,8. Dual-excitation ratio imaging was done on a Zeiss Axiovert microscope equipped with a cooled CCD camera (Visitron Systems, Puchheim, Germany), using a ×100/1.3 NeoFluar objective and 2 × 2 binning to achieve a final resolution of 134 nm per pixel. Images were acquired for 500 ms at λ = 490 ± 6 nm and for 1 s at λ = 440 ± 6 nm, using a DeltaRAM monochromator (PTI, Monmouth Junction, NJ), a 510 DRLP dichroic mirror (Omega Opticals, Brattleboro, VT), and a 535 ± 25 emission filter (Omega). Calibration, image processing, data analysis and statistics were done as described9.
ACKNOWLEDGEMENTS
We thank E. Kiyokawa, S. Pfeffer, M. Robinson, G. Nolan and J. Skowronski for providing various reagents and/or for helpful discussions; B. Wehrle-Haller for assistance with imaging; and M. Loche for the artwork. This study was supported by grants from the Gabriella Giorgi-Cavaglieri Foundation and from the Swiss National Science Foundation to D.T., and from the NIH to D.T. and G.T. V.P. was the recipient of an M.D.–Ph.D. scholarship from the Swiss National Science Foundation. Correspondence and requests for materials should be addressed to D.T. or G.T.
References
- 1.Schwartz O, Maréchal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nature Med. 1996;2:338–342. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- 2.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg ME, Iafrate AJ, Skowronski J. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. EMBO J. 1998;17:2777–2789. doi: 10.1093/emboj/17.10.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wan L, et al. PACS-1 defines a novel gene family of cytosolic sorting proteins required for transGolgi network localization. Cell. 1998;94:205–216. doi: 10.1016/s0092-8674(00)81420-8. [DOI] [PubMed] [Google Scholar]
- 5.Aiken C, Krause L, Chen YL, Trono D. Mutational analysis of HIV-1 Nef: identification of two mutants that are temperature-sensitive for CD4 downregulation. Virology. 1996;217:293–300. doi: 10.1006/viro.1996.0116. [DOI] [PubMed] [Google Scholar]
- 6.Lama J, Mangasarian A, Trono D. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking env incorporation in a nef- and vpu-inhibitable manner. Curr. Biol. 1999;9:622–631. doi: 10.1016/s0960-9822(99)80284-x. [DOI] [PubMed] [Google Scholar]
- 7.Ross TM, Oran AE, Cullen BR. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral nef protein. Curr Biol. 1999;9:613–621. doi: 10.1016/s0960-9822(99)80283-8. [DOI] [PubMed] [Google Scholar]
- 8.Aiken C, Konner J, Landau NR, Lenburg ME, Trono D. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell. 1994;76:853–864. doi: 10.1016/0092-8674(94)90360-3. [DOI] [PubMed] [Google Scholar]
- 9.Piguet V, et al. Nef-induced CD4 degradation: a diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of β-COP in endosomes. Cell. 1999;97:63–73. doi: 10.1016/s0092-8674(00)80715-1. [DOI] [PubMed] [Google Scholar]
- 10.Rhee SS, Marsh JW. Human immunodeficiency virus type 1 Nef-induced down-modulation of CD4 is due to rapid internalization and degradation of surface CD4. J. Virol. 1994;68:5156–5163. doi: 10.1128/jvi.68.8.5156-5163.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piguet V, et al. Mechanism of Nef induced CD4 endocytosis: Nef connects CD4 with the μ chain of adaptor complexes. EMBO J. 1998;17:2472–2481. doi: 10.1093/emboj/17.9.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Gall S, et al. Nef interacts with the μ subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC-I molecules. Immunity. 1998;8:483–495. doi: 10.1016/s1074-7613(00)80553-1. [DOI] [PubMed] [Google Scholar]
- 13.Greenberg M, et al. Co-localization of HIV-1 Nef with the AP-2 adaptor protein complex correlates with Nef-induced CD4 down-regulation. EMBO J. 1997;16:6964–6976. doi: 10.1093/emboj/16.23.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mangasarian A, et al. The HIV-1 Nef protein acts as a connector with sorting pathways in the Golgi and at the plasma membrane. Immunity. 1997;6:67–77. doi: 10.1016/s1074-7613(00)80243-5. [DOI] [PubMed] [Google Scholar]
- 15.Craig HM, Pandori MW, Guatelli JC. Interaction of HIV-1 nef with the cellular dileucine-based sorting pathway is required for CD4 down-regulation and optimal viral infectivity. Proc. Natl Acad. Sci. USA. 1998;95:11229–11234. doi: 10.1073/pnas.95.19.11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bresnahan PA, et al. A dileucine motif in HIV-1 Nef acts as an internalization signal for CD4 downregulation and binds the AP-1 clathrin adaptor. Curr. Biol. 1998;8:1235–1238. doi: 10.1016/s0960-9822(07)00517-9. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg M, DeTulleo L, Rapoport I, Skowronski J, Kirchhausen T. A dileucine motif in HIV-1 Nef is essential for sorting into clathrin-coated pits and for downregulation of CD4. Curr. Biol. 1998;8:1239–1242. doi: 10.1016/s0960-9822(07)00518-0. [DOI] [PubMed] [Google Scholar]
- 18.Demaurex N, Furuya W, D'Souza S, Bonifacino JS, Grinstein S. Mechanism of acidification of the trans-Golgi network (TGN). In situ measurements of pH using retrieval of TGN38 and furin from the cell surface. J. Biol. Chem. 1998;273:2044–2051. doi: 10.1074/jbc.273.4.2044. [DOI] [PubMed] [Google Scholar]
- 19.Mukherjee S, Ghosh RN, Maxfield FR. Endocytosis. Physiol. Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- 20.Mangasarian A, Piguet V, Wang JK, Chen Y, Trono D. Nef-induced CD4 and major histocompatibility complex class I (MHC-I) down-regulation are governed by distinct determinants: N-terminal alpha helix and proline repeat of Nef selectively regulate MHC-I trafficking. J. Virol. 1999;73:1964–1973. doi: 10.1128/jvi.73.3.1964-1973.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baur AS, et al. The N-terminus of Nef from HIV-1/SIV associates with a protein complex containing Lck and a serine kinase. Immunity. 1997;6:283–291. doi: 10.1016/s1074-7613(00)80331-3. [DOI] [PubMed] [Google Scholar]
- 22.Saksela K, Cheng G, Baltimore D. Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. EMBO J. 1995;14:484–491. doi: 10.1002/j.1460-2075.1995.tb07024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grzesiek S, Stahl SJ, Wingfield PT, Bax A. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry. 1996;35:10256–10261. doi: 10.1021/bi9611164. [DOI] [PubMed] [Google Scholar]
- 24.Ploegh HL. Viral strategies of immune evasion. Science. 1998;280:248–253. doi: 10.1126/science.280.5361.248. [DOI] [PubMed] [Google Scholar]
- 25.Brodsky FM, Lem L, Solache A, Bennett EM. Human pathogen subversion of antigen presentation. Immunol Rev. 1999;168:199–217. doi: 10.1111/j.1600-065x.1999.tb01294.x. [DOI] [PubMed] [Google Scholar]