

Abstract
Large DNA viruses of the herpesvirus family produce proteins that mimic host MHC-I molecules as part of their immunoevasive strategy. The m144 glycoprotein, expressed by murine cytomegalovirus, is thought to be an MHC-I homolog whose expression prolongs viral survival in vivo by preventing natural killer cell activation. To explore the structural basis of this m144 function, we have determined the three-dimensional structure of an m144/β2-microglobulin (β2m) complex at 1.9 Å resolution. This structure reveals the canonical features of MHC-I molecules including readily identifiable α1, α2, and α3 domains. A unique disulfide bond links the α1 helix to the b-sheet floor, explaining the known thermal stability of m144. Close juxtaposition of the α1 and α2 helices and the lack of critical residues that normally contribute to anchoring the peptide N and C termini eliminates peptide binding. A region of 13 amino acid residues, corresponding to the amino-terminal portion of the α2 helix, is missing in the electron density map, suggesting an area of structural flexibility that may be involved in ligand binding.
Keywords: X-ray structure, MHC-Iv, immunoevasin, NK recognition, viral virulence
Introduction
The dynamic interactions between molecules expressed by various infectious organisms and the immune systems of the host illustrate not only the power of vertebrate immunity but also the creative variations, both genetic and epigenetic, that microorganisms employ for their survival. Many mechanisms have evolved to allow infectious agents to avoid the immune response of their hosts, ranging from the rapid mutation of surface antigens to avoid recognition by host antibodies to the triggering of subtle molecular and cellular pathways that lead to viral latent states poised for reactivation under opportune conditions. Among the microorganisms that have achieved great success in inventing strategies for immune evasion are the herpesviruses and poxviruses,1,2 which, because of their large genomes, are capable of encoding many non-essential functions that can subvert recognition by innate and adaptive immune receptors.
Of particular interest are the cytomegaloviruses (CMV), members of the b-herpesvirus family, that are found in a wide range of vertebrate hosts, where they establish acute, latent, and persistent infections.3 Latent infection in the human may become associated with severe and even life-threatening disease in the clinical scenario of immunosuppression. The immune response to CMV infection in the mouse and human is mediated by natural killer (NK) cells,4 as well as CD8+ T cells5and antibodies,6 and the genomes of human and murine CMV encode a range of proteins that subvert the host’s immune response by interfering with both NK and T cell recognition as well as crucial steps in the pathways of antigen processing and presentation (reviewed by Tortorella et al.2). As part of this strategy to interfere with immune recognition, human and mouse CMV genomes encode proteins that are predicted, based on amino acid sequence comparisons and three-dimensional structure prediction algorithms, to be structural homologs of host major histocompatibility complex class I (MHC-I) molecules.7-9
Although scrutiny of the cytomegalovirus genomes has permitted the identification of a set of genes that might be expected to encode MHC-like molecules, only a few of these have been characterized functionally or biochemically, and to date, no MHC-I-like molecule encoded by a virus has been characterized structurally.
CMV-encoded putative MHC-I structural homologs potentially modulate host immunity, thus promoting viral survival and latency. The best-studied viral MHC-I homologs (which we shall refer to here in after as MHC-Iv (for “viral”)) are the human CMV (HCMV) UL18 protein, and murine CMV (MCMV) proteins m144, m145, m152, m155, and m157. At least two distinct mechanisms permit these CMV molecules to modulate immune recognition: UL18 and m157 directly interact, at the infected cell surface, with activating or inhibitory NK cell receptors; m145, m152 and m155 each down-regulate the surface expression of a stress-induced ligand for activating NK receptors. UL18 engages the inhibitory receptor LIR-1, widely expressed on lymphoid and myeloid cells.10-14 (Recently, another MHC-I-like molecule encoded in a clinical isolate of HCMV, UL142, has been shown to inhibit NK cell lysis.15) m157 targets the inhibitory NK receptor Ly49I, leading to reduced immunity and greater viral load.8,16,17 Conversely, mouse strains expressing Ly49H, an NK activating receptor that also binds m157, are protected from MCMV infection. Viruses with m157 mutations escape Ly49H-dependent immune surveillance.18m152, which encodes the gp40 glycoprotein, specifically down-regulates cell surface expression of RAE-1 proteins (another set of host MHC-Ib molecules), which are ligands for the NKG2D activating receptor.19 m145 reduces expression of the stress-induced MULT1 molecule,20 and m155 similarly down-regulates another NKG2D ligand, H60.21
Amino acid sequence similarity and structure prediction algorithms suggest that UL18, UL142, m144, m145, m152, m155, and m157 are true MHC-I homologs. However, their three-dimensional structures have not yet been reported. m144 is of particular interest, since both biochemical and functional studies, as well as amino acid sequence identity of 29% to classical MHC-I molecules throughout the putative extracellular region, suggest that it may interact with MHC-I sensing NK receptors. Fundamental questions concerning the function and evolution of m144 and other MHC-Iv molecules, and their potential for interactions with peptide, β2m, and ligand, may be addressed with knowledge of their structure. With this goal, we expressed, purified, crystallized, and determined, by X-ray diffraction at 1.9 Å resolution, the structure of the soluble extracellular domains of the MCMV protein m144 in complex with mouse β2-microglobulin (β2m). This first structure of an MHC-Iv molecule reveals that m144 has features indicative of the MHC-I family in addition to those unique to this particular molecule. We argue that a flexible region in the α2 domain may interact with, and be structurally stabilized by, host receptors targeted by m144.
Results
Peptide-independent expression of m144
Although considerable functional data indicating the role of m144 expression in resistance to NK-mediated cytolysis have been reported, and the analysis of m144 molecules expressed in a CHO expression system failed to indicate the presence of any bound peptide,22 we asked whether delivery of peptide via the transporter associated with antigen presentation, TAP, was required for cell surface expression of m144. We transfected both TAP deficient (RMA-S)23 and TAP sufficient (RMA) cells with a vector encoding the full length m144 as well the green fluorescent protein (GFP) (see Experimental Procedures). As shown in Figure 1, cell surface expression of m144 in transiently transfected cells represented the same proportion of cells when both RMA and RMA-S cells were transfected. In contrast, transfection of the classical MHC-I encoding gene, H-2Dd, revealed a profound effect of availability of peptide as conferred by TAP (Figure 1). This experiment confirms and extends previous studies revealing the surface expression of m144 in stable RMA-S transfectants.24 These experiments provided an experimental basis for our efforts to express m144 in Escherichia coli and to refold the protein from inclusion bodies in the absence of added peptide.
Figure 1.
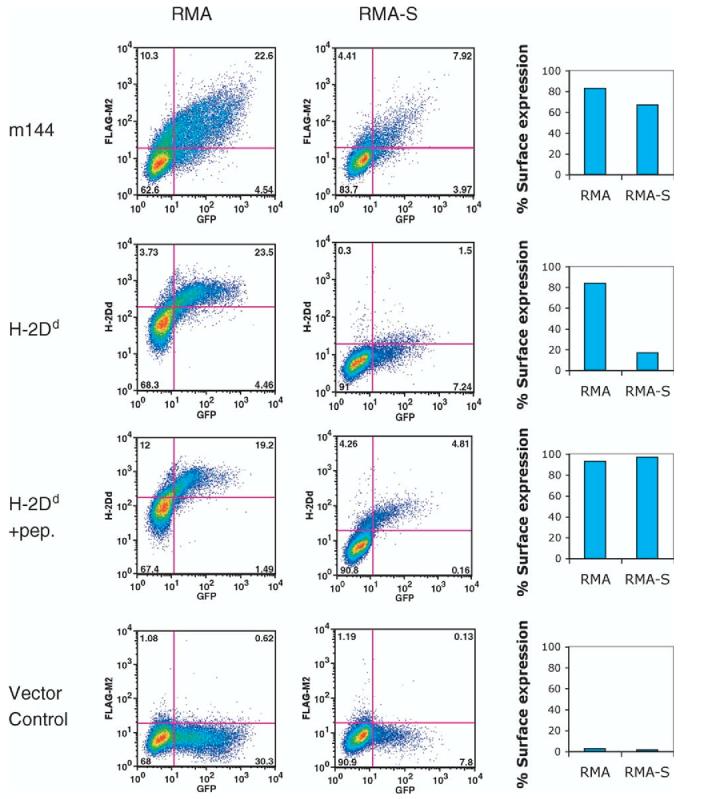
Cell surface expression of m144 is unaffected by TAP2 deficiency. RMA (TAP2+) or RMA-S (TAP2-) cells were transfected with a vector encoding m144 or H-2Dd as control as described in Experimental Procedures. Cell surface expression of FLAG-TAG or 34-5-8S (anti-H-2Dd) was evaluated by indirect immunofluorescence as described in Experimental Procedures.
Overall structure of m144
A fragment of the murine cytomegalovirus m144 protein encompassing the extracellular domain, residues 2-249, was expressed in E. coli as inclusion bodies, denatured in guanidine and refolded in vitro together with similarly expressed mouse β2m. The m144/β2m preparation was purified by gel filtration and ion exchange chromatography, which revealed two peaks of protein, one containing the m144/β2m heterodimer, the other the m144 heavy chain alone (see Supplementary Data, Figure 1). We screened crystallization conditions with material from both peaks, but only succeeded with that from the heterodimer-containing fraction. Synchrotron diffraction data to 1.9 Å were collected, the structure of m144 was solved by molecular replacement, and refined using all the available diffraction data (see Table 1). The 2FoKFc map of the m144 heavy chain shows continuous electron density from Glu8 to Lys242 except for a stretch of 13 residues (Asp115 to Asp127) in the amino-terminal portion of what would be expected to be the α2 helix.
Table 1.
Data collection and refinement statistics
| Data collection | |
| Space group | C121 |
| Unit cell dimensions | |
| a, b, c (Å) | 133.5, 51.2, 71.8 |
| α, β, γ (deg.) | 90.0, 105.7, 90.0 |
| Molecules per asymmetric unit | 1 |
| Resolution (Å) | 1.87 |
| Total observations | 136,195 |
| Unique reflections | 39,028 |
| Completeness (%)a | 96.7 (95.3) |
| I/σIa | 41.7 (10.07) |
| Rsym (%)a,b | 5.6 (17.1) |
| Refinement | |
| Resolution range (Å) | 41.7-1.9 |
| Reflections | |
| Working set | 38,443 |
| Test set | 1922 |
| Rcryst (%)c | 20.5 |
| Rfree (%)c | 23.0 |
| Number of non-H protein atoms | 2554 |
| Number of water molecules | 290 |
| r.m.s. deviations from ideality | |
| Bond lengths (Å) | 0.005 |
| Bond angles (deg.) | 1.26 |
| Average B values (Å2) | |
| Main-chain | 30.6 |
| Side-chain | 34.7 |
| Ramachandran plot statistics (%) | |
| Most favored | 88.7 |
| Allowed | 10.2 |
| Generous | 0.7 |
| Disallowed | 0 |
Values in parentheses are statistics for the highest resolution shell (1.94-1.87 Å).
, where Ij is the intensity of the jth observation of a reflection and ⟨I⟩ is the mean intensity from multiple measurements of that reflection.
, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. Rfree isas for Rcryst but calculated for a randomly selected 5.0% of reflections not included in the refinement.
The structure of m144 reveals a remarkable similarity to MHC-I molecules with readily identifiable α1; α2 and α3 domains and association with β2m (Figure 2). No extraneous electron density was observed in the potential groove region between the α1 and α2 helices. The relative dispositions of the α1α2 domain unit, the α3 domain, and the noncovalently associated β2m subunit are similar to those of MHC-I molecules. However, the angle between the α1α2 platform domain and the α3 immunoglobulin (Ig)-like domain, the hinge, is greater in m144 than in other MHC-I molecules (as summarized in Table 2). For m144, this is 988 as compared with 648 to 848 for a selection of both MHC-Ia and MHC-Ib molecules, which includes β2m-complexed, peptide-free molecules (Hfe, T22, rFcRn, and CD1a), β2m-complexed, peptide-containing molecules (H-2Kb, H-2Dd, HLA-A*0201, HLA-B*3501, and HLA-Cw4), and β2m-free, peptide-free molecules (MICA and Zn-α2 glycoprotein, ZAG). Only MICA has a larger hinge angle at 1148. Scrutiny of the amino acid sequence alignment (Figure 3) and the superposed structures of m144 and H-2Kb backbones (not shown) suggest that the lack of three amino acids corresponding to residues 173 to 175 of H-2Kb accounts in part for the relative domain displacement. Whether the wider hinge angles of m144 and MICA reflect any unique function or merely represent extreme possibilities available for MHC-I molecules as a group is not clear.
Figure 2.
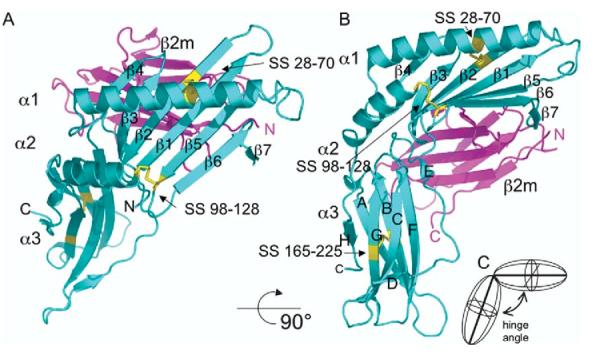
Overall structure of m144. Ribbon diagram of the m144 structure is shown in an MHC “standard view” (a) and rotated approximately 908 (b). m144 heavy chain is shown in cyan, and the β2m light chain in magenta. Disulfide bonds of the heavy chain are shown in yellow. Individual domains, α1; α2, α3, and β2m, and the polypeptide N and C termini are labelled. (c) A schematic illustrating the definition of the hinge angle between the α1α2 domain unit and α3 is shown.
Table 2.
Hinge angle between α1α2 and α3 domains in MHC-Ia, -Ib, and -Iv molecules
| Protein | PDB code | Angle (degrees) | Associated subunits |
|---|---|---|---|
| m144 | 1U58 | 98 | β2m, no peptide |
| Hfe | 1A6Z | 80 | β2m, no peptide |
| T22 | 1C16 | 71 | β2m, no peptide |
| RFcRn | 3FRU | 74 | β2m, no peptide |
| CD1a | 1ONQ | 77 | β2m, glycolipid |
| Zn α2 glycoprotein | 1ZAG | 72 | β2m, lipid |
| H-2Kb/OVA | 1VAC | 72 | β2m, peptide |
| H-2Dd/P18-I10 | 1DDH | 64 | β2m, peptide |
| HLA-A*0201 | 1DUY | 76 | β2m, peptide |
| HLA-B*3501 | 1A1N | 75 | β2m, peptide |
| HLA-Cw4 | 1QQD | 84 | β2m, peptide |
| MICA | 1B3J | 114 | No β2m, no peptide |
Hinge angles were calculated with the HINGE program as described in Experimental Procedures.
Figure 3.

Structure-based alignment of m144 and representative human, rat, and mouse MHC-Ia, -Ib, and -Iv molecules. Extracellular sequences of the indicated proteins were aligned with ClustalW (v1.4) as a first guide, and those molecules for which X-ray structures are available were aligned with ESPript 1.9.70,71 Citations are r144 (rat homolog of m144, from the rat cytomegalovirus complete sequence, NC_002512) and UL18 (from human cytomegalovirus (Herpesvirus 6) complete sequence, NC_001664). HFE, MICA, T22, rFcRn, ZAG, and HLA-B27, represent PDB structures: 1A6Z, 1B3J, 1C16, 3FRU, 1ZAG, 1K5N, respectively. Secondary structure elements for m144 and H-2Kb are indicated above and below the sequences, respectively. Cysteine residues involved in disulfide bonds are indicated by colored ovals, and contact residues to the β2m subunit are indicated for m144 by upward pointing magenta arrows and for H-2Kb by downward pointing blue arrows. Similarity scores, as indicated by the boxed and colored aligned residues, were calculated using the Risler72 matrix. Important salt bridges, for both m144 and H-2Kb, are also indicated. Carbohydrate addition sites for m144 are indicated by blue squares, and the region of m144 for which we observed no electron density, residues 115 to 127, is enclosed in a box.
As in MHC-Ia and MHC-Ib molecules, the Ig-like domains β2m and α3 are stabilized by conserved disulfide bonds. The conserved α3 domain disulfide links cysteine residues 165 and 225 in m144 spanning 60 residues, which is easily aligned with the structurally similar canonical disulfide bond of H-2Kb, linking residues 203 and 259 (spanning 56 residues) (see Figures 2 and 3). The α2 disulfide, linking Cys98 and Cys128, can easily be superposed on the canonical H-2Kb α2 disulfide (Cys101 to Cys164). Unlike MHC-Ia molecules and most MHC-Ib molecules, m144 stabilizes its α1 domain with an α1 helix to β2 strand disulfide, linking Cys28 and Cys70 (Figures 2 and 3). This likely contributes to the observed thermal stability of this peptide-free MHC-Iv molecule.22 The homologous rat CMV molecule r144 preserves cysteine residues 28 and 70, almost certainly possesses the same disulfide bond, and thus would be expected to exhibit similar peptide-free thermal stability. The only other example of an α1 intradomain disulfide bond in an MHC-Ia or MHC-Ib molecule is that between Cys36 and Cys41 of MICA (corresponding to residues 36 and 43 of H-2Kb; see Figure 3), which stabilizes the β3 to β4 loop of MICA.25
Association with β2m
Examination of the interaction of the m144 heavy chain with β2m, based on buried surface area, number of hydrogen bonds, and number of atomic contacts and salt bridges (see Supplementary Data, Table 1), suggests a relatively loose association between the two subunits. The m144/β2m interface buries 2094 Å2 of molecular surface, the lowest among MHC-I-like molecules, which range from 2300 Å2 for T22 to 2800 Å2 for the neonatal rat Fc receptor. The H-2Ld/β2m interface, representative of a weak heavy chain/β2m interaction,26 is 2480 Å2. The number of interchain hydrogen bonds, eight, is also small in the m144/β2m complex and is comparable to the nine found in H-2Ld and significantly fewer than the 14 recognized for H-2Kb. (Amino acid residues of m144 and of H-2Kb that form close contacts with β2m are indicated in the alignment shown in Figure 3.) When m144 heavy chain and β2m are refolded in vitro, the soluble molecules in native buffers are a mixture of the free m144 heavy chain and the heterodimeric m144/β2m complex that can be resolved by anion exchange chromatography (see Supplementary Data, Figure 1). Association of β2m with m144 or with H-2Kb is focused mainly on two regions of the MHC heavy chain: the b-strand floor of the α1α2 domain unit (including contacts from b-strands b1, β2, β3, β5, and β6) and contacts to the α3 domain, largely through strand E. Clearly, H-2Kb contacts in both locations are more extensive (Figure 3 and Supplementary Data, Table 1).
Secondary structure
The extracellular portion of the m144 heavy chain is 31 amino acid residues shorter than that of H-2Kb (see Figure 3). Relative to H-2Kb, the shorter length is due to deletions in the α1 and α2 domains. m144 has a tight turn connecting its β3 and β4 strands and includes four residues in the β4 strand, while H-2Kb uses eight residues to connect β3 with its two residue β4 strand. Unlike H-2Kb, which connects β4 to the α1 helix with two turns of 310 helix, m144 has several bend residues connecting β4 to its straight α1 helix. The α1 domain helix of m144 extends for 23 residues, while that of H-2Kb is 28 residues long. Deletions in the α2 domain, however, seem to have a greater impact on the MHC-like fold of m144. They lead to truncation of the β6 and β7 strands and deletion of b8, resulting in a platform consisting of only seven b strands in contrast to the eight found in H-2Kb and other MHC-I molecules. However, due to lack of electron density in this region, it was not possible to trace the polypeptide chain from Asp115 to Asp127, i.e. from the end of the truncated β7 strand to the beginning of the α2 helix. The missing 13 residues of electron density can be modeled if these are inserted between Asp115 and Asp127 in a relaxed helical conformation (see below). Electron density for m144 resumes with Cys128 and residues 129 to 140 are clearly in an a-helical configuration. This part of the α2 helix superposes quite well on the homologous stretch of H-2Kb, although three residues are missing in m144 as compared to H-2Kb in this region (Figure 3). The Ig-like α3 domain can be described as a b-sandwich, with a core of six extended strands, A, B, and F, packed against C, G, and H. m144 has two short b-strands, D and E. Strand D pairs with the carboxy-terminal portion of strand C, and strand E with the amino-terminal portion of strand F. The region of greatest difference in the α3 domains is that between the carboxyl-terminal end of strand C and the short strand E, where m144 has an insertion of four amino acid residues (Figures 2 and 3). A search of the CATH database27 for proteins with similar structural folds revealed close alignment of the structure of the m144 α3 domain with the β2 domain of the human MHC-II molecule, HLA-DM (see Supplementary Data, Figure 2).
Lack of peptide binding groove
m144 possesses a putative peptide binding groove formed by the apposition of two antiparallel a-helices supported by a platform of β-strands. However, we observed no additional electron density in this groove, an observation consistent with biochemical studies on m144 that failed to identify peptide associated with the purified molecule.22 In this respect, m144 shows a greater resemblance to those MHC-Ib molecules that lack bound peptide (such as Hfe and MICA) than to those MHC-Ia and MHC-Ib molecules that complex with peptides (e.g. HLA and H2-M3). Figure 4 shows a comparison of the groove of m144 with those of selected MHC-Ia and -Ib molecules. The m144 groove resembles those of peptide-lacking MHC-I-like molecules, such as Hfe, in being narrower than those of peptide-binding molecules. As an indication of the width of the different grooves, distances were measured between selected Cα atoms of the α1 and α2 helices. At the far left-hand side of the groove, m144 juxtaposes the two helices more closely than the peptide-complexed H-2Kb (Figure 4(a) and (g)), while the other peptide-free molecules also show a narrower groove at this end (Figure 4(c) and (e)). At the center and the far right-hand side of the cleft, our analysis is limited by a lack of structural information for residues Asp115 to Asp127 in the m144 α2 helix. Therefore we measured inter-helical distances in this region of the groove from the Cα of the conserved cysteine in the α2 helix (Cys128 in m144) to the Cα of Glu62 (in m144) or analogous residue for the other MHCs of the α1 helix. At this near-central location also, the inter-helical distance in m144 is smaller than that of H-2Kb (11.5 Å and 14.0 Å, respectively) and is similar to the inter-helical distances in non peptide-binding MHC-Ib molecules (Figure 4(c) and (e)). The superposition of the m144 α1α2 backbone with those of Hfe, MICA, and H-2Kb(Figure 4) emphasizes the similarities and differences of these related molecules.
Figure 4.

Comparison of the groove region of MHC-Ia, MHC-Ib, and MHC-Iv molecules. Structures of m144 (a), Hfe (c), MICA (e), and H-2Kb (g) see PDB designations in the legend to Figure 3) were displayed in PYMOL, and measurements were made as described in the text. Superposition of each of the molecules on to m144 was based on b-strands. Stereo views of m144 (b), and of m144 superposed onto HFE (d), MICA (f), and H-2Kb (h) are shown.
In addition to size constraints, the particular amino acid residues lining the groove of the m144 molecule are a further impediment to peptide binding. The electron density map of m144 at the left-hand side of the potential groove shows no bound ligand (Figure 5(a)). In peptide-complexed MHC molecules, peptides are anchored to the groove of the MHC through hydrogen bonding networks among conserved residues lining the groove, main-chain atoms at the termini of the bound peptide, and water molecules. These interactions serve both to bind peptide and enhance the stability of the MHC-I molecule. A set of tyrosine residues, Tyr7, Tyr59, Tyr159 and Tyr171 (see Figure 5(b)), conserved in both mouse and human MHC-Ia molecules, is oriented with each side-chain directed into the groove to form hydrogen bonds between their hydroxyl groups and main-chain atoms at the amino terminus of the bound peptide. Tyr7 is located in the platform strand b1, Tyr59 is in the α1 helix, and the remaining two are located in the α2 helix (Figure 5(b)). m144, however, conserves only two tyrosine residues from this cluster: Tyr13, situated in a position analogous to Tyr7 (H-2Kb) on platform strand b1, and Tyr135, analogous to Tyr171 (H-2Kb) towards the C-terminal region of the α2 helix. However, close juxtaposition of the α1 and α2 helices of m144 in this region results in hydrogen bonding of these two tyrosine residues to the side-chain of Glu62 of the α1 helix effectively closing off the groove. The bulky side-chain of Lys131 further constricts the groove (Figure 5(c)). Peptide-binding MHC-Ia molecules preserve Trp167 to accommodate the N terminus of the bound peptide.
Figure 5.
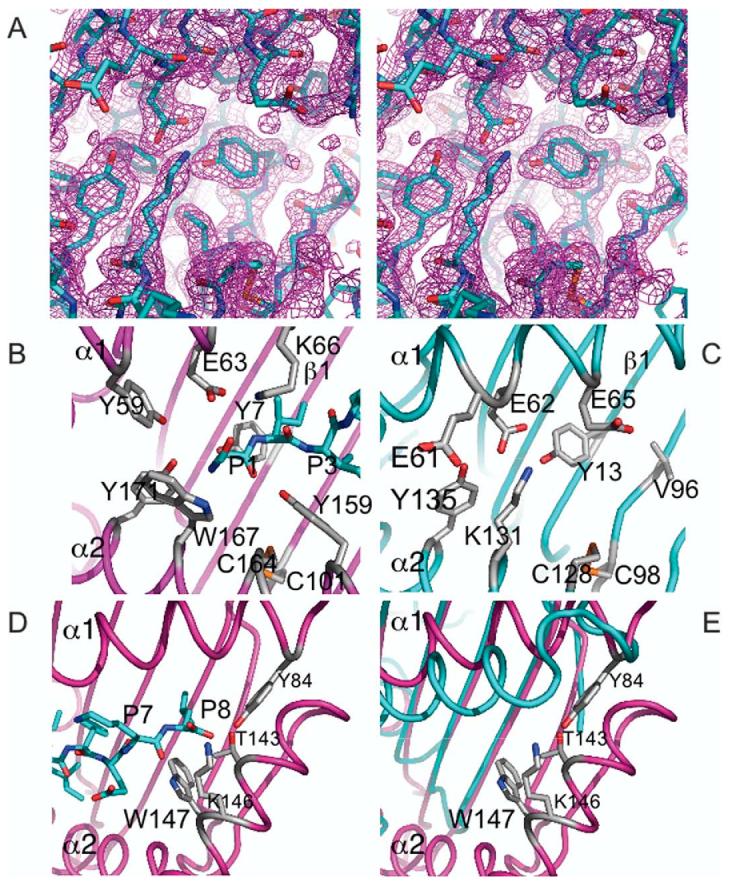
Region analogous to MHC-I “A” pocket shows loss of conserved peptide binding residues and no extraneous electron density. The region surrounding Tyr13 of m144 is shown in stereo, including the final 2FoKFc electron density map, contoured at 1.25s, is displayed (a). Residues lining the putative peptide binding cleft of m144 (b) and H-2Kb (c) are shown. The region around the H-2Kb F pocket is also illustrated (d). In (e), the m144/H-2Kb superposition, without the H-2Kb-bound peptide, is shown.
In the region where classical MHC-Ia molecules bind the C terminus of the peptide, conserved residues of the F pocket include Tyr84, Thr143, Lys146, and Trp147.28-30 These four residues are deleted in m144 (Figure 3). The equivalent to Tyr84 is part of a two residue deletion at the C-terminal region of the α1 helix (between m144 residues 82 and 83), and the equivalents of 143, 146, and 147 are contained within the large deletion including the end of the b8 strand and extending through the amino-terminal half of the α2 helix. The lack of these crucial conserved residues of the F pocket establishes a structural basis for the failure of m144 to bind peptides. (The location of these residues crucial for binding peptides in H-2Kb as compared to this region in m144 is illustrated in Figure 5(d) and (e).)
The understanding of the structural basis by which MHC-I molecules bind peptide is illustrated most clearly by the presence of pockets on the surface of the molecule that accommodate the amino acid side-chains of anchor residues of the peptide.28,31 In contrast to peptide-binding MHC-I molecules, members of the CD1 family that bind various lipids have a distinct binding cleft structure.32 In Figure 6, we compare the surface representation of m144 with that of H-2Kb and of CD1d. m144 lacks any well-defined pockets or channels that are clearly seen in the other two molecules.
Figure 6.
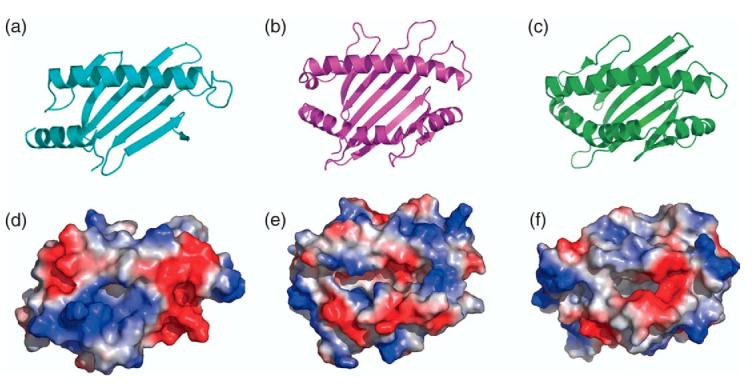
m144 shows no apparent binding groove. Ribbon illustrations of the α1α2 region are shown for: (a) m144, (b) H-2Kb, (c) murine CD1d (PDB designation 2AKR). Electrostatic surface representations (built with PYMOL, see Experimental Procedures), are shown in corresponding panels (d), (e), and (f).
Another characteristic of the classical MHC-Ia molecules is the conservation of a number of salt bridges that stabilize the MHC-I structure.28 The most important of these stabilize the interactions between strand and helix, and between the heavy chain and β2m. The conserved salt bridge stabilizing the α1 helix to the β4 strand, between Arg44 and Asp61 of classical MHC-I molecules (see Figure 3, H-2Kb) is missing in m144. Perhaps this is compensated for by the unique α1 helix to β2 strand disulfide bond (Cys70 to Cys28) found in m144.
Clues to ligand interaction: carbohydrate addition sites, CD8 binding site, NK recognition sites
Clues to the site where a ligand might interact with the m144 molecule, though speculative, maybe gathered from analysis of the location and conservation of predicted aspariginyl-carbohydrate addition sites. Classical human MHC-I molecules have a conserved carbohydrate addition site at position 86, at the carboxyl-terminal end of the α1 helix, and most murine classical MHC-I molecules have an additional site at the symmetrical position at the carboxyl-terminal part of the α2 helix at position 176. m144, by contrast, has N-linked carbohydrate addition sites at positions 45 (at the beginning of strand β4), 55 (at the beginning of the α1 helix), at position 82, past the end of the α1 helix (a site close to the conserved site, position 86, of MHC-I molecules), and at the beginning of strand β7 residue 111. The locations of the m144 N-linked carbohydrate addition sites, illustrated in Figure 7, do not appear to be in position to interfere sterically with interactions with a molecule that might bind m144 across the α1 and α2 helices in a manner similar to that of either an αβ T cell receptor (TCR),33to that of an Ig-like NK receptor such as a KIR2D molecule,34 or to that of a C-type lectin-like NK activating receptor such as NKG2D.35 Either of these modes of interaction would seem to be permissible for m144. However, the carbohydrate addition site at position 111 would appear to provide a degree of steric hindrance to interactions in the region that serves as the CD8 binding site of MHC-Ia and MHC-Ib molecules,36-38 or that serves as the binding site for NK receptors of the Ly49 family, such as Ly49A and Ly49C, which interact with MHC-I in this region.34,39,40
Figure 7.
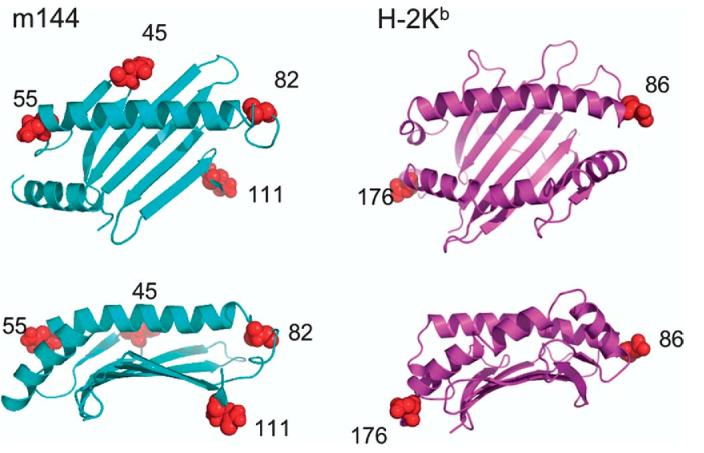
Location of N-asparaginyl carbohydrate addition sites. Location of asparagine residues expected to be glycosylated in the mature m144 and H-2Kb proteins are indicated as red spheres. No sites exist in the α3 domain of these molecules.
MHC-Ia and some MHC-Ib molecules (notably TL) interact with the CD8 coreceptor largely through α3 domain interactions.36-38,41 In particular, the region from H-2Kb 220 to 227 (NGEELIQD) and the highly conserved sequence in TL are structurally important in the CD8 interaction. It is notable, then, that m144 introduces an additional four residues in this extended loop, providing a structural suggestion that this molecule does not interact with CD8.
Does m144 conserve the amino acid residues of the classical MHC-I molecules, H-2Dd and H-2Kb that interact with their respective Ly49A and Ly49C ligands, as determined crystallographically and confirmed by mutational analysis? The major residues of the MHC-Ia molecules shown to contact Ly49 ligands and confirmed by mutagenesis and binding studies are: Arg6, Asp122, Lys243, and β2m residues Gln29 and Lys58 for the H-2Dd/Ly49A interaction.42-44 For Ly49C, the H-2Kb interaction has been analyzed structurally, implicating Asp122 and Lys243 as well as a number of other residues.39It is interesting to note that these most crucial residues for Ly49 interaction, Arg6, Asp122, Lys243 are conserved in the m144 residues Arg12, Glu112, and Lys209 (Figure 3).
Discussion
The classical description of MHC molecules as cell surface receptors that bind peptide ligands to generate complexes detected by TCR and NK receptors has steadily given way to a more general view that the MHC fold may serve as a structural scaffold for a number of different functions, some immunological and some not. The identification of MHC-like genes in the DNA viruses and the demonstration that several of these encode molecules that behave as immunoevasins has provided the impetus for a more complete biochemical and structural analysis. Although ligands for some of the MHC-Iv molecules have been identified, detailed structural studies have been lacking. The description of the m144 structure reported here offers a tangible view of the evolution of an MHC-Iv molecule that contributes to viral virulence.
Molecules that contain an MHC fold serve a wide variety of functions, and their ligands belong to a number of different molecular families. Although no ligand for m144 has yet been identified, the known function of this molecule with respect to recognition by NK cells suggests that its primary molecular mechanism may be one of two: to be expressed at the cell surface of the MCMV infected cell to serve directly as a ligand for a NK cell inhibitory molecule; or to be expressed intracellularly in the infected cell to bind and sequester molecules, induced by the stress of viral infection, that would serve as ligands for NK activating receptors. It has been shown that the MHC-Iv molecule, m157, functions by the first mechanism,8,16 and m145, m152 and m155 function by the second.19-21,45
m144 can be detected at the surface of MCMV infected cells shortly after infection,46 and functional data suggest that it interferes with NK recognition.24,47,48 A viral mutant bearing an m144 deletion is cleared more efficiently in vivo than its wild-type counterpart.47 This phenotype is dependent upon NK cells of the host, and can be reversed by treating the host with NK cell-depleting anti-GM1 antibodies. In addition, the tumorigenicity of the RMA-S cell line can be enhanced by transfected expression of m144,24 and m144 transfection of target cells partially blocks antibody-dependent cellular cytoxicity by mouse NK cells.48 Cultured myoblasts, normally acutely rejected upon transplantation into normal syngeneic mice, can be tolerated if expressing transfected m144.49
The structure of m144 reveals several features that were predicted by amino acid sequence alignment and three-dimensional structure prediction programs: m144 preserves the MHC fold and it associates with the light chain β2m in a manner similar to other MHC-I molecules. Our biochemical analysis of the in vitro refolded m144 indicates that its assembly with β2m is weak. The structure also confirms the experimental observation that no peptides copurify with the recombinant molecule.22TAP function, which provides peptides for stable assembly of MHC-I molecules, was not required for cell surface expression of m144. Also, no peptide addition was required for proper refolding of the E. coli expressed molecules. The lack of electron density in the putative peptide binding cleft, and the substitution of several amino acid residues that structurally are required for binding the amino and carboxyl termini of the peptide are further evidence for the lack of peptide ligands. We cannot formally rule out the possibility that other non-peptide ligands such as lipids or carbohydrate might be bound by m144. The lack of electron density in the cleft region and the lack of well defined pockets or channels in the molecule argue against this possibility.
Additional unexpected features of m144 have been elucidated by the determination of its three-dimensional structure. The unique intradomain disulfide bond linking the α1 helix with the β2 strand adds structural stability, and almost certainly explains the observed peptide-free thermal stability of m144.22 Our efforts to directly assess the importance of this unique disulfide bond using in vitro mutagenesis have been thwarted by the decreased solubility of mutants lacking either or both α1 domain cysteine residues. A large deletion encompassing what would be the b8 strand and the amino-terminal end of the region that would be the α2 helix, a unique characteristic of m144, eliminates residues that contribute to the F pocket (which anchors the peptide carboxyl terminus in MHC-I molecules). The lack of bound peptide might suggest that m144 has no physiological requirement to interact with components of the peptide loading complex that facilitate this process for MHC-Ia molecules.50,51 A crucial step in the loading of peptide into MHC-I and the selection of peptide ligands is the bridging of unfolded MHC-I with the peptide transporter, TAP, by tapasin. Amino acid residues of MHC-I that influence the interaction of the peptide-free MHC-I molecule in the endoplasmic reticulum with tapasin include residues 70, 86, 115, 116, 122, 128-136, and 151 in the α1α2 domain, and, in the α3 domain, 219-233.52 Although m144 conserves residues equivalent to MHC-I 70, 86, 116, and 122, its deletion of the b8 strand (133-135 in H-2Kb) would appear to significantly impair tapasin interaction. In addition, the insertion in m144 of four residues into the region equivalent to the MHC-Ia α3 domain 219-233 loop would also be expected to impede tapasin interaction.
Lack of similarity of m144 in its α3 domain to the CD8 binding site of MHC-I molecules leads to the prediction that this viral molecule cannot bind the CD8 coreceptor. Consideration of the location of the four carbohydrate addition sites of m144, in particular the lack of positions that might block interaction across the α1 and α2 helices, and the presence of the carbohydrate at position 111, suggest that NK receptors of either the Ig or C-type lectin-like families might bind through the α1 and α2 helices. The possibility of an interaction involving the b-sheet floor, the α3 domain, and β2m, similar to the site of the Ly49/H-2 interaction, cannot be eliminated, though we would consider this unlikely.
The region of m144 not visualized in the electron density map, extending from amino acid residues Asp115 to Asp127, most likely represents a region of structural flexibility, and thus would be a candidate for the site of NK receptor interaction. Lack of electron density reveals the absence of a canonical structure in the repeating asymmetric units as propagated in the crystal, and suggests that, despite being tethered at both ends by amino acid residues of well-defined structure, this part of the molecule differs from one instance to another. The best example of an MHC-Ib molecule that has a specific region lacking electron density is that of MICA, which, remarkably, shows no density in a similar region of its α2 helix.53 However, in the complex of MICA with the NKG2D activating receptor, this region of the molecule tightens up, presumably as a result of this interaction, and is then revealed in electron density.25 We suggest that the undefined region between residues 115 and 127 is a flexible region, primarily a-helical, that in the absence of the m144 ligand explores a variety of positions in conformational space, and in the presence of its ligand favors one of these.
Another line of evidence is also consistent with the involvement of this flexible region with a host ligand. Comparison of the amino acid sequence of m144 with its rat viral homolog, r144 (Figure 3), indicates that this molecule, with 36% identity to m144 over the extracellular domains, preserves the novel disulfide bond between the α1-helix and β2-strand, but has unique predicted N-asparaginyl carbohydrate addition sites (r144 has lost sites at 45, 55, and 111, preserved the one at 82, and has a new site at 121 (m144 numbering)). Strikingly, m144 and r144 differ the most in the region from residue 100 to 132 (only 13% identical, with two residues deleted in r144), the same region that lacks electron density in m144. Because of the apparent rapid evolution of these MHC-Iv molecules, in the context of rapidly evolving host responses,54 such variability further supports the view that this region may be involved in interaction with host NK receptors.
In summary, we describe the X-ray structure of an MHC-Iv molecule, m144, encoded by the murine CMV. Despite low (29%) amino acid sequence identity with MHC-Ia molecules, m144 preserves the MHC-I fold and β2m association, but lacks bound peptide or other identifiable small molecule ligand. A unique disulfide bond between the α1-helix and the β2-strand stabilizes the molecule. Further efforts to identify murine host ligand(s) for m144, and the comparative structure of other MHC-Iv molecules will be helpful in revealing not only the mechanism of action of these molecules, but also the nature of the evolutionary forces that have molded their structure.
Experimental Procedures
Tansfection and analysis of m144 cell surface expression
RMA and RMA-S cell lines,23 maintained in RPMI medium supplemented with heat inactivated fetal calf serum (10%, v/v) were transfected with pIRES-hr-GFP II vector (Strategene) encoding either H-2Dd or N-terminally FLAG-tagged m144 using the Amaxa nucleofector (3×106 cells were transfected with 5 mg DNA in solution T, using program A30). Cells were incubated at 37 8C in RPMI without serum for 18 h. Surface expression of m144 was detected by staining with the anti-FLAG antibody M2 (Sigma) and anti-mouse IgG1-PE (Southern Biotech). H-2Dd expression was detected by staining with anti-mouse H-2Dd monoclonal antibody 34-5-8S conjugated to PE (BD Pharmingen). The cells were analyzed by flow cytometry in a FACScan (Becton Dickinson). Cells transfected with the parental vector were used as a control for background staining. As a positive control for peptide dependence, H-2Dd transfected cells were incubated with or without H-2Dd-specific peptide P18-I10 (RGPGRAFVTI). Dead cells were excluded from the analysis using propidium iodide gating.
Bacterial expression, in vitro folding, and purification of the m144/β2m complex
DNA encoding the full-length m144 protein was obtained by polymerase chain reaction (PCR) amplification performed directly on supernatants derived from mouse salivary gland cultures infected with the Smith strain of MCMV (obtained from the American Type Culture Collection, Manassas, VA, catalog no. VR-1399). The forward and reverse primers were: 5’-ATGGACTCGGCGAGACGAAACACGTCTCCAGG and 5’-AATGCTGGGATCCGGGACCGTGACGATCGGACC, respectively, and were based on the m144 sequence of the Smith strain in GenBank, accession no. U68299.55 The amplified fragment was cloned into pCR4-TOPO (Invitrogen, Carlsbad, CA) for sequence verification and to serve as template for sub-cloning. DNA encoding the extracellular portion of m144 encompassing Gly2 to Gly249 (numbering according to Chapman et al.22), was obtained by PCR amplification with forward and reverse primers incorporating NdeI and XhoI sites, respectively, and cloned into the corresponding sites of pET21b (Novagen, Madison, WI) in frame with the C-terminal hexahistidine tag. Although the histidine tag was included to provide flexibility in purification and concentration, it was not utilized in the steps described below. The bacterial expression vector encoding mouse β2m has been described.56
Exponentially growing bacterial cultures harboring m144 and β2m plasmids were induced with 0.5 mM isopropyl-thio-D-galactoside (IPTG) for 3 h at 37 8C, and inclusion bodies were prepared by overnight lysis of bacterial pellets with lysozyme, sonication, and repeated washes with buffer containing 100 mM Tris (pH 8), 2 mM EDTA, 0.1% (w/v) sodium deoxycholate. Inclusion bodies containing m144 and β2m protein were denatured separately in 6 M guanidine-HCl, 0.1 mM DTT in the above Tris buffer lacking deoxycholate for 2 h at room temperature, and insoluble debris was removed by centrifugation. Denatured β2m was first added to chilled refolding buffer (0.4 M arginine-HCl, 100 mM Tris (pH 8), 2 mM EDTA, 3 mM reduced glutathione, 0.3 mM oxidized glutathione) at a final concentration of approximately 50 mg/ml and placed at 4 8C overnight. After 24 h, denatured m144 was added and left at 4 8C for an additional four days. The refolding mixture was dialyzed against eight volumes of TBS (25 mM Tris (pH 8), 150 mM NaCl), purified by gel-filtration chromatography on a Superdex 75 HR10/30 column (Amersham Biosciences, Uppsala) equilibrated in TBS, followed by anion exchange chromatography on a mono Q HR 5/5 column (Amersham Biosciences), eluted with a linear gradient of 0 to 0.5 M NaCl in 25 mM Tris (pH 8). Peak fractions were pooled, concentrated, and brought to Tris buffer containing 100 mM NaCl. Two major peaks were identified and analyzed further by SDS-PAGE, the first consisted only of the m144 heavy chain, the second of a non-covalent complex of equimolar amounts of m144 and β2m (Supplementary Data, Figure 1). For both peaks, binding to the m144 specific antibody 15C622 was confirmed by surface plasmon resonance on a BIAcore™ 2000 (data not shown). Successful crystallization was accomplished with protein from the peak containing both chains.
Crystallization and X-ray data collection
Selected solutions from the Crystal Screen 1 and 2 kits from Hampton Research (Aliso Viejo, CA) were screened for initial crystallization conditions in hanging drops at 4 8C by mixing 0.5 ml of m144/β2m at 12 mg/ml with an equal volume of reservoir buffer. Crystals grew in two days in solution 22 of the Hampton Crystal Screen 2 kit, 12% (w/v) PEG 20,000 in 0.1 M morpholinoethanesulfonic acid (Mes) (pH 6.5). Reduction of the PEG 20,000 concentration to 10% yielded larger crystals suitable for diffraction analyses. Crystals were cryoprotected in reservoir solution containing 15% ethylene glycol, flash-frozen in liquid nitrogen, and shipped to NSLS, Brookhaven. Data were collected at beamline X26C equipped with an ADSC Quantum 4 Detector. Crystals belong to space group C121 with unit cell dimensions a=133.5 Å, b=51.2 Å, c=71.8 Å, a=γ=90.08, and b=105.78 and contain one m144/β2m molecule per asymmetric unit. Data from a single crystal were indexed, integrated, and scaled with HKL2000,57 yielding a dataset that was 95.3% complete at 1.91 Å (see Table 1). Data were not collected to a higher resolution due to the orientation of the long axis, which required a re-orientation of the crystal, a maneuver not feasible at the time.
Structure determination and refinement
The structure of m144 was solved by molecular replacement using AMoRe58 as implemented in the CCP4 suite.59 The search model was the human hemochromatosis protein Hfe,60 Protein Data Bank (PDB61) accession code 1A6Z, which was modified by replacing amino acids of the human β2m subunit with those of mouse β2m. Additionally, the Hfe heavy chain was deleted of those residues absent in alignment with m144 and was substituted with alanine at those positions where the equivalent amino acid was not identical. Amino acid replacements were performed in XtalView.62 The search with this modified Hfe probe using data from 8-4 Å yielded a solution with a correlation coefficient (CC)Z23.6 and RZ51.1. The second best solution had Cc=18.4. Rigid body refinement carried out in REFMAC563 using data from 20-3 Å and splitting the molecule into three domains (α1α2, α3, and β2m) dropped Rfree to 46.6% (Rfree was calculated using 5% of the data). The rigid-body refined solution served as the starting point for automatic model building in ARP/wARP,64 yielding a model in which 307 of 347 residues were successfully traced. This model was then subjected to 100 cycles of energy minimization in CNS65 which resulted in RZ29.6% and Rfree Z32.9%. Further rounds of manual building in XtalView, alternating with standard crystallographic refinement protocols such as energy minimization, simulated annealing, and group B-factor refinement in CNS, provided the final model of m144 which has RZ20.5% and RfreeZ23.0% for all data to 1.9 Å with no s cutoff applied. Amide flips of Asn, Gln, and His were chosen following addition of hydrogen atoms with REDUCE and analysis with MOLPROBITY.66 Rotamers were selected based on the library available†. The model geometry was examined with PROCHECK.67 No electron density was observed for residues Asp115 to Asp127 of the m144 heavy chain and these were omitted from the crystallographic model. The carboxyl-terminal Met99 of β2m was also not visualized. (In an effort to visualize additional electron density, TLS refinement68 was also used. Although modest improvement in Rcryst and Rfree was achieved, geometrical parameters as assessed with PROCHECK66 were somewhat less favorable, so we report the final model without TLS refinement. No additional electron density was seen.) Model refinement statistics are listed in Table 1. Analysis of structural features was performed with programs from CCP4 and CNS, as well as HINGE‡. HINGE calculates an ellipsoid (defined by axes a, b, and c) for each indicated domain, and reports the angle between the long axes of the adjacent domains as the hinge angle. This is illustrated schematically in Figure 2(c). Graphic displays were generated with PYMOL§. Computational prediction of protein secondary structure was accomplished with Predict Protein.69
Protein Data Bank accession codes
Atomic coordinates have been deposited in the RCSB Protein Data Bank61 under accession code 1U58. (Following completion of the refinement of our structure, a similar structure of the same molecule, determined to 2.1 Å, was released from the PDB as 1PQZ.)
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
We thank L. Boyd, R. Carey, K. Larabee, D. Levin, M. Mage, M. Paskow, and J. Revilleza for comments on the manuscript. R.G. and R.A.M. are supported by National Institutes of Health grant AI47990. This research was supported in part by the Intramural Research Program of the NIAID,NIH.
Footnotes
- MHC-I
- major histocompatibility complex encoded class I molecule
- MHC-Iv
- viral MHC-I-like molecule
- β2m
- β2-microglobulin
- CMV
- cytomegalovirus
- MCMV
- murine cytomegalovirus
- HCMV
- human cytomegalovirus
- NK
- natural killer
- Ig
- immunoglobulin
- PDB
- Protein Data Bank.
References
- 1.Harrison SC, Alberts B, Ehrenfeld E, Enquist L, Fineberg H, McKnight SL, et al. Discovery of antivirals against smallpox. Proc. Natl Acad. Sci. USA. 2004;101:11178–11192. doi: 10.1073/pnas.0403600101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tortorella D, Gewurz BE, Furman MH, Schust DJ, Ploegh HL. Viral subversion of the immune system. Annu. Rev. Immunol. 2000;18:861–926. doi: 10.1146/annurev.immunol.18.1.861. [DOI] [PubMed] [Google Scholar]
- 3.Roizman B, Whitley RJ, Lopez C. The Human Herpesviruses. Raven Press; New York: 1993. [Google Scholar]
- 4.Arase H, Lanier LL. Specific recognition of virus-infected cells by paired NK receptors. Rev. Med. Virol. 2004;14:83–93. doi: 10.1002/rmv.422. [DOI] [PubMed] [Google Scholar]
- 5.Moss P, Khan N. CD8(C) T-cell immunity to cytomegalovirus. Hum. Immunol. 2004;65:456–464. doi: 10.1016/j.humimm.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 6.Sester M, Gartner BC, Sester U, Girndt M, Mueller-Lantzsch N, Kohler H. Is the cytomegalovirus serologic status always accurate? A comparative analysis of humoral and cellular immunity. Transplantation. 2003;76:1229–1230. doi: 10.1097/01.TP.0000083894.61333.56. [DOI] [PubMed] [Google Scholar]
- 7.Farrell H, Degli-Esposti M, Densley E, Cretney E, Smyth M, Davis-Poynter N. Cytomegalovirus MHC class I homologues and natural killer cells: an overview. Microbes Infect. 2000;2:521–532. doi: 10.1016/s1286-4579(00)00315-4. [DOI] [PubMed] [Google Scholar]
- 8.Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl Acad. Sci. USA. 2002;99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basta S, Bennink JR. A survival game of hide and seek: cytomegaloviruses and MHC class I antigen presentation pathways. Viral Immunol. 2003;16:231–242. doi: 10.1089/088282403322396064. [DOI] [PubMed] [Google Scholar]
- 10.Saverino D, Ghiotto F, Merlo A, Bruno S, Battini L, Occhino M, et al. Specific recognition of the viral protein UL18 by CD85j/LIR-1/ILT2 on CD8CT cells mediates the non-MHC-restricted lysis of human cytomegalovirus-infected cells. J. Immunol. 2004;172:5629–5637. doi: 10.4049/jimmunol.172.9.5629. [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Botet M, Llano M, Ortega M. Human cytomegalovirus and natural killer-mediated surveillance of HLA class I expression: a paradigm of host-pathogen adaptation. Immunol. Rev. 2001;181:193–202. doi: 10.1034/j.1600-065x.2001.1810116.x. [DOI] [PubMed] [Google Scholar]
- 12.Cosman D, Fanger N, Borges L. Human cytomegalovirus, MHC class I and inhibitory signalling receptors: more questions than answers. Immunol. Rev. 1999;168:177–185. doi: 10.1111/j.1600-065x.1999.tb01292.x. [DOI] [PubMed] [Google Scholar]
- 13.Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, Hsu ML. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity. 1997;7:273–282. doi: 10.1016/s1074-7613(00)80529-4. [DOI] [PubMed] [Google Scholar]
- 14.Vitale M, Castriconi R, Parolini S, Pende D, Hsu ML, Moretta L, et al. doi: 10.1093/intimm/11.1.29. [DOI] [PubMed] [Google Scholar]
- 15.Wills MR, Ashiru O, Reeves MB, Okecha G, Trowsdale J, Tomasec P, et al. Human cytomegalovirus encodes an MHC class I-like molecule (UL142) that functions to inhibit NK cell lysis. J. Immunol. 2005;175:7457–7465. doi: 10.4049/jimmunol.175.11.7457. [DOI] [PubMed] [Google Scholar]
- 16.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 17.Voigt V, Forbes CA, Tonkin JN, Degli-Esposti MA, Smith HR, Yokoyama WM, Scalzo AA. Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc. Natl Acad. Sci. USA. 2003;100:13483–13488. doi: 10.1073/pnas.2233572100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.French AR, Pingel JT, Wagner M, Bubic I, Yang L, Kim S, et al. Escape of mutant double-stranded DNA virus from innate immune control. Immunity. 2004;20:747–756. doi: 10.1016/j.immuni.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, Lanier LL. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J. Exp. Med. 2003;197:1245–1253. doi: 10.1084/jem.20021973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krmpotic A, Hasan M, Loewendorf A, Saulig T, Halenius A, Lenac T, et al. NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J. Exp. Med. 2005;201:211–220. doi: 10.1084/jem.20041617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lodoen MB, Abenes G, Umamoto S, Houchins JP, Liu F, Lanier LL. The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J. Exp. Med. 2004;200:1075–1081. doi: 10.1084/jem.20040583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapman TL, Bjorkman PJ. Characterization of a murine cytomegalovirus class I major histocompatibility complex (MHC) homolog: comparison to MHC molecules and to the human cytomegalovirus MHC homolog. J. Virol. 1998;72:460–466. doi: 10.1128/jvi.72.1.460-466.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ljunggren HG, Stam NJ, Ohlen C, Neefjes JJ, Hoglund P, Heemels MT, et al. Empty MHC class I molecules come out in the cold. Nature. 1990;346:476–480. doi: 10.1038/346476a0. [DOI] [PubMed] [Google Scholar]
- 24.Cretney E, Degli-Esposti MA, Densley EH, Farrell HE, Davis-Poynter NJ, Smyth MJ. m144, a murine cytomegalovirus (MCMV)-encoded major histocompatibility complex class I homologue, confers tumor resistance to natural killer cell-mediated rejection. J. Exp. Med. 1999;190:435–444. doi: 10.1084/jem.190.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li P, Morris DL, Willcox BE, Steinle A, Spies T, Strong RK. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nature Immunol. 2001;2:443–451. doi: 10.1038/87757. [DOI] [PubMed] [Google Scholar]
- 26.Ribaudo RK, Margulies DH. Polymorphism at position nine of the MHC class I heavy chain affects the stability of association with beta 2-microglobulin and presentation of a viral peptide. J. Immunol. 1995;155:3481–3493. [PubMed] [Google Scholar]
- 27.Pearl F, Todd A, Sillitoe I, Dibley M, Redfern O, Lewis T, et al. The CATH Domain Structure Database and related resources Gene3D and DHS provide comprehensive domain family information for genome analysis. Nucl. Acids Res. 2005;33:D247–D251. doi: 10.1093/nar/gki024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saper MA, Bjorkman PJ, Wiley DC. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 Å resolution. J. Mol. Biol. 1991;219:277–319. doi: 10.1016/0022-2836(91)90567-p. [DOI] [PubMed] [Google Scholar]
- 29.Doytchinova I, Flower D. The HLA-A2supermotif: a QSAR definition. Org. Biomol. Chem. 2003;1:2648–2654. doi: 10.1039/b300707c. [DOI] [PubMed] [Google Scholar]
- 30.Matsumura M, Fremont DH, Peterson PA, Wilson IA. Emerging principles for the recognition of peptide antigens by MHC class I molecules. Science. 1992;257:927–934. doi: 10.1126/science.1323878. [DOI] [PubMed] [Google Scholar]
- 31.Garrett TP, Saper MA, Bjorkman PJ, Strominger JL, Wiley DC. Specificity pockets for the side chains of peptide antigens in HLA-Aw68. Nature. 1989;342:692–696. doi: 10.1038/342692a0. [DOI] [PubMed] [Google Scholar]
- 32.Zajonc DM, Maricic I, Wu D, Halder R, Roy K, Wong CH, et al. Structural basis for CD1d presentation of a sulfatide derived from myelin and its implications for autoimmunity. J. Exp. Med. 2005;202:1517–1526. doi: 10.1084/jem.20051625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rudolph MG, Luz JG, Wilson IA. Structural and thermodynamic correlates of T cell signaling. Annu. Rev. Biophys. Biomol. Struct. 2002;31:121–149. doi: 10.1146/annurev.biophys.31.082901.134423. [DOI] [PubMed] [Google Scholar]
- 34.Natarajan K, Dimasi N, Wang J, Mariuzza RA, Margulies DH. Structure and function of natural killer cell receptors: multiple molecular solutions to self, nonself discrimination. Annu. Rev. Immunol. 2002;20:853–885. doi: 10.1146/annurev.immunol.20.100301.064812. [DOI] [PubMed] [Google Scholar]
- 35.Strong RK, McFarland BJ. NKG2D and related immunoreceptors. Advan. Protein Chem. 2004;68:281–312. doi: 10.1016/S0065-3233(04)68008-9. [DOI] [PubMed] [Google Scholar]
- 36.Kern PS, Teng MK, Smolyar A, Liu JH, Liu J, Hussey RE, et al. Structural basis of CD8 coreceptor function revealed by crystallographic analysis of a murine CD8alphaalpha ectodomain fragment in complex with H-2Kb. Immunity. 1998;9:519–530. doi: 10.1016/s1074-7613(00)80635-4. [DOI] [PubMed] [Google Scholar]
- 37.Gao GF, Tormo J, Gerth UC, Wyer JR, McMichael AJ, Stuart DI, et al. Crystal structure of the complex between human CD8alpha(alpha) and HLA-A2. Nature. 1997;387:630–634. doi: 10.1038/42523. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Xiong Y, Naidenko OV, Liu JH, Zhang R, Joachimiak A, et al. The crystal structure of a TL/CD8alphaalpha complex at 2.1 Å resolution: implications for modulation of T cell activation and memory. Immunity. 2003;18:205–215. doi: 10.1016/s1074-7613(03)00027-x. [DOI] [PubMed] [Google Scholar]
- 39.Dam J, Guan R, Natarajan K, Dimasi N, Chlewicki LK, Kranz DM, et al. Variable MHC class I engagement by Ly49 natural killer cell receptors demonstrated by the crystal structure of Ly49C bound to H-2Kb. Nature Immunol. 2003;4:1213–1222. doi: 10.1038/ni1006. [DOI] [PubMed] [Google Scholar]
- 40.Tormo J, Natarajan K, Margulies DH, Mariuzza RA. Crystal structure of a lectin-like natural killer cell receptor bound to its MHC class I ligand. Nature. 1999;402:623–631. doi: 10.1038/45170. [DOI] [PubMed] [Google Scholar]
- 41.Devine L, Rogozinski L, Naidenko OV, Cheroutre H, Kavathas PB. The complementarity-determining region-like loops of CD8 alpha interact differently with β2-microglobulin of the class I molecules H-2Kb and thymic leukemia antigen, while similarly with their alpha 3 domains. J. Immunol. 2002;168:3881–3886. doi: 10.4049/jimmunol.168.8.3881. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto N, Yokoyama WM, Kojima S, Yamamoto K. The NK cell MHC class I receptor Ly49A detects mutations on H-2Dd inside and outside of the peptide binding groove. J. Immunol. 2001;166:4422–4428. doi: 10.4049/jimmunol.166.7.4422. [DOI] [PubMed] [Google Scholar]
- 43.Matsumoto N, Mitsuki M, Tajima K, Yokoyama WM, Yamamoto K. The functional binding site for the C-type lectin-like natural killer cell receptor Ly49A spans three domains of its major histocompatibility complex class I ligand. J. Exp. Med. 2001;193:147–158. doi: 10.1084/jem.193.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Whitman MC, Natarajan K, Tormo J, Mariuzza RA, Margulies DH. Binding of the natural killer cell inhibitory receptor Ly49A to its major histocompatibility complex class I ligand. Crucial contacts include both H-2Dd and β2-microglobulin. J. Biol. Chem. 2002;277:1433–1442. doi: 10.1074/jbc.M110316200. [DOI] [PubMed] [Google Scholar]
- 45.Bubic I, Wagner M, Krmpotic A, Saulig T, Kim S, Yokoyama WM, et al. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J. Virol. 2004;78:7536–7544. doi: 10.1128/JVI.78.14.7536-7544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andrews DM, Andoniou CE, Granucci F, Ricciardi-Castagnoli P, Degli-Esposti MA. Infection of dendritic cells by murine cytomegalovirus induces functional paralysis. Nature Immunol. 2001;2:1077–1084. doi: 10.1038/ni724. [DOI] [PubMed] [Google Scholar]
- 47.Farrell HE, Vally H, Lynch DM, Fleming P, Shellam GR, Scalzo AA, Davis-Poynter NJ. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature. 1997;386:510–514. doi: 10.1038/386510a0. [DOI] [PubMed] [Google Scholar]
- 48.Kubota A, Kubota S, Farrell HE, Davis-Poynter N, Takei F. Inhibition of NK cells by murine CMV-encoded class I MHC homologue m144. Cell. Immunol. 1999;191:145–151. doi: 10.1006/cimm.1998.1424. [DOI] [PubMed] [Google Scholar]
- 49.Hodgetts SI, Spencer MJ, Grounds MD. A role for natural killer cells in the rapid death of cultured donor myoblasts after transplantation. Transplantation. 2003;75:863–871. doi: 10.1097/01.TP.0000053754.33317.4B. [DOI] [PubMed] [Google Scholar]
- 50.Pamer E, Cresswell P. Mechanisms of MHC class I—restricted antigen processing. Annu. Rev. Immunol. 1998;16:323–358. doi: 10.1146/annurev.immunol.16.1.323. [DOI] [PubMed] [Google Scholar]
- 51.Turnquist HR, Vargas SE, Schenk EL, McIlhaney MM, Reber AJ, Solheim JC. The interface between tapasin and MHC class I: identification of amino acid residues in both proteins that influence their interaction. Immunol. Res. 2002;25:261–269. doi: 10.1385/ir:25:3:261. [DOI] [PubMed] [Google Scholar]
- 52.Bouvier M. Accessory proteins and the assembly of human class I MHC molecules: a molecular and structural perspective. Mol. Immunol. 2003;39:697–706. doi: 10.1016/s0161-5890(02)00261-4. [DOI] [PubMed] [Google Scholar]
- 53.Li P, Willie ST, Bauer S, Morris DL, Spies T, Strong RK. Crystal structure of the MHC class I homolog MIC-A, a gammadelta T cell ligand. Immunity. 1999;10:577–584. doi: 10.1016/s1074-7613(00)80057-6. [DOI] [PubMed] [Google Scholar]
- 54.Nylenna O, Naper C, Vaage JT, Woon PY, Gauguier D, Dissen E, et al. The genes and gene organization of the Ly49 region of the rat natural killer cell gene complex. Eur. J. Immunol. 2005;35:261–272. doi: 10.1002/eji.200425429. [DOI] [PubMed] [Google Scholar]
- 55.Rawlinson WD, Farrell HE, Barrell BG. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996;70:8833–8849. doi: 10.1128/jvi.70.12.8833-8849.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shields MJ, Moffat LE, Ribaudo RK. Functional comparison of bovine, murine, and human beta2-microglobulin: interactions with murine MHC I molecules. Mol. Immunol. 1998;35:919–928. doi: 10.1016/s0161-5890(98)00083-2. [DOI] [PubMed] [Google Scholar]
- 57.Otiwinowski Z, Minor W. AMoRe: an automated package for molecular replacement. Acta Crystallog. sect. A. 1994;50:157–163. [Google Scholar]
- 58.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallog. sect. A. 1994;50:157–163. [Google Scholar]
- 59.Collaborative Computational Project, No. 4 The CCP4 suite: programs for protein crystallography. Acta Crystallog. sect. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 60.Lebron JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, et al. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell. 1998;93:111–123. doi: 10.1016/s0092-8674(00)81151-4. [DOI] [PubMed] [Google Scholar]
- 61.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucl. Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McRee DE. XtalView/Xfit—Aa versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 63.Murshudov GN. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallog. sect. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 64.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nature Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 65.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallog. sect. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 66.Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins: Struct. Funct. Genet. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 67.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 68.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallog. sect. D. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 69.Rost B, Yachdav G, Liu J. The Predict Protein server. Nucl. Acids Res. 2004;32:W321–W326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 71.Gouet P, Robert X, Courcelle E. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucl. Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Risler J, Delorme M, Delacroix H, Henaut A. Amino acid substitutions in structurally related proteins. A pattern recognition approach. Determination of a new and efficient scoring matrix. J. Mol. Biol. 1988;204:1019–1029. doi: 10.1016/0022-2836(88)90058-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.