

Abstract
We examined the chemical reactions influencing dissolved concentrations, speciation, and transport of naturally occurring arsenic (As) in a shallow, sand and gravel aquifer with distinct geochemical zones resulting from land disposal of dilute sewage effluent. The principal geochemical zones were: (1) the uncontaminated zone above the sewage plume [350 μM dissolved oxygen (DO), pH 5.9]; (2) the suboxic zone (5 μM DO, pH 6.2, elevated concentrations of sewage-derived phosphate and nitrate); and (3) the anoxic zone [dissolved iron(II) 100–300 μM, pH 6.5–6.9, elevated concentrations of sewage-derived phosphate]. Sediments are comprised of greater than 90% quartz but the surfaces of quartz and other mineral grains are coated with nanometer-size iron (Fe) and aluminum (Al) oxides and/or silicates, which control the adsorption properties of the sediments. Uncontaminated groundwater with added phosphate (620 μM) was pumped into the uncontaminated zone while samples were collected 0.3 m above the injection point. Concentrations of As(V) increased from below detection (0.005 μM) to a maximum of 0.07 μM during breakthrough of phosphate at the sampling port; As(III) concentrations remained below detection. These results are consistent with the hypothesis that naturally occurring As(V) adsorbed to constituents of the coatings on grain surfaces was desorbed by phosphate in the injected groundwater. Also consistent with this hypothesis, vertical profiles of groundwater chemistry measured prior to the tracer test showed that dissolved As(V) concentrations increased along with dissolved phosphate from below detection in the uncontaminated zone to approximately 0.07 and 70 μM, respectively, in the suboxic zone. Concentrations of As(III) were below detection in both zones. The anoxic zone had approximately 0.07 μM As(V) but also had As(III) concentrations of 0.07–0.14 μM, suggesting that release of As bound to sediment grains occurred by desorption by phosphate, reductive dissolution of Fe oxides, and reduction of As(V) to As(III), which adsorbs only weakly to the Fe-oxide-depleted material in the coatings. Results of reductive extractions of the sediments suggest that As associated with the coatings was relatively uniformly distributed at approximately 1 nmol/g of sediment (equivalent to 0.075 ppm As) and comprised 20%-50% of the total As in the sediments, determined from oxidative extractions. Quartz sand aquifers provide high-quality drinking water but can become contaminated when naturally occurring arsenic bound to Fe and Al oxides or silicates on sediment surfaces is released by desorption and dissolution of Fe oxides in response to changing chemical conditions.
Introduction
Biogeochemical reactions in aquifers can mobilize naturally occurring arsenic (As) from sediments that are not enriched in As compared to its average abundance in crustal rocks[1,2] Aquifers with dissolved As concentrations significantly higher than 0.67 micromoles per liter (μM), which equals 50 micrograms per liter (μg/l), that supply large populations with drinking water have received considerable attention. [1-3] With the recent lowering of the maximum contaminant level (MCL) for As in drinking water in the United States to 0.13 μM (10 μg/l), interest in understanding the fate and transport of As in potential drinking-water supplies is likely to intensify. [4-7] In addition to its potential impact on human health, scientific interest in As in groundwater has been stimulated by the potential for using it to gain insight into oxidation-reduction reactions[8] and, more recently, the role of microbial communities in As cycling. [9] The ultimate source of naturally occurring As in most aquifers is likely to be sulfide minerals because the As content of these minerals greatly exceeds those of other rock-forming minerals that typically comprise aquifer sediments. [1] Arsenic in pyrite and other metal sulfide minerals exists in low oxidation states, primarily – 1 or O. [10] In contrast, the dominant oxidation states of As in groundwater are As(III) (As in the plus 3 oxidation state) and As(V). [1] The biogeochemical reactions and hydrologic processes leading to the oxidation of As in primary sedimentary minerals to As(III) and As(V), as well as those reactions that influence the fate and transport of As(III) and As(V) are subjects of continued research. [1,11] Improved understanding of these processes can contribute to improved understanding of subsurface biogeochemistry and could provide important information for maintaining critical drinking water supplies. [12]
The objective of this paper is to analyze the chemical reactions influencing dissolved As concentrations and speciation in a shallow aquifer whose mineralogy is dominated by quartz, feldspars, and other silicate minerals. These minerals react with groundwater mainly through the mineral dissolution and precipitation reactions associated with chemical weathering, which tend to be slow, and, therefore, tend to impart low concentrations of solutes to the groundwater with which they are in contact. [13] However, as a result of these weathering reactions, nanometer-size precipitates of iron (Fe) and aluminum (Al) oxides and silicates can form on the surfaces and interiors of primary mineral grains (e.g., Refs. 14–16). Arsenic released as a result of dissolution of pyrite and other sulfide minerals may be incorporated into these precipitates by adsorption or other processes. Arsenic adsorbed onto these precipitates could be desorbed and, therefore, mobilized in response to changes in chemical conditions. This was examined by determining dissolved As concentrations and speciation in different zones of an aquifer characterized by different chemical conditions owing to land-disposal of dilute sewage effluent to the aquifer. In addition, the results of a field experiment are presented in which the mobilization of As in uncontaminated groundwater above the sewage plume was examined by injecting phosphate, which is a strongly adsorbing anion capable of des-orbing As through competition for adsorption sites. The study was carried out at a field site where previous investigations have made significant contributions to characterizing the site's hydrogeology, [17-23] the biogeochemistry of the sewage plume, [24-27] and the chemical properties of the sediments. [15,28,29] Drawing on this literature in interpreting the results of our investigation, we discuss the impact of groundwater chemistry on As concentrations in the aquifer.
Methods
Site description
The study was carried out in a shallow, unconfined aquifer at the U. S. Geological Survey research site on Cape Cod, MA[17] (Fig. 1). The permeable, glacial outwash sediments consist of coarse-sand and gravel, with lesser fine-sand and silt. [19] Mineralogical analyses have focused on the less-than-2-millimeter size-fractions (< 2 mm), in which approximately 90% of the material is comprised of quartz;[14,15] the remainder consists of potassium and plagioclase feldspars, magnetite, hematite, goethite, glauconite, and lithic fragments. [15,28,30] Material greater than 2 mm in diameter comprises approximately 25% of the sediments and consists of quartz grains and lithic fragments, most of which are granitic in composition. [30] Locally, the hydraulic gradient direction has varied over approximately 15° during the past 15 yr when it has been measured frequently[31] (Fig. 1). The groundwater flow direction follows closely the hydraulic gradient direction[22] and the groundwater velocity has been steady at approximately 0.4 m per day.
Figure 1.
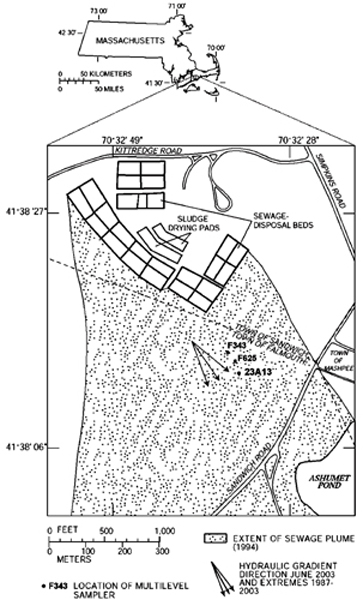
Map, showing locations of sampling sites (23A13, F625, F343), sewage disposal beds, extent of historically sewage-contaminated groundwater (Ref. 32), the hydraulic gradient direction in June 2003 (middle arrow), and, for comparison, extremes in hydraulic gradient direction measured between 1987 and 2003.
Land disposal of dilute, secondary sewage effluent at a sewage-treatment facility upgradient of the site over a 60-yr period resulted in a plume of sewage-contaminated groundwater approximately 6 km long, 1 km wide, and 50 m thick. [17,32] Effluent disposal ceased in December of 1995 and dissolved salt concentrations have steadily decreased since that time. However, the distribution of several characteristic redox-reactive species [dissolved oxygen, nitrate, ammonium, and dissolved Fe(II)], pH values, and distributions of strongly adsorbing sewage contaminants like phosphate and zinc have changed little in the region between the upper boundary and the core of the sewage plume. [27,33-35] Concentrations of dissolved organic carbon are 1 to 2 mg carbon per liter;[36] concentrations of particulate organic carbon on sediments downgradient of the disposal beds are less than 0.01 %. [28,30] The boundary between the upper region of the sewage plume and uncontaminated groundwater above it is marked by steep vertical gradients in groundwater chemistry that are temporally and spatially persistent. [25,37,38]
Water sample collection and analysis
Samples were collected with peristaltic pumps. [32] After purging, a 50 milliliter (ml) filtered [0.45 micrometer (μm) nominal pore size] sample was collected. Approximately 20 ml were transferred to a high-density polyethylene scintillation vial and acidified to pH 2 with trace-metal-grade nitric acid. The remaining 30 ml sample was preserved by adding ethylenediaminetetraacetic acid (EDTA), which sequesters Fe thereby preventing oxidation and precipitation of hydrous ferric oxide, to a concentration of approximately 600 μM and passed through a syringe packed with 500 mg of a strong anion exchanger (SAX, tetramethylammonium on styrene-divinylbenzene base) pretreated with methanol and water. [39] At a pH of 4 (resulting from addition of EDTA), As(V) oxyanions are retained by the SAX whereas the neutral As(III)-hydroxo species passes through. [39] Additional unfiltered samples were collected for determining field parameters, including turbidity, specific conductance,[32] and pH. [34] Dissolved oxygen concentrations below 30 μM were determined in the field;[32] dissolved oxygen concentrations greater than 30 μM were determined with a dissolved oxygen probe on samples collected in thoroughly purged biological-oxygen-demand bottles. [32]
Concentrations of major, minor, and many trace metals and metalloids were determined on the nitric-acid preserved samples using inductively coupled plasma-atomic emission spectrometry (ICPAES) as described elsewhere. [40] Relative precision (2 times the standard deviation divided by the average concentration determined analytically) and accuracy (ratio of the average concentration determined analytically to the known concentration), determined by repeated analyses of multielement standards, for all elements reported here were 5% and 90%–110%, respectively, or better. Concentrations of total dissolved As were determined on nitric-acid-preserved samples using ICP-mass spectrometry (ICPMS). Concentrations of As(III) were determined on EDTA-preserved samples passed through SAX using ICPMS after processing to replace chloride with nitrate because high concentrations of chloride can cause an interference in the determination of As. Processing involved evaporating samples of known volume to dryness. The precipitate was reconstituted in 1 ml of concentrated nitric acid (trace metal grade) and once again evaporated to dryness, driving off chloride as hydrogen chloride vapors. This was repeated at least one more time, after which the sample was reconstituted in a known volume of 0.15 moles per liter (M) nitric acid (trace metal grade). Concentrations of As(V) were determined by difference. In some cases, As(V) concentrations were also determined by eluting the SAX column with 0.15 M nitric acid, processing the eluate as described above to diminish the chloride concentration, and analyzing using ICPMS. In all cases, concentrations of As(V) determined by difference were the same as those determined on the nitric acid eluates of the SAX columns within analytical error. The relative precision and accuracy of the As determinations by ICPMS were 3% and 103%, respectively, at a concentration of 0.033 μM; the limit of quantitation was determined to be 0.005 μM (0.4 μg/l). Analytical errors for the As(V) determinations in the SAX eluates were significantly higher than those for other samples largely because of uncertainties in the volume of sample passed through the SAX.
Tracer test
The tracer test involved pumping 445 1 of groundwater from the uncontaminated zone with added sodium phosphate and sodium chloride (NaCl) into a single port of multilevel sampler (MLS), 23A13 (Fig. 1) at an altitude of 13.2 m. A detailed description of the construction and installation of MLS has been presented elsewhere. [19] The added tracers brought the measured specific conductance to 441 ± 6 micro-siemens per centimeter (μS/cm), the phosphate concentration to 620 ± 30 μM, and the pH to 6.22 ± 0.03. The wells pumped to collect groundwater for the injectate and the pumping rate were chosen to have a minimal impact on the hydraulic gradient near the injection port. Samples were collected as described above.
Sediment sample collection and analysis
Sediment samples were collected using a wire-line coring apparatus as described elsewhere[41] and frozen within 4 h of collection. Sediment samples designated R23AWC2 and R23AWC3 were collected approximately 3.6 and 4.8 m west of 23A13, respectively. Subsequently, sediments were removed from the vinyl core liners, dried in a laminar flow hood, and dry-sieved to separate out gravel-sized material (greater than 2 mm in diameter).
Two types of chemical extractions of sediment samples were conducted. Sediment samples were extracted in 0.25 M hydroxylamine hydrochloride in 0.25 M hydrochloric acid at 50°C for 96 h. [15] For the < 2 mm size fraction, approximately 5 g of sediment were extracted in 25 ml of solution. For the > 2 mm size fraction, approximately 3 g of sediment were extracted in 20 ml of solution. After 96 h, samples were allowed to cool, filtered (0.45 μm), and evaporated to dry-ness. In order to diminish the chloride concentration prior to determining arsenic concentrations by ICPMS, samples were repeatedly dissolved in 1 to 2 ml of trace-metal grade concentrated nitric acid and evaporated to dryness. After 8 cycles, each sample was dissolved in 20 ml of 0.15 M nitric acid. Sediment samples were also extracted using hot, concentrated nitric acid and 30% hydrogen peroxide following Environmental Protection Agency (EPA) method 3050B. Approximately 2 g of the < 2 mm size fraction and 3 g of the > 2 mm size fraction were extracted using this method. After evaporating to dryness, samples were brought up in 10 ml 0.15 M nitric acid, heated for approximately 15 min to help dissolve precipitates formed during evaporation, and filtered (0.45 μm). Procedural blanks had dissolved concentrations of As, Al, Fe, and manganese (Mn) less than 0.1% of concentrations measured in samples.
Results
Groundwater chemistry
Vertical profiles showing groundwater chemistry (prior to conducting the tracer test) at the top of the sewage-contaminated zone are shown in Fig. 2. Just below the water table there is a zone of uncontaminated groundwater (above approximately 13 m to sea level in Fig. 2), which has high concentrations of dissolved oxygen, pH values near 5.8, and low concentrations of dissolved salts, indicated by the low specific conductance of approximately 20 μS/cm. Steep vertical gradients in groundwater chemistry mark the transition zone between uncontaminated groundwater and the upper part of the sewage plume (between approximately 11 and 13 m to sea level in Fig. 2). Across this transition zone there is a decrease in dissolved oxygen concentrations, a net increase in pH, an increase in specific conductance, and increases in concentrations of sewage contaminants like phosphorus. The upper part of the sewage plume, referred to as the suboxic zone[37] (below approximately 11 m to sea level Fig. 2), is characterized by low but measurable concentrations of dissolved oxygen (approximately 5 μM), pH values near 6.2, and elevated concentrations of phosphorus and dissolved salts (the latter indicated by specific conductance values above 100 μS/cm). Concentrations of dissolved Mn below an altitude of 11 m were 2 to 3 μM (data not shown), typical of values in the suboxic zone at this distance (approximately 300 m) downgradient of the sewage-disposal beds. [37,38] Previous comparisons between total dissolved phosphorus concentrations (determined by ICPAES) and dissolved reactive phosphate concentrations (determined colorimetrically) have shown that phosphate accounts for all of the dissolved phosphorus, within analytical error. [42,43]
Figure 2.
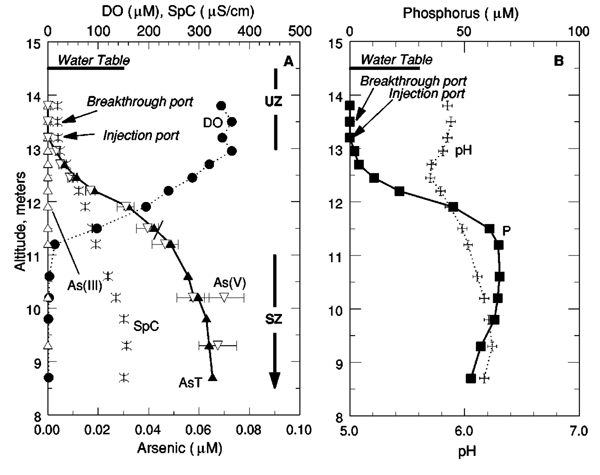
Groundwater chemistry at 23A13, sampled May 23, 2003 (prior to the phosphate injection), (a) Dissolved oxygen (DO) in micromoles per liter (μM), specific conductance (SpC) in microsiemens per centimeter (μS/cm), and concentrations of total dissolved arsenic (AsT), arsenic(III) [As(III)], and arsenic(V), [As(V)] in μM. (b) Phosphorus (P) in μM and pH. Bars show error limits for measured As(V) concentrations. Also shown are locations of the water table at the time of sampling, the injection and breakthrough-curve-sampling ports used for the tracer test, and the uncontaminated (UZ) and suboxic zones (SZ).
The groundwater-chemical features shown in Fig. 2 are typical of those observed within 500 m of the former sewage disposal beds, a region that has been sampled frequently over the past 20 yr (e.g., Refs. 25, 26, 32, 37, 38, 44–46). The vertical locations of the dissolved oxygen, pH, and phosphorus gradients are virtually identical to those observed in 1993 at the same location (cf., Fig. 2 in Ref. 47).
Concentrations of dissolved As increased across the upper boundary of the sewage plume (Fig. 2). Speciation determinations showed that, within analytical errors, all of the dissolved As was present as As(V). Concentrations of As(V) in the upper part of the sewage plume were 0.06 to 0.07 μM, which is below the MCL for As in drinking water in the United States of 0.13 μM. [48]
Arsenic concentrations were also determined in samples from the anoxic zone of the sewage plume (Table I). None of the MLS ports in or adjacent to 23A13 extend deep enough to reach the anoxic zone. Therefore data are presented for samples obtained from MLS F625 and F343, which are located approximately 30 and 50 m, respectively, upgradient of 23A13 (Fig. 1). At F625, which was sampled approximately 2 weeks after 23A13, Fe(II) concentrations of 69–236 μM, phosphorus concentrations of 18–93 μM, and pH values of 6.34–6.78 were observed. Total dissolved As concentrations were 0.04–0.20 μM (Table I). Arsenic(III) concentrations were 0.02–0.14 μM (Table I). At most depths, concentrations of As(V), determined by difference, were similar to those observed in the suboxic zone of the sewage plume. The total dissolved As exceeded the MCL for As in drinking water in the United States at every depth where the Fe(II) concentration exceeded 100 μM.
Table I.
Arsenic concentrations and related groundwater chemical conditions from sampling points in the anoxic zone of the sewage plume.
| Altitude (meters to sea level) | Turbidity (NTU)a | PH | Fe (μM) | P (μM) | Ast (μM) | As(III) (μM) | As(V)b (μM) |
| F625 sampled June 4, 2003 | |||||||
| 3.1 | 0.6 | 6.34 | 69 | 18.3 | 0.038 | 0.018 | 0.020 |
| 2.2 | 0.7 | 6.66 | 244 | 61 | 0.203 | 0.143 | 0.060 |
| 1.4 | 0.7 | 6.66 | 234 | 84 | 0.196 | 0.131 | 0.065 |
| 0.6 | 0.7 | 6.71 | 207 | 91 | 0.189 | 0.062 | 0.127 |
| -0.2 | 2.8 | 6.70 | 175 | 64 | 0.166 | 0.114 | 0.052 |
| -1.0 | 1.4 | 6.73 | 102 | 34.9 | 0.158 | 0.100 | 0.058 |
| -1.5 | 5.4 | 6.75 | 148 | 44.0 | 0.157 | 0.111 | 0.046 |
| -1.9 | 1.2 | 6.78 | 141 | 51 | 0.159 | 0.111 | 0.048 |
| -2.7 | 0.9 | 6.63 | 236 | 93 | 0.192 | 0.135 | 0.057 |
| F343 sampled June 18, 2002 | |||||||
| 2.9 | 0.3 | 6.48 | 250 | 24.3 | 0.174 | 0.101 | 0.073 |
| 2.0 | 0.3 | 6.54 | 330 | 90 | 0.197 | ||
| 1.1 | 0.8 | 6.56 | 280 | 77 | 0.165 | ||
| 0.2 | 0.3 | 6.50 | 255 | 49.2 | 0.143 | 0.073 | 0.070 |
| -0.7 | 1.0 | 6.58 | 214 | 41.2 | 0.152 | ||
aNephelometric turbidity units.
bBy difference between AsT and As(III).
The MLS at F625 had not been sampled previously. In order to compare the groundwater chemical conditions in the anoxic zone determined in June 2003 with historical data we also present data from the anoxic zone at a nearby MLS, F343 (Fig. 1), which was sampled in June 2002 (Table I). Arsenic concentrations and other groundwater quality parameters from the anoxic zone at F343 are similar to those at F625. The Fe(II) concentrations of 214–330 μM, phosphorus concentrations of 24–90 μM, and pH values of 6.48–6.58 were within the range of values determined previously in the anoxic zone at this location. [32,34,37,44] Dissolved sulfide concentrations were less than 3 μM (the analytical detection limit).
Gschwend and Reynolds[39] observed ferrous phosphate colloids approximately 0.1 μm in diameter and Fe and phosphate concentrations supersaturated with respect to vivianite [Fe3(PO4)2· 8H2O] in a nearby well sampled in 1985. Chemical compositions in Table I indicate supersaturation with respect to vivianite (thermodynamic data from Al-Borno and Tomson[50]). If present, ferrous phosphate colloids 0.1 μm in diameter would have passed through the 0.45 μm filters and contributed to the observed Fe and P concentrations. However, in contrast to the light scattering results reported by Gschwend and Reynolds,[49] turbidity values for these samples were low (Table I) and similar to those of samples from the uncontaminated and suboxic zones. Furthermore, previous sampling at this MLS showed similar Fe and P concentrations but no difference between Fe and P concentrations in unfiltered samples and those filtered through 0.1, 0.45, and 8 μm filters. [32] Why precipitation of vivianite would be inhibited in the anoxic zone of the sewage plume is unknown, but these observations suggest that it is unlikely that ferrous phosphate colloids contributed significantly to the Fe, P, and, by extension, As concentrations reported in Table I.
Tracer test
Groundwater to which phosphate had been added was pumped into a port in the uncontaminated zone and the port 0.3 m above it was sampled periodically (altitudes of these ports are indicated in Fig. 2). Chloride was also added to increase the specific conductance, which was used as a conservative, nonreactive tracer. Previous experience has shown that, during the injection (which lasted 7.7 h), the tracer cloud (injected groundwater with added tracers) displaces the ambient groundwater with minimal mixing as it expands away from the injection port. After the injection, solutes are transported horizontally at the ambient groundwater flow rate (e.g., Ref. 37). Assuming that the tracer cloud expanded spherically at the pumping rate (1 1 per min) and the aquifer into which it was injected had an effective porosity of 0.39,[19] the leading edge of the tracer cloud was expected to arrive at the sampling port 0.7 h after the beginning of the injection. In reasonable agreement with this, specific conductance reached half of the value in the injectate between 0.5 and 1 h after the beginning of the injection (Fig. 3). Phosphate concentrations reached half of the injectate concentration between 1 and 2 h after beginning the injection, indicating that adsorption of phosphate onto the sediments[35,44,51] caused phosphate to move more slowly than chloride and other ions contributing to the specific conductance. Values of pH increased but did not achieve 6.2, the pH of the injectate, by the end of the injection. This is consistent with the results of previous tracer tests, which have shown that groundwater pH values evolve slowly as a result of ion exchange and adsorption reactions driven by differences in chemical composition between the injected solution and ambient groundwater. [52]
Figure 3.
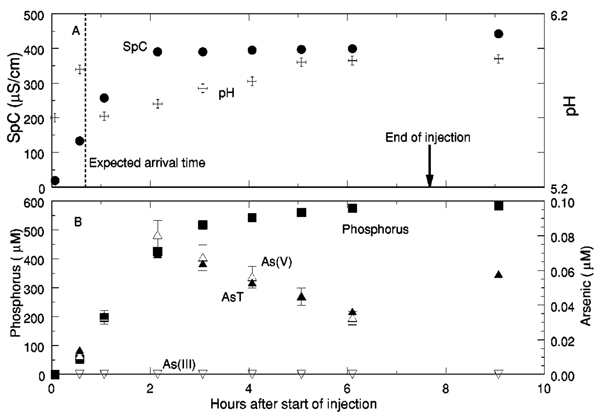
Tracer test breakthrough curves, (a) Specific conductance (SpC) in microsiemens per centimeter (μS/cm) and pH.Expected arrival time of the tracers, calculated assuming spherical symmetry of the injected tracer cloud and an effective porosity of 0.39, and the end of the injection are indicated, (b) Concentrations of phosphorus, total dissolved arsenic (AsT), arsenic(III) [As(III)], and arsenic(V) [As(V)] in micromoles per liter (μM). Bars show error limits for measured As(V) concentrations. Sampling 14.5 h after completing the injection showed 0.078 μM As(V), 435 μM phosphorus, and a pH of 5.91.
Arsenic concentrations increased as phosphate concentrations increased over the first 2 h of the injection (Fig. 3). After reaching a maximum of 0.07 μM, As concentrations decreased through the end of the injection. All of the As was present as As(V) within analytical errors. Sampling conducted 1.5 h after completing the injection showed that As concentrations increased to approximately 0.06 μM but phosphate concentrations and pH values remained constant. Sampling conducted 14.5 h after completing the injection showed an As(V) concentration of 0.078 μM, phosphorus concentration of 435 μM, and a pH of 5.91.
The injection port was sampled 1.4 and 14.4 h after completing the injection (Table II). Phosphate concentrations, specific conductance, and pH values 1.4 h after completing the injection were the same as those in the injectate. Arsenic(V) was present at a lower concentration at the injection port than at the port 0.3 m above it (0.0215 μM, Table II, as compared to 0.06 μM, Fig. 3). Thirteen hours later, the As(V) concentrations had increased to 0.059 μM and the phosphate concentration had decreased to 530 μM. Specific conductance was not determined but likely remained equal to that in the injectate. As observed elsewhere in the uncontaminated and suboxic zones, As(III) concentrations were below the detection limit.
Table II.
Selected water chemical data from the injection port, sampled after completing the injection.
| Hours after injection | SpC (μS/cm)a | pH | P (μM) | AsT (μM) | As (III) (μM) | As(V)b (μM) |
| 1.4 | 447 | 6.23 | 640 | 0.0215 | 0 | 0.0215 |
| 14.4 | ndc | 6.17 | 530 | 0.0593 | 0 | 0.0593 |
aSpecific conductance, microSiemens per centimeter.
bBy difference between AsT and As(III).
cNot determined.
Chemical extractions of sediments
Chemical extractions of the sediments were used to estimate the quantity of As associated with coatings on sediment grains and the total quantity of As in the sediments. Reductive extractions, using hydroxylamine hydrochloride at 50°C for 4 days, were used to estimate the quantity of As associated with the coatings. Previous work with Cape Cod sediments has shown that this method removes visible coatings containing Fe, Al, Mn, and silicon (Si)[15,29] as well as other elements, like zinc and phosphorus, likely to be adsorbed onto constituents of the coatings. [42,44] However, neither Fe nor Al is removed quantitatively from sediment-grain surfaces and it is likely that the extraction procedure leaches some Al and Fe associated with silicate minerals. [15,29] Oxidative extractions, using hot concentrated nitric acid and 30% hydrogen peroxide (EPA method 3050B) were used to estimate the total quantity of As in the sediments. Previous studies have shown that this method extracts As from Fe oxides, metal sulfides, and As sulfides. [53,54]
Reductive extractions leached 0.9–1.1 nanomoles As per gram of sediment dry weight (nmol/g) from the < 2 mm size-fraction of sediments collected adjacent to the tracer test site and from the < 1 mm size-fraction of a composite sample comprised of sediments from the uncontaminated zone (Table III). Preparation of this composite sample has been described elsewhere. [47] Quantities of As extracted from duplicate samples from the same core varied by less than 10% (Table III). Quantities of extractable Fe (17–23 μmol/g), Al (14–20 μmol/g), and Mn (0.16–0.23 μmol/g) (Appendix 1) were in good agreement with values determined previously on sediments from this site. [15] Gravel-size (> 2 mm) material had significant quantities of reductively extractable As (0.9 to 1.5 nmol/g). Gravel-size material was comprised of individual quartz grains and rock fragments, most of which were granitic or gneissic in composition. The individual quartz grains, most of which had surface coatings ranging in color from white to yellow-brown, were separated from the rock fragments by hand and extracted separately. The quartz grains also had significant quantities of extractable As (0.7–1.8 nmol/g).
Table III.
Extractable arsenic content of sediments.
| Arsenic(nmol/g) | ||||
| Core number | Altitude (m)a | Mineral fraction | Oxidativeb | Reductivec |
| < 2 mm size fraction | ||||
| R23AWC02d | 13.4 ± 0.2 | whole | 4.0 ± 0.2 | 1.08 ± 0.03 |
| R23AWC02e | 12.9 ± 0.2 | whole | 2.3 ± 0.7 | 1.11 ± 0.01 |
| R23AWC03f | 13.4 ± 0.2 | whole | 3.2 ± 1.1 | 0.98 ± 0.04 |
| R23AWC03g | 12.9 ± 0.2 | whole | 2.0 ± 0.6 | 0.97 ± 0.02 |
| 94UZComposite | wholeh | 5.18 ± 0.02 | 0.91 ± 0.02 | |
| > 2 mm size fraction | ||||
| R23AWC03 | 13.4 ± 0.2 | whole except quartz | 1.54 | 0.90 |
| R23AWC03i | 13.4 ± 0.2 | quartz grains | nd | 0.66 |
| R23AWC02 | 13.4 ± 0.2 | whole except quartz | 0.33 | 1.49 |
| R23AWC02j | 13.4 ± 0.2 | quartz grains | nd | 1.77 |
nd=not determined.
aAltitude (meters to sea level) of middle of 0.4-m-long cored interval.
bHot, concentrated nitric acid and hydrogen peroxide.
cHydroxylamine hydrochloride at 50°C for 96 h.
d< 2 mm size fraction comprised 97% by weight of sample.
e< 2 mm size fraction comprised 90% by weight of sample.
f< 2 mm size fraction comprised 98% by weight of sample.
g<2 mm size fraction comprised 91% by weight of sample.
h< 1 mm size fraction.
iQuartz grains comprised 36% by weight of >2 mm size fraction.
JQuartz grains comprised 22% by weight of >2 mm size fraction.
The oxidative extractions leached 2.0 to 4.0 nmol/g of As from the < 2 mm size fractions of sediments adjacent to 23A13 and 5.1 nmol/g from the < 1 mm size fraction of the composite sample (Table III). Greater variability was observed in the quantity of As leached by the oxidative extractions than by the reductive extractions. The quantity of As leached from the gravel-sized material was less than that leached from the <2 mm size-fraction from the same core. A separate gravel-sized sample of the whole-except-quartz fraction from core R23AWC02 at 13.4 m to sea level was ground to < 0.125 mm and then extracted using the same procedure. The quantity of As leached from the ground sample was 0.56 nmol/g, as compared to 0.33 nmol/g for the sample that had not been ground. This difference is smaller than the difference in As extracted from the two samples of gravel-size sediment. Thus, whether this difference results from greater accessibility of As-containing minerals to the extracting solution owing to grinding or from variability in the As content of different samples of gravel-size sediment is unknown.
Discussion
Arsenic and phosphate adsorption on hydrous ferric oxide
Previous studies have shown that adsorption onto iron oxides and other phases present at sediment-grain surfaces plays an important role in controlling the concentrations and, therefore, mobility of As in sediments. [54-57] The potential role of adsorption in controlling As concentrations at the Cape Cod site was first examined by computing compositions at adsorptive equilibrium with hydrous ferric oxide and either As(III) or As(V) in the presence and absence of phosphate (Fig. 4). These computations used the generalized diffuse layer surface complexation model and database for adsorption onto hydrous ferric oxide of Dzombak and Morel. [58] The total concentration of adsorption sites was set equal to 4.77 mM (millimoles per liter), the estimated total concentration of adsorption sites in the aquifer. [59] For the purposes of these computations, the total As and total phosphate concentrations were set equal to 0.6 μM and 3.1 mM, respectively, which yield a concentration of As(V) equal to 0.06 μM and dissolved phosphate concentration equal to 50 μM at pH 6.1. These are typical values for the suboxic zone of the sewage plume (cf., Fig. 2). Parameter values used in the computations are summarized in Appendix 2.
Figure 4.
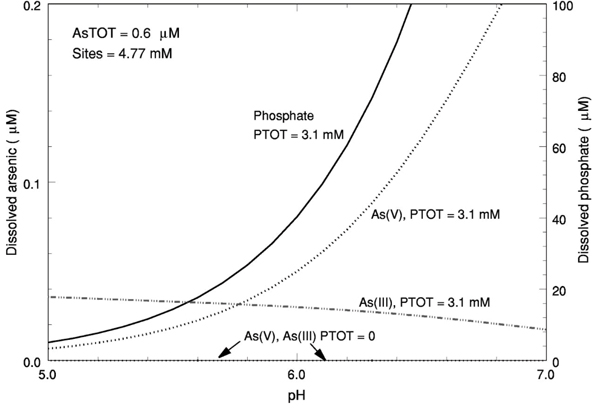
Dissolved concentrations of arsenic(III) [As(III)] or arsenic(V) [As(V)] and phosphate in ad-sorptive equilibrium with hydrous ferric oxide plotted against pH computed using the adsorption model of Dzombak and Morel (Ref. 58) at the total arsenic (dissolved plus adsorbed, AsTOT), total phosphate (PTOT), and adsorption site (Sites) concentrations indicated. Values used in the computations are presented in Appendix 2.
Results of the computations illustrate that, in the absence of phosphate, As(III) and As(V) adsorb extensively onto hydrous ferric oxide so that concentrations of both species are maintained at extremely low levels (well below the analytical detection limit) throughout the pH range 5–7 (Fig. 4). In the presence of phosphate, detectable concentrations of As(III) and As(V) are observed throughout the pH range 5–7 (Fig. 4). Adsorbed phosphate comprises greater than 90% of total phosphate throughout the pH range 5–7 and dissolved phosphate concentrations are not affected significantly by the adsorption of either As species at the low As concentrations used in the computations. The trends illustrated in Fig. 4 are consistent with those observed experimentally for the influence of phosphate on adsorption of As(III)[60,61] and As(V) (e.g., Refs. 61–65) on amorphous and crystalline hydrous ferric oxides.
In addition to phosphate, groundwater in the sewage-contaminated zone had sulfate and dissolved silica at concentrations of 100–200 μM and bicarbonate at concentrations of 50–250 μM. Experimental studies have shown that the effect of phosphate greatly exceeds the effects of such low concentrations of these other solutes on As(III) and As(V) adsorption onto hydrous ferric oxide in the pH range 5–7 [60,66-68]
Adsorption properties of the sediments and the distribution of arsenic
Adsorption properties of Cape Cod aquifer sediments are controlled by coatings on the surfaces of quartz and other minerals that make up the sediments. [15] Iron is an important constituent of these coatings but quantitative differences between the adsorption properties of the coatings and those of hydrous ferric oxide have been noted. [52,69,70] The coatings range in thickness from approximately 10 nanometers (nm) to 30 μm and are comprised of Al, Si, and Fe. [15,71] High-resolution transmission electron microscopy (HRTEM) of the material coating quartz grains revealed nanometer-size hematite in contact with nanometer-size particles comprised of Al and/or Si. [15,72] HRTEM studies of coatings on quartz grains in Atlantic coastal plain sediments also revealed isolated, nanometer- sized Fe oxide crystallites (goethite in that case) encapsulated in aluminosilicate material, which resembled allophane or halloysite. [16] Aluminum- and Si-rich material in coatings on quartz grains from the Cape Cod site has not been identified but kaolinite, illite, and other clay minerals, not detectable in assays of bulk sediments, have been isolated from feldspar grains. [14]
Hydrous Al oxides and clay minerals have a much lower affinity for adsorption of As(III) than do amorphous and crystalline hydrous Fe oxides. [73-75] In addition, As(V) adsorbs less extensively on kaolinite, illite, and other clay minerals than onto hydrous Fe or Al oxides at equal site concentrations. [74,76] The presence of phosphate at concentrations higher than As causes As (III) to adsorb very little onto hydrous Al oxide, kaolinite, illite, and other clay minerals and greatly decreases the extent of adsorption of As(V). [73,74,76]
These trends in adsorption of As(III) and As(V) onto hydrous Fe and Al oxides and silicates indicate that, if adsorbed As(III) were present at significant concentrations in the sediments, it would have desorbed in response to increasing concentrations of phosphate during the tracer test. The observed increase in As(V) concentrations with increasing phosphate concentrations is consistent with the hypothesis that there is naturally occurring As present as As(V) adsorbed to constituents of the coatings on mineral grains in the sediments and that it is released as a result of competition with phosphate for adsorption sites. This mechanism also probably accounts for the cooccurrence of As(V) and phosphate in the suboxic zone of the sewage plume (Fig. 2).
Arsenic concentrations in the anoxic, Fe(II)-containing zone of the sewage plume were higher than those in the suboxic zone owing to the presence of As(III) in addition to As(V) (Table I). Reductive dissolution of Fe oxides in the coatings would promote the release of As(V) adsorbed to or occluded in Fe oxides and would lead to the greater importance of hydrous Al oxides and silicates in controlling adsorption of As species. Reduction of As(V) to As(III) in the anoxic zone of the sewage plume has been observed but whether the reduction was carried out by As (V)-respiring microorganisms[77,78] or one or more abiotic reactions has not yet been determined. [79] As discussed above, hydrous Al oxides and silicates adsorb As(III) very little, especially in the presence of high concentrations of phosphate, and have a lower affinity for adsorption of As(V) than Fe oxides. Therefore As(III) is likely to be more mobile and present at higher concentrations than As(V) under the chemical conditions in the anoxic zone of the aquifer. Greater mobility of As(III) than As(V) in anoxic, Fe(II)-containing groundwater is consistent with results of previously conducted field experiments at the Cape Cod site[79,80] as well as results from other studies. [1] Thus it was the combination of sewage-derived phosphate, reduction of As(V) to As(III), and reductive dissolution of iron oxides associated with the sediments that caused concentrations of dissolved As in the anoxic zone to exceed the 0.13 μM drinking water standard.
Rate and reversibility of arsenic and phosphate adsorption reactions
During the phosphate injection, As(V) concentrations increased steadily as phosphate concentrations increased (Fig. 3). This demonstrates that at least some of the As(V) adsorbed within the coatings on sediment-grain surfaces was desorbed rapidly. This is interesting in light of findings of experimental studies on Fe oxide and clay minerals that the fraction of As(V) taken up by the solid that can be rapidly desorbed decreases with increasing contact time. [74,81-83] Thus these results suggest that significant concentrations of As(V) can be desorbed from these sediments over short periods of time in response to changing chemical conditions.
The decrease in As(V) concentrations observed prior to the complete breakthrough of phosphate (Fig. 3) cannot be interpreted unambiguously. It likely results, at least in part, from a chromatographic effect, sometimes called the "snow plow" effect,[59,84] whereby increased mobility of As(V) within the cloud of elevated dissolved and adsorbed phosphate concentrations as compared to the surrounding aquifer causes the accumulation of As(V) near the advancing front of elevated phosphate concentrations. However, slow rates of adsorption and desorption reactions may have played a role in producing a similarly shaped As(V) breakthrough curve obtained in a column experiment where As(V) adsorbed to sand-sized quartz and feldspar grains was displaced by elevated concentrations of phosphate. [85,86] The possible contribution of rate-limited adsorption/desorption reactions in our experiment is suggested by the observed increase in As concentration 1.4 h after completing the injection (Fig. 3). Further increases in As concentrations observed 14 to 15 h after the end of the injection at the breakthrough curve sampling port (Fig. 3) and the injection port (Table II) could have resulted from continued progress toward adsorptive equilibrium between phosphate and As(V) but may be a result of horizontal transport of different solute concentrations from upgradient in the injected cloud. Studies of adsorption of As(V) onto hydrous iron oxide have shown that slow rates of As(V) adsorption and desorption result from slow rates of diffusion through aggregates. [87] The coatings on Cape Cod sediment-grains consist of intimately aggregated networks of nanometer-size particles,[15,72] a structure that is compatible with a possible limitation to the rate of adsorption by diffusion. Experimental studies have shown that ion exchange reactions responsible for lithium adsorption on Cape Cod sediments are limited by intragranular diffusion. [14]
Origin and chemical forms of arsenic in the sediments
Reductive extractions of the sediments yielded a narrow range of As concentrations (0.9–1.1 nmol/g, Table III). This narrow range of values is consistent with the hypothesis that the As leached by the reductive extractions was primarily associated with coatings on sediment grains, which are ubiquitous and relatively uniformly distributed. [15,29] At the average solid/liquid ratio in the aquifer of 4145 g/l,[59] an As concentration in the coatings of 1 nmol/g corresponds to a concentration of 4.1 μM. Concentrations of As(V) observed in the suboxic zone and released from the sediments in the uncontaminated zone during the tracer test were 0.03–0.08 μM, which corresponds to 0.7%-2% of the total As in the coatings. Some of the As leached by the reductive extractions may be occluded in Fe oxides or in some other form that is not readily desorbable. [83] Nevertheless, the results of these calculations suggest that the As(V) concentrations observed during the tracer test and in the suboxic zone of the sewage plume represent a small percentage of the As associated with the coatings.
Oxidative extractions, which are assumed to leach essentially all of the As from the sediments, yielded a wider range of As concentrations on the sediments (2.0–5.2 nmol/g, Table III). The wider range of total As concentrations on the sediments is consistent with the hypothesis that As not associated with coatings on sediment grains is in the interior of mineral grains and that the range of values reflects heterogeneity in the distribution of these grains in the sediments. Studies of As in drinking water supplies elsewhere in New England[88,89] concluded that, in aquifers where elevated As concentrations were observed, As was ultimately derived from pyrite and other sulfide minerals, which, in contrast to silicate minerals, typically have significant concentrations of As. [1] Thus the dominant As-containing minerals are likely to be pyrite and other sulfide minerals, and, possibly, minerals derived from chemical weathering of these minerals. Sulfide minerals have not been identified in mineralogical studies of the sediments[30] but their abundance is likely to be low. Based on the maximum As concentration determined in the oxida-tive extractions of the sediments and assuming that the As content of sulfide minerals present in the sediments exceeds 1.3 μmol/g (100 ppm), the lower limit for pyrite,[1] sulfide minerals should be present at less than 0.4% by weight of the sediment. Sulfide minerals would be difficult to identify at such low concentrations.
The abundance of As in these sediments is lower than the estimated average abundance of As in crustal rocks of approximately 27 nmol/g (2 ppm), but similar to the range of As concentrations determined in granitic rocks from outcrops at various places in New England of 0.5–9 nmol/g (0.04–0.7 ppm). [89] Thus the As concentrations in these rocks may be representative of As concentrations of source rocks for the aquifer sediments.
Arsenic associated with coatings on sediment grains likely resulted from adsorption and other reactions between constituents of the coatings and As released as a result of chemical weathering of pyrite and other sulfide minerals under the oxic, mildly acidic conditions characteristic of uncon-taminated groundwater in the aquifer. Results of experimental studies suggest that arsenic associated with pyrite is readily released in contact with oxic water and is oxidized to As(III) and As(V) at a rate similar to the rate at which Fe(II) is oxidized to hydrous ferric oxide. [90] In his examination of the sediments from the site, Barber[30] identified grains comprised of amorphous and crystalline iron oxyhydroxides, likely products of chemical weathering of pyrite exposed to oxic, mildly acidic groundwater. [13,90,91] A field experiment carried out in the uncontaminated zone showed that As(III) was oxidized to As(V) over short transport distances. [80] Thus chemical weathering of pyrite would lead to the production of As(V), which is extensively adsorbed by the hydrous Fe and Al oxides and silicates that comprise the coatings on sediment grains. Arsenic leached by the reductive extractions constitutes 18%–49% of the total As in the sediments (Table III), which suggests that a significant fraction of the As originally associated with pyrite and other As-containing minerals at the time the sediments were deposited has been transferred to the coatings on sediment grains.
Conclusion
Based on these results we propose a working hypothesis for the occurrence of As in this aquifer. Chemical weathering reactions driven by oxic, mildly acidic groundwater over thousands of years resulted in the mass transfer of As from reduced forms in the interior of primary minerals of these glacial outwash sediments to As(V) adsorbed onto the hydrous Fe and Al oxide and silicate particles that comprise the coatings on sediment-grain surfaces. Adsorbed As(V) responds readily to changes in groundwater chemistry. Sewage-derived phosphate, transported away from the sewage-effluent disposal beds, caused desorption of As(V) resulting in concentrations up to 0.07 μM in the suboxic zone of the sewage plume. Deeper in the sewage plume, biodegradation of organic compounds transported away from the disposal beds resulted in reductive dissolution of Fe oxides associated with the coatings; As(V) was reduced to As(III) either by As-respiring microorganisms or abiotic reactions. Reductive dissolution of hydrous Fe oxides likely contributed to causing As concentrations in excess of the 0.13 μM MCL by releasing As not otherwise desorbable by phosphate, such as As occluded in Fe oxides, and by decreasing the solid-phase concentration of Fe oxides, which have a higher affinity for adsorption of As, especially As(III), than other constituents of the coatings. The combination of reduction of As(V) to As(III) and reductive dissolution of Fe oxides has been shown to be a likely cause of elevated As concentrations at other sites. [2,54,55,92-94]
Further research remains to be done to test this hypothesis, but the results have implications for management of shallow, unconfined, quartz-sand aquifers like the one on Cape Cod. As a shallow, unconfined aquifer, water quality is readily influenced by human activities on the land surface. The hydrous Fe and Al oxide and silicates on surfaces of quartz and feldspars, which dominate the mineralogy of the sediments, react with groundwater mainly through adsorption-desorption reactions. [14,15,35,59,69,71,95] The comparatively low level of reactivity of these minerals provides high-quality drinking water[13] but it also provides only a limited capacity for taking up contaminants and buffering against changes in groundwater chemistry. Arsenic occurs naturally and its presence, even at low concentrations, can render groundwater unfit as a source of drinking water. Maintaining an aquifer as a sustained source of drinking water requires preventing the development and spreading of conditions, like those described here, favorable to the mobilization of As. Given the dominance of adsorption reactions to the overall reactivity of the sediments with As and other groundwater contaminants, improved understanding of the chemical properties of the nanoparticulate material that comprises the coatings on sediment grain surfaces will also contribute to managing these types of aquifers as a sustained source of drinking water.
Note
a)Presented at the ACS Division of Geochemistry Symposium, "The Impact of Nanoparticle Growth and Transformation Processes on Contaminant Geochemical Cycling," New Orleans, March 2003.
APPENDIX 1.
EXTRACTABLE Al, Fe, AND Mn ON SEDIMENTS
| Core number | Altitude m | Mineral fraction | Al μmol/g) | Fe (μ mol/g) | Mn μmol/g | |||
| Oxidative | Reductive | Oxidative | Reductive | Oxidative | Reductive | |||
| <2 mm size fraction | ||||||||
| R23AWC02 | 13.4 ± 0.2 | whole | 33.8 ± 0.2 | 14.6 ± 0.5 | 52 ± 2 | 17.8 ± 0.6 | 0.30 ± 0.02 | 0.199 ± 0.017 |
| R23AWC02 | 12.9 ± 0.2 | whole | 33 ± 7 | 17.5 ± 0.4 | 49 ± 11 | 22.1 ± 0.9 | 0.27 ± 0.13 | 0.236 ± 0.001 |
| R23AWC03 | 13.4 ± 0.2 | whole | 32 ± 6 | 15.8 ± 0.2 | 45 ± 10 | 19.5 ± 0.4 | 0.23 ± 0.07 | 0.173 ± 0.001 |
| R23AWC03 | 12.9 ± 0.2 | whole | 22 ± 8 | 17.6 ± 1.8 | 31 ± 12 | 21 ± 2 | 0.13 ± 0.04 | 0.215 ± 0.020 |
| 94UZComposite | whole | 31 ± 1 | 20 ± 4 | 56 ± 5 | 23 ± 4 | 0.33 ± 0.01 | 0.29 ± 0.04 | |
| >2 mm size fraction | ||||||||
| R23AWC03 | 13.4 ± 0.2 | whole except quartz | 143 | 74 | 162 | 71.1 | 1.39 | 0.87 |
| R23AWC03 | 13.4 ± 0.2 | quartz grains | 22 | 2.37 | 0.050 | |||
| R23AWC02 | 13.4 ± 0.2 | whole except quartz | 120 | 87 | 86 | 98.2 | 1.31 | 1.07 |
| R23AWC02 | 13.4 ± 0.2 | quartz grains | 3.5 | 4.84 | 0.066 | |||
APPENDIX 2.
PARAMETER VALUES USED IN SURFACE COMPLEXATION MODEL CALCULATIONS IN FIG. 4
| Parameter | Value |
| Ionic Strength | 0.003 |
| m2/la | 1240 |
| Surface species equilibrium expressions and log K valuesb | |
| >FeOH =>FeO- + H+ | -8.93 |
| >FeOH + H+=> FeO |
7.29 |
| >FeOH+ H3AsO3 = >FeH2AsO3 + H2O | 5.41 |
| >FeOH+As |
29.31 |
| >FeOH+As |
23.51 |
| >FeOH+As |
10.58 |
| >FeOH+P |
31.29 |
| >FeOH+P |
25.39 |
| >FeOH+P |
17.72 |
| Solution species equilibrium expressions and log K valuesb | |
| H++H2As |
9.29 |
| H++As |
11.50 |
| 2H++As |
18.46 |
| 3H++As |
20.70 |
| H++P |
12.35 |
| 2H+ = P |
19.55 |
| 3H++P |
21.70 |
aSurface area-to-volume ratio.
bAll log K values from Dzombak and Morel (Ref. 58). Activity coefficients for aqueous species calculated using Davies equation.
Acknowledgments
Acknowledgements
Jessica Cochrane, John Fitzpatrick, Jennifer Joye, and Jonathan King made significant contributions to the field sampling and analytical work. Kathryn Hess and Denis Le-Blanc made significant contributions to designing and conducting the tracer test. Critical reviews by Yuji Arai, John Coleman, Jim Davis, Ken Stollenwerk, and Dimitri Vlasso-poulos improved the quality of the manuscript. This research was funded by the National Research Program and Toxic Substances Hydrology Program of the U. S. Geological Survey.
References
- Smedley PL, Kinniburgh DG. Appl Geochem. 2002. p. 517.
- Harvey CF, Swartz CH, Badruzzaman ABM, et al. Science. 2002;298:1602. doi: 10.1126/science.1076978. [DOI] [PubMed] [Google Scholar]
- van Geen A, et al. Water Resour Res. 2003. 101029/2002WR001617.
- NRC . Arsenic in Drinking Water. National Academy Press, Washington, D.C; 1999. p. 310. [Google Scholar]
- NRC . Arsenic in Drinking Water 2001 Update. National Academy of Sciences, Washington DC; 2001. p. 225. [Google Scholar]
- WHO . Guidelines for Drinking Water. 2. World Health Organization, Geneva, Switzerland; 1994. p. 188. [Google Scholar]
- Smith AH, Lopipero PA, Steinmaus CM. Science. 2002;296:2145. doi: 10.1126/science.1072896. [DOI] [PubMed] [Google Scholar]
- Cherry JA, Shaikh AU, Tallman BE, Nicholson RV, Hydrol J. 1979. p. 373.
- Oremland RS, Stolz JF. Science. 2003;300:939. doi: 10.1126/science.1081903. [DOI] [PubMed] [Google Scholar]
- Foster AL. In: Arsenic in Ground Water. Welch AH, Stollenwerk KG, editor. Kluwer Boston; 2003. pp. 27–65. [Google Scholar]
- Welch AH, Stollenwerk KG, editor. Arsenic in Groundwater. Kluwer, Norwell, MA; 2003. p. 475. [Google Scholar]
- Nordstrom DK. Science. 2002;296:2143. doi: 10.1126/science.1072375. [DOI] [PubMed] [Google Scholar]
- Appelo CAJ, Postma B. In: Geochemistry Groundwater and Pollution. Balkema AA, editor. Roterdam, The Netherlands; 1993. p. 536. [Google Scholar]
- Wood WW, Kraemer TF, Hearn PP. Science. 1990. p. 1569. [DOI] [PubMed]
- Coston JA, Fuller CC, Davis JA. Geochim Cosmochim Acta. 1995. p. 3535.
- Penn RL, Zhu C, Xu H, Veblen BR. Geology. 2001. p. 843.
- LeBlanc DR, Geol US. Survey Water-Supply Pap 2218. 1984. p. 28.
- Dunkle-Shapiro S, LeBlanc BR, Schlosser P, Ludin A. Ground Water. 1999. p. 861.
- LeBlanc DR, Garabedian SP, Hess KM, Gelhar LW, Quadri RB, Stollerwerk KG, Wood WW. Water Resour Res. 1991. p. 895.
- Garabedian SP, LeBlanc DR, Gelhar LW, Celia MA. WaterResour Res. 1991. p. 911.
- Hess KM, Wolf SH, Celia MA. Water Resour Res. 1992. p. 2011.
- Hess KM, Davis JA, Kent DB, Coston JA. Water Resour Res. 2000. 101029/2001WR000945.
- Masterson JP, Stone BD, Walter DA, Savoie JG, Geol US. Survey Hydrologic-Investigations Atlas HA. 1997. p. 1.
- Thurman EM, Barber LB, LeBlanc DR, Contam J. Hydrol. 1986. p. 143.
- Smith RL, Harvey RH, LeBlanc DR, Contam J. Hydrol. 1991. p. 285.
- Smith RL, Howes BL, Duff JH. Geochim Cosmochim Acta. 1991. p. 1815.
- LeBlanc DR, Hess KM, Kent BB, Smith RL, Barber LB, Stollerwerk KG, Campo K. In: US Geol Survey Water-Resourc Invest Report 4018C. Morganwalp BW, Buxton HT, editor. Vol. 3. 1999. pp. 245–260. [Google Scholar]
- Barber LB, Thurman EM, Runnells DD, Contam J. Hydrol. 1992. p. 35.
- Fuller CC, Davis JA, Coston JA, Dixon E, Contam J. Hydrol. 1996. p. 165.
- Barber LB. Ph.D. dissertation, Univ Colorado, Boulder, CO; 1990. p. 237. [Google Scholar]
- McCobb TM, LeBlanc DR, Hess KM. In: US Geol SurveyWater-Resourc Invest Report 4018C. Morganwalp BW, Buxton HT, editor. Vol. 3. 1999. pp. 349–360. [Google Scholar]
- Savoie JG, LeBlanc DR. US GeolSurvey Water-Resource Invest Report 97-4269. 1998. p. 208.
- Smith RL, Rea-Kumler BA, Peacock TR, Miller BN. In: U S Geol Survey Water-Resourc Invest Report 4018C. Morganwalp BW, Buxton HT, editor. Vol. 3. 1999. pp. 285–291. [Google Scholar]
- Kent DB, Maeder V. In: U S Geol Survey Water-Resourc Invest Report 4018C. Morganwalp BW, Buxton HT, editor. Vol. 3. 1999. pp. 293–304. [Google Scholar]
- Parkhurst DL, Stollenwerk KG, Colman JA. U S Geol Survey Water-Resourc Invest Report 03-4017. 2003. p. 33.
- Barber LB, Keefe SH. In: U S Geol Survey Water-Resourc Invest Report 4018C. Morganwalp BW, Buxton HT, editor. Vol. 3. 1999. pp. 261–270. [Google Scholar]
- Kent DB, Davis JA, Anderson JCD, Rea BA, Waite TD. Water Resour Res. 1994. p. 1099.
- Davis JA, Kent DB, Coston JA, Hess KM, Joye JL. Water Resour Res. 2000. p. 119.
- Bednar AJ, Garbarino JR, Ranville JF, Wildeman TR. Environ Sci Technol. 2002;36:2213. doi: 10.1021/es0157651. [DOI] [PubMed] [Google Scholar]
- Savoie JG, Kent BB, Smith RL, LeBlanc BR, Hubble BW. US Geol Surv Water Resourc Invest Report 03-4309. 2004. p. 84.
- Anderson LB, Kent DB, Davis JA. Environ Sci Technol. 1994. p. 178. [DOI] [PubMed]
- Rea BA, Kent BB, Anderson LCB, Davis JA, LeBlanc BR. In: US Geol Survey Water-Resourc Invest Report94-4015. Marganwalp BW, Aronson BA, editor. Vol. 1. 1994. pp. 191–198. [Google Scholar]
- McCobb TD, LeBlanc BR, Walter BA, Hess KM, Kent BB, Smith RL. U S Geol Survey Water-Resourc Invest Report 02-4306. 2003. p. 69.
- Walter DA, Rea BA, Stollenwerk KG, Savoie JG. U S Geol Survey Water-Supply Pap 2463. 1996. p. 89.
- Bussey KW, Walter DA. U S Geol Survey Open-File Report. 1996. p. 44.
- Stollenwerk KG. Water Resour Res. 1998. p. 2727.
- Davis JA, Coston JA, Kent BB, Hess KM, Joye JL, Brienen P, Campo KW. U S Environ: Protect Agency Report EPA/600/R-01/ 007b. 2001. p. 47.
- United States Environmental Protection Agency EPA 815-F-00-015. 2001.
- Gschwend PM, Reynolds MB, Contam J. Hydrol. 1987. p. 309.
- Al-Borno A, Tomson MB. Geochim Cosmochim Acta. 1994. p. 5373.
- Stollenwerk KG. Appl Geochem. 1996. p. 317.
- Kent DB, Davis JA, Anderson LCB, Rea BA, Coston JA. Geochim Cosmochim Acta. 2002. p. 3017.
- Keon NE, Swartz CH, Brabander DJ, Harvey CF, Hemond HF. Environ Sci Technol. 2001;35:2778. doi: 10.1021/es001511o. [DOI] [PubMed] [Google Scholar]
- Kneebone PE, O'Day PA, Jones N, Hering JG. Environ Sci Technol. 2002;36:381. doi: 10.1021/es010922h. [DOI] [PubMed] [Google Scholar]
- Belzile N, Tessier A. Geochim Cosmochim Acta. 1990. p. 103.
- Appelo CAJ, van der Weiden MJ, Tournassat C, Charlet L. Environ Sci Technol. 2002;36:3096. doi: 10.1021/es010130n. [DOI] [PubMed] [Google Scholar]
- Stollenwerk KG. In: Arsenic in Groundwater. Welch AH, Stollenwerk KG, editor. Kluwer, Norwell, MA; 2003. pp. 67–100. [Google Scholar]
- Dzombak DA, Morel FMM. Surface Chemical Modeling: Hydrous Ferric Oxide. Wiley, New York; 1990. p. 393. [Google Scholar]
- Kent DB, Abrams RH, Davis JA, Coston JA, LeBlanc BR. Water Resour Res. 2000. p. 3411.
- Jain A, Loeppert RH. J Environ Qual. 2000. p. 1422.
- Dixit S, Hering JG. Environ Sci Technol. 2003;37:4182. doi: 10.1021/es030309t. [DOI] [PubMed] [Google Scholar]
- Hingston FJ, Posner AM, Quirk JP. Discuss Faraday Soc. 1971. p. 334.
- Manning BA, Goldberg S. Soil Sci Soc Am J. 1996. p. 121.
- Hiemstra T, van Riemsdijk WH, Colloid J. Interface Sci. 1999. p. 182. [DOI] [PubMed]
- Gao Y, Mucci A. Geochim Cosmochim Acta. 2001. p. 2361.
- Wilkie JA, Hering JG. Colloids Surf A. 1996. p. 97.
- Swedlund PJ, Webster JG. Water Res. 1999. p. 3413.
- Meng X, Bang S, Korfiatis GP. Water Res. 2000. p. 1255.
- Stollenwerk KG. Water Resour Res. 1995. p. 347.
- Kent DB, Davis JA, Anderson LCD, Rea BA. Water Resour Res. 1995. p. 1041.
- Davis JA, Coston JA, Kent DB, Fuller CC. Environ Sci Technol. 1998. p. 2820.
- Banfield JF, Hamers RJ. In: Advances in Mineralogy. Banfield JF, Nealson KH, editor. Vol. 35. Mineralogical Society of America, Washington, D.C; 1997. pp. 81–122. [Google Scholar]
- Manning BA, Goldberg S. Environ Sci Technol. 2005. 1997.
- Lin Z, Puls RW. Environ Geol. 2000. p. 753.
- Arai Y, Elzinga EJ, Sparks DL, Colloid J. Interface Sci. 2001. p. 80. [DOI] [PubMed]
- Manning BA, Goldberg S. Clays Clay Miner. 1996. p. 609.
- Zobrist J, Dowdle PR, Davis JA, Oremland RS. Environ Sci Technol. 2000. p. 4747.
- Oremland RS, Newman DK, Kail BW, Stolz JF. In: Environmental Chemistry of Arsenic. Frankenberger WT, editor. Marcel De-kker, New York; 2001. pp. 273–295. [Google Scholar]
- Hoehn R, Isenbeck-Schröter M, Niedan V. In: Water-Rock Interaction. Cidu R, editor. Balkema, Lisse, Italy; 2001. pp. 1099–1102. [Google Scholar]
- Stadler S, Jann S, Holm R. In: Water-Rock Interaction. Cidu R, editor. Balkema, Lisse, Italy; 2001. pp. 1013–1016. [Google Scholar]
- Puls RW, Powell RM. Environ Sci Technol. 1992. p. 614. [DOI] [PubMed]
- Fuller CC, Davis JA. U S Geol Survey Prof Pap 1681. 2004. pp. 27–48.
- Ford RG. Environ Sci Technol. 2002. p. 2459. [DOI] [PubMed]
- Starr JL, Parlange JY. Soil Sci Soc Am J. 1979. p. 448.
- Darland JE, Inskeep WP. J Environ Qual. 1997. p. 1133.
- Darland JE, Inskeep WP. Environ Sci Technol. 1997. p. 704.
- Fuller CC, Davis JA, Waychunas GA. Geochim Cosmochim Acta. 1993. p. 2771.
- Peters SC, Blum JD, Karagas MR. Environ Sci Technol. 1999. p. 1328.
- Ayotte JD, Montgomery DL, Flanagan SM, Robinson KW. Environ Sci Technol. 2003;37:2075. doi: 10.1021/es026211g. [DOI] [PubMed] [Google Scholar]
- Nesbitt HW, Muir IJ, Pratt AR. Geochim Cosmochim Acta. 1995. p. 1773.
- Nicholson RV, Gillham RW, Reardon EJ. Geochim Cosmo chim Acta. 1990. p. 395.
- Cummings DE, Caccavo F, Jr, Fendorf S, Rosenzweig RF. Environ Sci Technol. 1999. p. 723.
- Welch AH, Westjohn DB, Helsel DR, Wanty RB. Ground Water. 2000. p. 589.
- McCreadie H, Blowes DW, Ptacek CJ, Jambor JL. Environ Sci Technol. 2000. p. 3159.
- Ceazan ML, Thurman EM, Smith RL. Environ Sci Technol. 1989. p. 1402.