

Abstract
Since its introduction as a new clinical entity in 1992, the Brugada syndrome has attracted great interest because of its high incidence in many parts of the world and its association with high risk for sudden death in infants, children, and young adults. Recent years have witnessed an exponential rise in the number of reported cases and a striking proliferation of articles serving to define the clinical, genetic, cellular, ionic, and molecular aspects of the disease. A consensus report published in 2002 delineated diagnostic criteria for the syndrome. A second consensus conference was held in September 2003. This review provides an in-depth overview of the clinical, genetic, molecular, and cellular aspects of the Brugada syndrome, incorporating the results of the two consensus conferences, and the numerous clinical and basic publications on the subject. The proposed terminology, diagnostic criteria, risk stratification schemes, and device and pharmacologic approach to therapy discussed are based on available clinical and basic studies and should be considered a work-in-progress that will without doubt require fine-tuning as confirmatory data from molecular studies and prospective trials become available.
Since its introduction as a new clinical entity in 1992,1 the Brugada syndrome has attracted great interest because of its high incidence in many parts of the world and its association with high risk for sudden death in infants, children, and young adults. Recent years have witnessed an exponential rise in the number of reported cases and a striking proliferation of articles serving to define the clinical, genetic, cellular, ionic, and molecular aspects of the disease.2 A consensus report published in 2002 delineated diagnostic criteria for the syndrome.3,4 A second consensus conference was held in September 2003. This review provides an in-depth overview of the clinical, genetic, molecular, and cellular aspects of the Brugada syndrome, incorporating the results of the two consensus conferences, and the numerous clinical and basic publications on the subject. The proposed terminology, diagnostic criteria, risk stratification schemes, and device and pharmacologic approach to therapy discussed are based on available clinical and basic studies and should be considered a work-inprogress that will without doubt require fine-tuning as confirmatory data from molecular studies and prospective trials become available.
ClINICAL CHARACTERISTICS
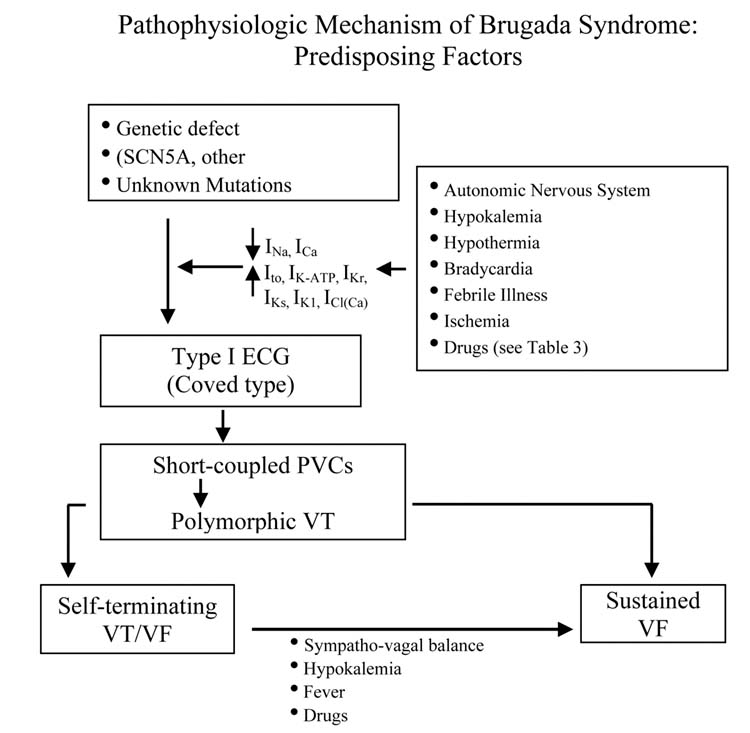
The Brugada syndrome is characterized by ST segment elevation in the right precordial ECG leads and a high incidence of sudden death in patients with structurally normal hearts. The syndrome manifests primarily during adulthood, with a mean age of sudden death of approximately 40 years. The youngest patient diagnosed with the syndrome was 2 days of age, and the oldest was 84. The syndrome is thought to be responsible for 4-12% of all sudden deaths and at least 20% of deaths in patients with structurally normal hearts. The incidence of the disease is on the order of 5 per 10,000 inhabitants and, apart from accidents, is the leading cause of death of men under the age of 40 in regions of the world where the syndrome is endemic. Because the ECG is so dynamic and often concealed, it is difficult to estimate the true incidence of the disease in the general population.5
M. M. Scheinman: The true incidence of the Brugada syndrome is not known but estimates by the authors that this syndrome is responsible for 4-12% of all sudden deaths and at least 20% of deaths in patients with structurally normal hearts are difficult to substantiate. It is my impression that the Brugada syndrome is a relatively uncommon cause of aborted sudden death in the United States, but is more common as detailed by the authors in the Asian population.
The electrocardiographic manifestations of the Brugada syndrome when concealed can be unmasked by sodium channel blockers, a febrile state, or vagotonic agents.6-9 Three types of repolarization patterns in the right precordial leads are recognized (Table 1 and Fig 1)3,4 Type 1 is diagnostic of Brugada syndrome and is characterized by a coved ST-segment elevation ≥2 mm (0.2 mV) followed by a negative T-wave. Type 2 has a saddleback appearance with a high take-off ST-segment elevation of ≥2 mm followed by a trough displaying ≥1 mm ST elevation followed by either a positive or a biphasic T-wave. Type 3 has either a saddleback or a coved appearance with an ST-segment elevation of <1 mm. These three patterns may be observed sequentially in the same patient or following the introduction of specific drugs. Type 2 and Type 3 ECGs should not be considered diagnostic of the Brugada syndrome.
TABLE 1.
Diagnostic criteria for Brugada syndrome (from 1st consensus document) (ST segment abnormalities in leads V1–V3)
| Type 1 | Type 2 | Type 3 | |
|---|---|---|---|
| J-point | ≥2 mm | ≥2 mm | ≥2 mm |
| T-wave | Negative | Positive or biphasic | Positive |
| ST-T configuration | Coved type | Saddleback | Saddleback |
| ST segment (terminal portion) | Gradually descending | Elevated ≥1 mm | Elevated <1 mm |
1 mm = 0.1 mV. The terminal portion of the ST segment refers to the latter half of the ST segment.
Fig 1.
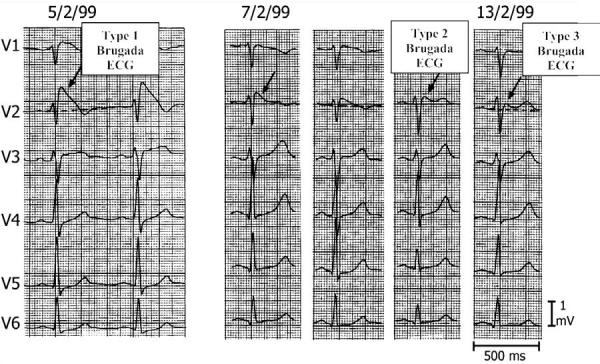
Three types of ST segment elevation generally observed in patients with the Brugada syndrome. Shown are precordial leads recorded from a patient diagnosed with the Brugada syndrome. Note the dynamic ECG changes occurring over a period of 2 days. The left panel shows a clear Type 1 ECG, which is diagnostic of the Brugada syndrome. A saddleback ST segment elevation (Type 2) was observed on 2-7-99. The ST segment is further normalized on 2-13-99 showing a Type 3 ECG. Modified from ref. 4, with permission.
Placement of the right precordial leads in a superior position (up to the 2nd intercostal spaces above normal) can increase the sensitivity of the ECG for detecting the Brugada phenotype in some patients, in the presence or absence of a drug challenge (Fig 2),10,11 although this may compromise specificity of the diagnosis.
Fig 2.
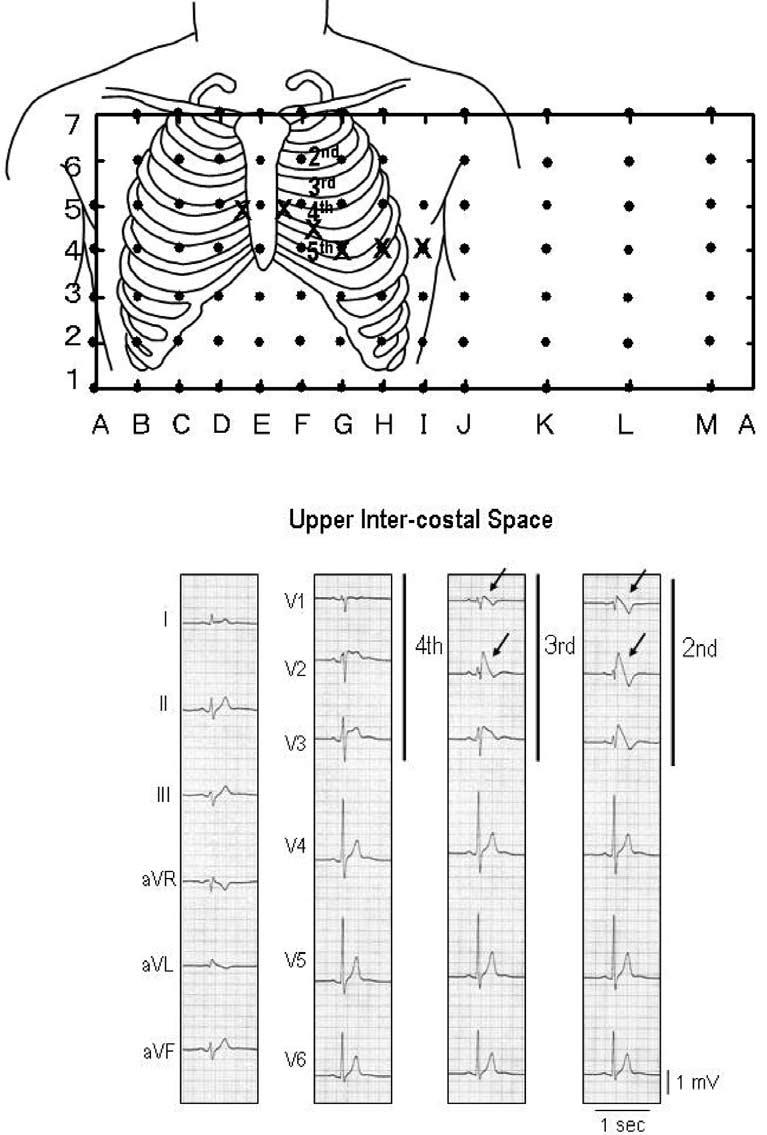
Shift of right precordial leads to 2nd and 3rd intercostal space unmasks a type 1 Brugada ECG. Top: Plot of 87 unipolar electrode sites (dots) and 6 precordial electrocardiograms (ECG) (crosses). Eighty-seven lead points are arranged in a lattice-like pattern (13 × 7 matrix), except for four lead points on both midaxillary lines, and covered the entire thoracic surface. V1 and V2 leads of the ECG are located between D5 and E5, and between E5 and F5, respectively, whereas V4, V5, and V6 are coincident with G4, H4, and I4, respectively. Bottom: Twelve-lead electrocardiograms (ECG) in a patient with Brugada syndrome. Type 2 saddle-back type segment elevation was observed in V1 and V2 of the standard 12-lead ECG (4th intercostal space), whereas typical type 1 coved type ST-segment elevation was apparent in V1 and V2 recorded from the 2nd and 3rd intercostal space (arrows). Modified from reference 159 with permission.
Of note, a Brugada-like ECG can occasionally be recorded within the first few hours following DC cardioversion. Therefore the electrocardiogram after cardioversion should be interpreted with caution in that the DC shock may contribute to ST-segment elevation.4
A slight prolongation of the QT interval is sometimes observed associated with the ST segment elevation.12-14 The QT interval is prolonged more in the right vs. left precordial leads, presumably due to a preferential prolongation of action potential duration (APD) in right ventricular (RV) epicardium secondary to accentuation of the action potential notch.15 Depolarization abnormalities including prolongation of P-wave duration, PR, and QRS intervals are frequently observed, particularly in patients linked to SCN5A mutations.16 PR prolongation likely reflects HV conduction delay.12
Recent reports indicate that approximately 20% of Brugada syndrome patients develop supraventricular arrhythmias.17 It is as yet unknown whether atrial vulnerability is correlated with ventricular inducibility of arrhythmias or whether atrial arrhythmias may serve as triggering events for VT/VF, although the latter seems unlikely based on current knowledge. Slowed atrial conduction as well as atrial standstill have been reported in association with the syndrome.18
Observations from Japan suggest that in some cases arrhythmia initiation is bradycardia-related.19 This may contribute to the higher incidence of sudden death at night in individuals with the syndrome and may account for the success of pacing in controlling the arrhythmia in isolated cases of the syndrome.20 However, not all patients die at night and not all the cases are controlled with rapid ventricular pacing. South Asian patients who have the ECG pattern usually develop VT/VF during sleep at night.
VT/VF often terminates spontaneously in patients with the Brugada syndrome, as first reported by Bjerregaard et al.21 This may explain why patients wake up at night after episodes of agonal respiration caused by the arrhythmia.
Sudden unexplained nocturnal death syndrome (SUNDS, also known as SUDS), a disorder most prevalent in Southeast Asia, and Brugada syndrome have recently been shown to be phenotypically, genetically, and functionally the same disorder.22 Sudden and unexpected death of young adults during sleep, known in the Philippines as bangungut (“to rise and moan in sleep”), was first described in Philippine medical literature in 1917. In Japan, this syndrome, known as pokkuri (“sudden and unexpectedly ceased phenomena”), was reported as early as 1959.23 In 1997, Nademanee et al24 reported that among 27 Thai men referred for aborted cases of what was known in Thailand as Lai Tai (“death during sleep”), as many as 16 had the ECG pattern of Brugada syndrome. In their review of the literature in 1999, Alings and Wilde found that, of the 163 patients who met the criteria for Brugada syndrome, 58% were of Asian origin.12
Relationship with Structural Heart Disease
A subpopulation of arrhythmogenic right ventricular cardiomyopathy (ARVC) patients have been found to display an ST-segment elevation and polymorphic VT characteristic of the Brugada syndrome.25 In addition one case has been reported in which a patient with a Brugada phenotype required heart transplantation due to untreatable arrhythmias26 and in whom severe fibrosis of the right ventricle was subsequently reported.
These facts notwithstanding, the vast majority of Brugada patients possess a structurally normal heart, consistent with the notion that this is a primary electrical heart disease.27 It is not unreasonable to speculate that fibrosis and myocarditis, however mild, may occur and may exacerbate or indeed trigger events in patients with the Brugada syndrome, although definitive evidence in support of this hypothesis is lacking. It is noteworthy that recent studies suggest that some SCN5A defects may be capable of causing fibrosis in the conduction system and ventricular myocardium.28
ARVC and Brugada syndromes are distinct clinical entities with respect to both the clinical presentation and the genetic predisposition (Table 2).4 The only gene thus far linked to the Brugada syndrome is SCN5A, the gene that encodes for the α-subunit of the cardiac sodium channel, whereas ARVC has been linked to seven different chromosomal loci and three putative genes independent of those responsible for the Brugada syndrome.29 Only the ARVC5 locus has been mapped to a region overlapping with the second locus for Brugada syndrome, but neither gene has been identified as yet.30,31 In Brugada syndrome, imaging techniques such as echocardiography, angiography, magnetic resonance imaging, and radionuclide scintigraphy show no evidence of overt structural heart disease, whereas ARVC patients characteristically display right ventricular morphological and functional changes (such as global dilatation, bulgings/aneurysms, and wall motion abnormalities). Ventricular arrhythmias in ARVC are most commonly monomorphic VT (LBBB type), often precipitated by catecholamines or exercise, accounting for sudden death of young competitive athletes. In contrast, ST-segment elevation and arrhythmias in Brugada patients are enhanced by vagotonic agents or β-adrenergic blockers, and polymorphic VT most commonly occurs during rest or sleep. Unlike Brugada syndrome, the ECG abnormalities in ARVC are not dynamic, displaying a constant T-wave inversion, epsilon waves and, in the progressive stage, reduction of the R amplitude, which are largely unaffected by sodium channel blocker administration.
TABLE 2.
Differential diagnosis between ARVC and Brugada syndrome
| Clinical characteristics | ARVC | Brugada syndrome |
|---|---|---|
| Age | 25–35 | 35–40 |
| Sex (male/female) | M > F (3:1) | M > F (8:1) |
| Distribution | Worldwide (Italy) | Worldwide (southeast Asia) |
| Inheritance | AD (AR) | AD |
| Chromosomes | 1, 2, 3, 10, 1417 | 3 |
| Gene | hRYR2, plakobin, desmoplakin | SCN5A |
| Symptoms | Palpitations | Syncope |
| Syncope | Cardiac arrest | |
| Cardiac arrest | ||
| Circumstances | Effort | Rest |
| Imaging | Morpho-functional RV (and LV) abnormalities | Normal |
| Pathology | Fibrofatty replacement | Normal |
| ECG repolarization | Inverted T-waves in right precordial leads | High take-off ST-segment V1–V3 |
| ECG depolarization | Epsilon-waves | RBBB/LAD |
| QRS prolongation | Late potentials | |
| Late potentials | ||
| AV conduction | Normal | 50% abnormal PR/HV intervals |
| Atrial arrhythmias | Late (secondary) | Early (primary 10–25%) |
| ECG changes | Fixed (mostly) | Variable |
| Ventricular arrhythmias | Monomorphic VT/VF | Polymorphic VT/VF |
| Mechanism of arrhythmias | Scar-related reentry | Phase 2 reentry |
| Drug effect Class I | ↓ | ↑ |
| Drug effect Class II | ↓ | ↑ |
| Drug effect Class III | ↓ | −/↑ |
| Drug effect Class IV | −/↓ | – |
| Beta-stimulation | ↑ | ↓ |
| Natural history | Sudden death | Sudden death |
| Heart failure |
Arrows denote changes in ST segment elevation (↑, increased; ↓, decreased; −/, small change, if any).
Modified from (4), with permission.
M. M. Scheinman: The authors' clearly and cogently separate Brugada syndrome from ARVC in both the text and Table 2. It is still clear from the work of Nave et al from northern Italy that the Brugada ECG may be recorded in patients with ARVC. Rather than suggest the coexistence of two relatively rare diseases, it would make more sense to hypothesize that disease of the RV muscle might accentuate the Brugada sign, similar to that of ischemia, electrolyte disorders, or drugs.
Electron beam computed tomography has uncovered wall motion abnormalities in a series of Brugada patients tested.32 Although such contractile abnormalities are commonly considered pathognomonic of structural disease, recent studies33,34 suggest that such contractile dysfunction can result from loss of the action potential dome in regions of right ventricular epicardium, and, thus, may be unrelated to any type of morphological defect. Loss of the dome leads to contractile dysfunction because calcium entry into the cells is greatly diminished and sarcoplasmic reticulum calcium stores are depleted. Signal-averaged ECG (SAECG) recordings have demonstrated late potentials in patients with the Brugada syndrome, especially in the anterior wall of the right ventricular outflow tract (RVOT)35,36 and recording from the epicardial surface of the anterior wall of the RVOT have revealed delayed potentials.37 Although these types of potentials are commonly considered to be representative of delayed activation of the myocardium secondary to structural defects, recent studies suggest that in the case of the Brugada syndrome these late and delayed potentials may represent the delayed second upstroke of the epicardial action potential or local phase 2 reentry.34
Diagnostic Criteria
ST-segment elevation is associated with a wide variety of benign as well as malignant pathophysiologic conditions. A differential diagnosis is at times difficult, particularly when the degree of ST-segment elevation is relatively small and the specificity of sodium channel blockers such as flecainide, ajmaline, procainamide, disopyramide, propafenone, and pilsicainide7,14,38 to identify patients at risk is uncertain. The recommended dosage regimens are listed in Table 3. The test should monitored with a continuous ECG recording (speed 10 mm/sec and interposed 50 mm/sec) and should be terminated when (1) the diagnostic Type 1 Brugada ECG develops; (2) ST-segment in Type 2 ECG increases by ≥2 mm; (3) premature ventricular beats or other arrhythmias develop; or (4) QRS widens to ≥130% of baseline. These sodium channel blockers should be used with particular caution in the presence of atrial and/or ventricular conduction disease (suspected cases of Lev-Lenègre disease or in the presence of wide QRS, wide P-waves, or prolonged PR intervals). Mechanoelectrical dissociation has been encountered in isolated cases. Isoproterenol and sodium lactate may be effective antidotes.
TABLE 3.
Drugs used to unmask the Brugada syndrome
| Ajmaline: 1 mg/kg/5 min, iv |
| Flecainide: 2 mg/kg/10 min, iv (400 mg, po) |
| Procainamide: 10 mg/kg/10 min, iv |
| Pilsicainide: 1 mg/kg, iv/10 min |
Three types of repolarization patterns have been identified, as discussed above (Table 1 and Fig 1). Brugada syndrome is definitively diagnosed when a Type 1 ST-segment elevation is observed in more than one right-precordial leads (V1-V3), in the presence or absence of sodium channel block, and in conjunction with one of the following: documented ventricular fibrillation, self-terminating polymorphic ventricular tachycardia, a family history of SCD (<45 years), coved-type ECGs in family members, inducibility of VT with programmed electrical stimulation, syncope, or nocturnal agonal respiration. Importantly, any confounding factor(s) that could account for the ECG abnormality should be carefully excluded (see Table 4). Striking ST-segment elevation is sometimes observed for a brief period following DC cardioversion, although it is not known whether these patients are gene carriers for Brugada syndrome.39 Another prominent confounding factor is the ST-elevation encountered in well-trained athletes (Fig 3). Distinguishing features include the fact that, in the athlete, the ST-segment elevation is upsloping rather than downsloping and is largely unaffected by challenge with a sodium channel blocker. In addition, a variety of drugs have been reported to produce a Brugada-like ST-segment elevation (Table 5), although it is not as yet clear whether or to what extent a genetic predisposition may be involved.
TABLE 4.
ECG abnormalities that can lead to ST segment elevation in the right precordial leads
| Atypical right bundle branch block |
| Left ventricular hypertrophy |
| Early repolarization |
| Acute pericarditis |
| Acute myocardial ischemia or infarction |
| Pulmonary embolism |
| Prinzmetal's angina109 |
| Dissecting aortic aneurysm110 |
| Various central and autonomic nervous system abnormalities111,112 |
| Duchenne muscular dystrophy113 |
| Thiamine deficiency114 |
| Hyperkalemia110,115,116 |
| Hypercalcemia117,118 |
| Arrhythmogenic right ventricular dysplasia/cardiomyopathy25,119 |
| Hypothermia120,121 |
| Mechanical compression of right ventricular outflow tract as with mediastinal tumor122 |
| Hemopericardium123 |
Fig 3.
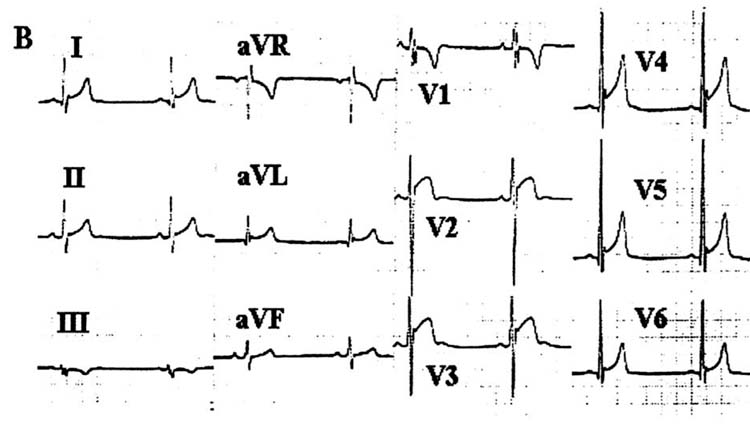
Twelve-lead ECG of a well-trained athlete without the Brugada syndrome.
TABLE 5.
Drug-induced Brugada-like ECG patterns
|
Modified from reference 159, with permission.
The diagnosis of Brugada syndrome is also considered positive when a Type 2 (saddleback pattern) or Type 3 ST-segment elevation is observed in more than one right precordial lead under baseline conditions and conversion to the diagnostic Type 1 pattern occurs after sodium channel blocker administration (ST-segment elevation should be ≥2 mm). One or more of the clinical criteria described above should also be present.
Drug-induced conversion of Type 3 to Type 2 ST-segment elevation is considered inconclusive for diagnosis of Brugada syndrome.
While most cases of Brugada syndrome display right precordial ST-segment elevation, isolated cases of inferior lead40 or left precordial lead41 ST-segment elevation have been reported in Brugada-like syndromes, in some cases associated with SCN5A mutations.
Risk Stratification
Identification of patients at risk for sudden death is an important goal of investigators worldwide.42,43 Brugada et al42 found that patients initially presenting with aborted sudden death are at the highest risk for a recurrence (69%), whereas those presenting with syncope and a spontaneously appearing Brugada ECG sign have a recurrence rate of 19%. An 8% occurrence of cardiac events was observed in initially asymptomatic patients. Among asymptomatic patients, those at highest risk displayed the Brugada sign spontaneously; those in whom ST segment elevation appeared only after provocation with sodium channel blockers appear to be at minimal or no risk for arrhythmic events. Brugada patients at highest risk are (1) males with (2) inducible VT/VF and (3) a spontaneously elevated ST segment.
Recent studies have suggested that combined electrocardiographic markers may be helpful in risk stratification. Atarashi et al used the width of the S-wave and the ST segment elevation magnitude, whereas Morita et al combined ST-segment elevation and the presence of late potentials.44,45
A recent study by Brugada et al42 suggested that among asymptomatic patients inducibility of VT during electrophysiologic study (EPS) may forecast risk. Studies by Priori et al,43 Kanda et al,46 and Eckardt et al,47 however, failed to find an association between inducibility and recurrence of VT/VF among Brugada patients (both asymptomatic and symptomatic). These discrepancies may be due to differences in patient characteristics and the use of multiple testing centers with nonstandardized or noncomparable stimulation protocols.47 Additional studies are needed to further define risk stratification strategies for asymptomatic patients.
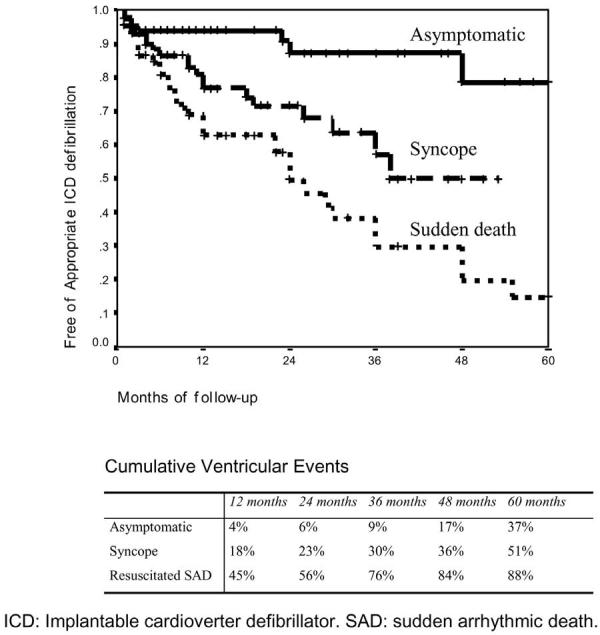
Brugada et al48 recently reported on 547 individuals diagnosed with Brugada syndrome and no previous cardiac arrest. In 124 patients the abnormal electrocardiogram was identified after one or multiple episodes of syncope and in 423 individuals during routine electrocardiographic screening or during study because they were family members of patients with the syndrome. Structural disease was ruled out in all patients. This study, evaluating the clinical outcome of the largest population of Brugada patients thus far reported, concluded that (1) patients have a high risk for sudden arrhythmic death, even in the absence of a history of prior cardiac arrest: 8.2% experienced sudden death or at least one documented episode of ventricular fibrillation during a mean follow-up of 24 ± 33 months. A spontaneously abnormal Type I ECG carried a 7.7-fold higher risk of developing an arrhythmic event during a lifetime as compared to individuals in whom the electrocardiogram diagnostic of Brugada syndrome was evident only after sodium channel blocker challenge; (2) male gender is another risk factor for sudden death, because males had a 5.5 higher risk of sudden death as compared to females; (3) programmed electrical stimulation resulting in inducibility of a sustained ventricular arrhythmia is the strongest marker of risk, associated with a 8-fold higher risk of (aborted) sudden death than noninducible patients; and (4) familial forms of the disease are not associated with a worse prognosis than sporadic cases, because a positive family history of Brugada syndrome did not predict outcome. They found a 27.2% probability of an event by logistic regression analysis in a patient with a spontaneously abnormal ECG, a previous history of syncope, and inducible sustained ventricular arrhythmias. Thus, inducibility of ventricular arrhythmias and a previous history of syncope are suggested to be markers of a poor prognosis in Brugada syndrome. Symptomatic patients require protective treatment even when they are not inducible. Asymptomatic patients can be reassured if they are noninducible.
M. M. Scheinman: The authors have nicely summarized the attempts to stratify risk for patients with the Brugada ECG sign. It should be emphasized that the study cited (ref. 48) is retrospective and hence subject to all the problems inherent in a nonrandomized prospective study. The chief risk factor was laboratory inducibility of VT or VF but, unfortunately, this risk factor was not replicated by others.43,46,47 Conceivably, as others follow their Brugada patients for a greater duration, the results will become more homogenous. Alternately, it is possible that the original Brugada observations were biased toward a more malignant cohort and not truly representative of the group as a whole.
Genetic Factors Underlying the Brugada Syndrome
Inheritance of the Brugada syndrome is via an autosomal dominant mode of transmission. The first and only gene to be linked to the Brugada syndrome is SCN5A, the gene encoding for the α-subunit of the cardiac sodium channel gene.49 Figure 4 shows a schematic illustration of SCN5A highlighting the various mutations thus far associated with the Brugada syndrome. Mutations in SCN5A are also responsible for the LQT3 form of the long QT syndrome and cardiac conduction disease. A number of mutations have been reported to cause overlapping syndromes.
Fig 4.
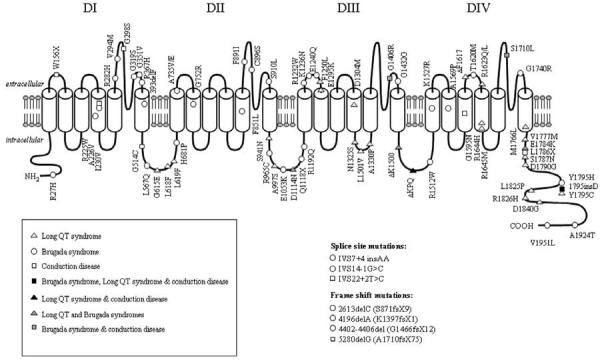
Diagrammatic representation of the human cardiac sodium channel displaying locations of mutations associated with Long QT syndrome type 3, Brugada syndrome, isolated cardiac conduction disease, and overlap syndromes. DI: domain I; DII: domain II; DIII: domain III; DIV: domain IV. From Tan157 with permission.
Approximately five dozen mutations in SCN5A have been linked to the syndrome in recent years.43,50-52 (Also see http://www.brugada.org.) About two dozen of the these mutations have been studied in expression systems and shown to result in loss of function due either to (1) a failure of the sodium channel to express; (2) a shift in the voltage- and time-dependence of INa activation, inactivation, or reactivation; (3) an entry of the sodium channel into an intermediate state of inactivation from which it recovers more slowly, or (4) an accelerated inactivation of the sodium channel (Table 6). The premature inactivation of the sodium channel is sometimes observed at physiological temperatures, but not at room temperature.53 Because this characteristic of the mutant channel was exaggerated at temperatures above the physiological range, it was suggested that the syndrome may be unmasked, and that patients with the Brugada syndrome may be at an increased risk, during a febrile state.53 A number of Brugada patients displaying fever-induced polymorphic VT have been identified since the publication of this report (see ref. 9 for references).
TABLE 6.

Biophysical changes of SCN5A mutations in Brugada syndrome
Another locus on chromosome 3, close to but distinct from SCN5A, has recently been linked to the syndrome30 in a large pedigree in which the syndrome is associated with progressive conduction disease, a low sensitivity to procainamide, and a relatively good prognosis.
SCN5A mutations account for approximately 18-30% of Brugada syndrome cases. A higher incidence of SCN5A mutations has been reported in familial than in sporadic cases.54 Of note, negative SCN5A results generally do not rule out causal gene mutations, since in general the promoter region, cryptic splicing mutations or presence of gross rearrangements is not part of routine investigation.
Based on findings to date, knowledge of a specific mutation may not provide guidance in formulating a diagnosis or determining a prognosis. Mutations have been reported throughout the SCN5A gene, and it is not as yet clear which of these if any are associated with a greater risk of arrhythmic events or sudden death. Genetic testing is recommended for support of the clinical diagnosis, for early detection of relatives at potential risk, and particularly for the purpose of advancing research and consequently our understanding of genotype–phenotype relations.
M. M. Scheinman: The phenotypic expressions of mutation of a single gene (ie, SCN5A) continue to fascinate. For example, loss of function mutations can lead to Brugada syndrome, idiopathic cardiac conduction system disease, and most recently, congenital sinus node dysfunction. Mutations associated with gain in function can produce the Long QT3 syndrome. In addition, families with phenotypic characteristics of both the prolonged QT as well as Brugada syndrome have also been described. Clinicians can only watch with great excitement as the etiology of these syndromes is discussed.
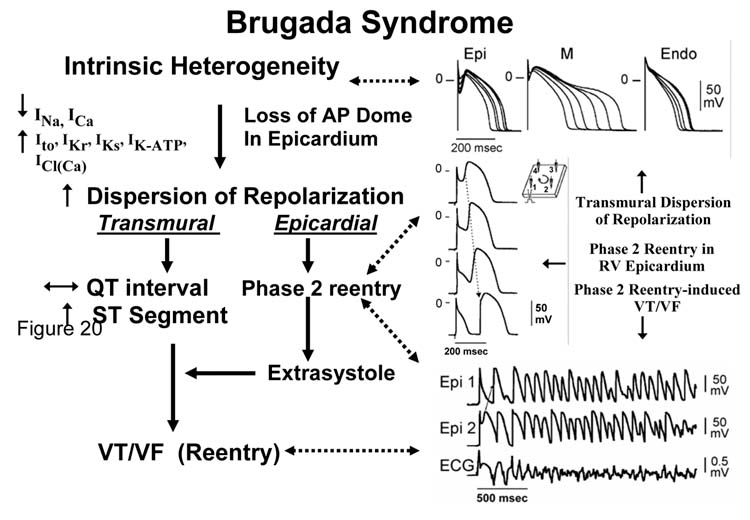
Cellular and Ionic Mechanisms
Phase 2 reentry and other characteristics of strong sodium channel blockade, which give rise to a Brugada-like syndrome, were described in the early 1990's and evolved in parallel with the clinical syndrome.2,55-57 Studies conducted over the past decade suggest that rebalancing of the currents active at the end of phase 1, leading to an accentuation of the action potential notch in right ventricular epicardium, is responsible for the accentuated J-wave or ST segment elevation associated with the Brugada syndrome (see ref. 50 for references).
Genetic defects, pathophysiologic factors, and pharmacologic influences capable of amplifying the intrinsic electrical heterogeneities present in the early phases of the action potential among the three principal ventricular cell types leads to accentuation of the J-wave and eventually loss of the action potential dome, giving rise to extrasystolic activity in the form of phase 2 reentry. Activation of Ito leads to a paradoxical prolongation of APD in canine ventricular tissues,58 but to the traditional abbreviation of APD in ventricular tissues that normally exhibit brief action potentials (eg, mouse and rat).59 Pathophysiologic conditions (eg, ischemia, metabolic inhibition) and some pharmacologic interventions (eg, INa or ICa blockers or IK-ATP, Ito, IKr, or IKs activators) can lead to marked abbreviation of the action potential in canine and feline60 ventricular cells where Ito is prominent (Fig 5). Under these conditions, canine ventricular epicardium exhibits an all-or-none repolarization as a result of the shift in the balance of currents flowing at the end of phase 1 of the action potential. When phase 1 reaches approximately −30 mV, all-or-none repolarization of the action potential ensues leading to loss of the dome as the outward currents overwhelm the inward currents. Loss of the action potential dome typically occurs at some epicardial sites but not others, resulting in the development of a marked dispersion of repolarization within the epicardium as well as between epicardium and endocardium. Propagation of the action potential dome from the epicardial site at which it is maintained to sites at which it is abolished can cause local re-excitation of the preparation. This mechanism, termed phase 2 reentry, produces extrasystolic beats capable of initiating circus movement reentry61 (Figs 6 and 7). Phase 2 reentry has been shown to occur when right ventricular epicardium is exposed to (1) K+ channel openers such as pinacidil62; (2) sodium channel blockers such as flecainide57; (3) increased [Ca2+]o 63; (4) calcium channel blockers such as verapamil; (5) metabolic inhibition64; and (5) simulated ischemia.61
Fig 5.
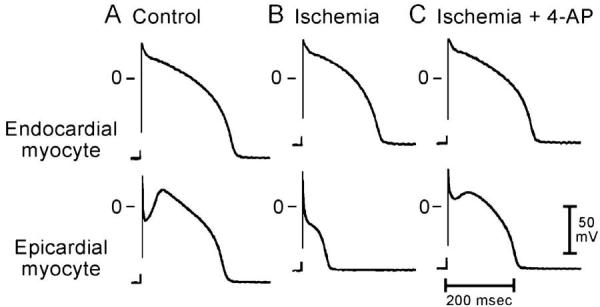
Differential effects of ischemia on action potentials recorded from endocardial and epicardial myocytes dissociated from the canine left ventricle. A: Control. B: Responses recorded after 30 min of superfusion with ischemic solution. “Ischemia” caused a marked abbreviation of APD90 in the epicardial cell and a modest shortening in the endocardial cell. C: Responses obtained 5 min after switching to an ischemic solution containing 4-aminopyridine (4-AP: 1 mM). 4-AP reversed the ischemia-induced shortening of APD90 in the epicardial myocyte and also prolonged APD90 in the endocardial cell. These effects of 4-AP were similar to those observed in syncytial tissue preparations from the corresponding ventricular layer. BCL of 800 msec. (From ref. 68 with permission.)
Fig 6.
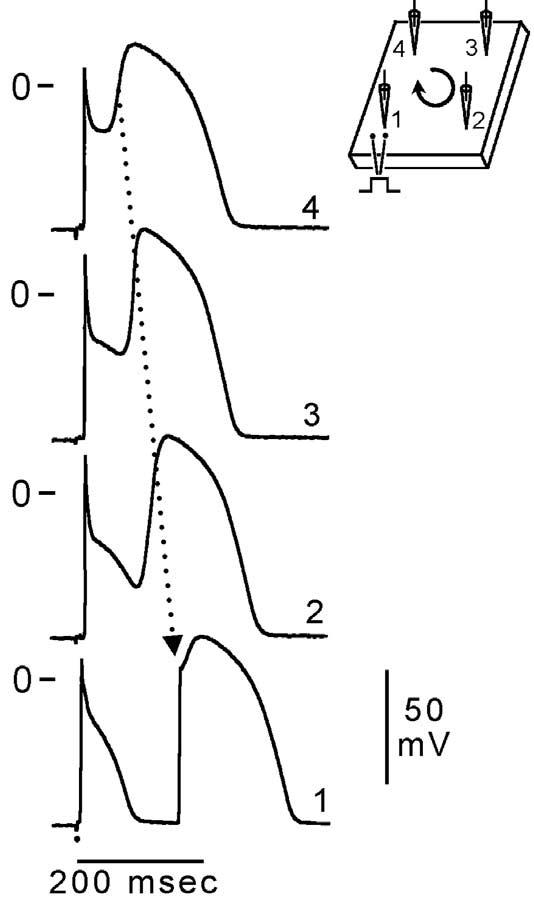
Phase 2 reentry. Reentrant activity induced by exposure of a canine ventricular epicardial preparation (0.7 cm2) to simulated ischemia. Microelectrode recordings were obtained from four sites as shown in the schematic (upper right). After 35 min of ischemia, the action potential dome develops normally at site 4, but not at sites 1, 2, or 3. The dome then propagates in a clockwise direction re-exciting sites 3, 2, and 1 with progressive delays, thus generating a closely coupled reentrant extrasystole (156 msec) at site 1. In this example of phase 2 reentry, propagation of the dome occurs in a direction opposite to that of phase 0, a mechanism akin to reflection. BCL = 700 msec. (Modified from ref. 61 with permission.)
Fig 7.
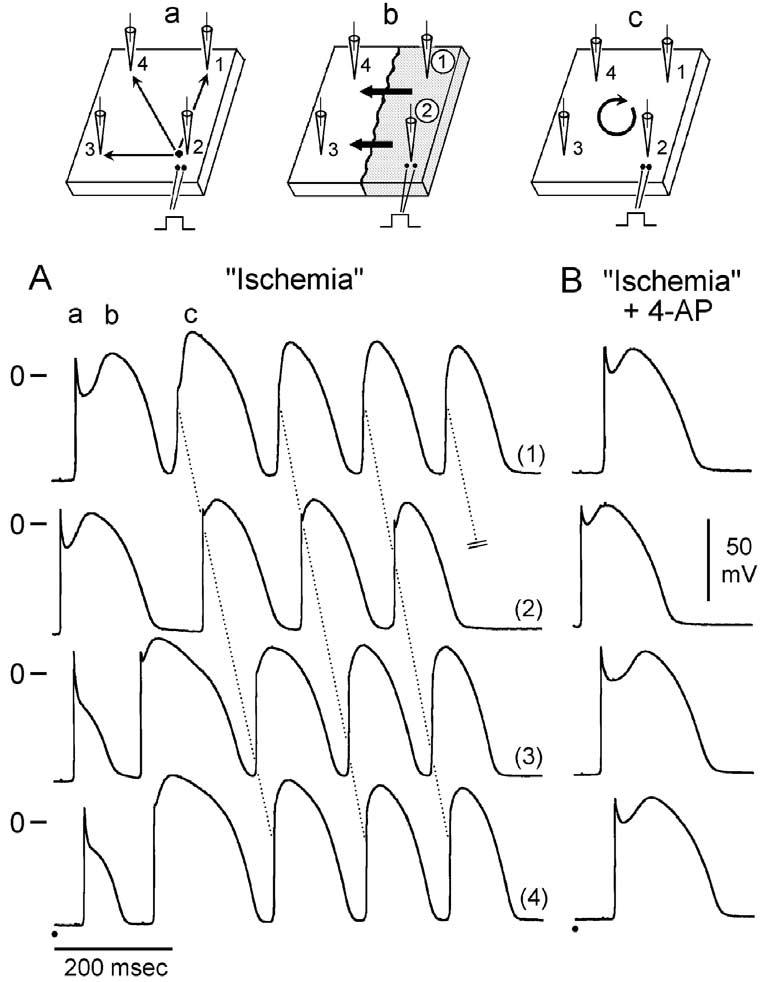
Phase 2 reentrant extrasystole triggers circus movement reentry. A: Exposure of a relatively large canine right ventricular epicardial sheet (6.3 cm2) to simulated ischemia results in loss of the dome at sites 3 and 4 but not at sites 1 and 2 (BCL = 1100 msec). Conduction of the basic beat proceeds normally from the stimulation site (site 2; see schematic a). Propagation of the action potential dome from the right half of the preparation caused re-excitation of the left half via a phase 2 reentry mechanism (see schematic b). The extrasystolic beat generated by phase 2 reentry then initiates a run of tachycardia that is sustained for four additional cycles via typical circus movement reentry. The proposed reentrant path is shown in schematic c. Note that phase 2 reentry provides an activation front roughly perpendicular to that of the basic beat. This type of cross-field activation has previously been shown to predispose to the development of vortex-like reentry in isolated epicardial sheets. B: Recorded after addition of 1 mM 4-AP, an inhibitor of the transient outward current. In the continued presence of ischemia, 4-AP restored the dome at all epicardial recording sites within 3 min. Thus electrical heterogeneity was restored and all reentrant activity abolished. (Modified from ref. 61 with permission.)
Accentuated or otherwise abnormal J-waves have long been linked to idiopathic ventricular fibrillation and the Brugada syndrome.1,21,65,66 The Brugada syndrome is characterized by exaggerated J-wave that manifests as an ST segment elevation in the right precordial leads.1 A number of studies have highlighted the similarities between the conditions that predispose to phase 2 reentry and those that attend the appearance of the Brugada syndrome.
Loss of the action potential dome in epicardium, but not endocardium, generates a transmural current that manifests on the ECG as an ST segment elevation, similar to that encountered in patients with Brugada syndrome.64,67,68 Autonomic neurotransmitters like acetylcholine facilitate loss of the action potential dome69 by suppressing ICa and/or augmenting potassium current. β-Adrenergic agonists restore the dome by augmenting ICa. Sodium channel blockers also facilitate loss of the canine right ventricular action potential dome via a negative shift in the voltage at which phase 1 begins.56,57 These findings are consistent with accentuation of ST segment elevation in patients with Brugada syndrome following vagal maneuvers or Class I antiarrhythmic agents as well as normalization of the ST segment elevation following β-adrenergic agents and phosphodiesterase III inhibitors.8,67,70 The appearance of an ST segment elevation only in the right precordial leads in Brugada patients is consistent with the finding that loss of the action potential dome is much more commonly encountered in right vs. left canine ventricular epicardium.64,68,71 The Brugada syndrome is a right ventricular disease presumable because Ito density is intrinsically much larger in the right vs. left ventricular epicardium.
Thus accentuation of the action potential notch and eventual loss of the dome of the right ventricular epicardial action potential dome is the basis for the ST segment elevation. The vulnerable window created transmu-rally as well as within epicardium serves as the substrate and phase 2 reentry provides the extrasystole that serves as the a trigger for episodes of ventricular tachycardia and fibrillation in the Brugada syndrome. Evidence in support of this hypothesis was recently provided in an arterially perfused canine right ventricular experimental model of the Brugada syndrome (Fig 8).72 The VT and VF generated in these preparations is often polymorphic, resembling a very rapid form of Torsade de Pointes (TdP). This activity may be mechanistically related to the migrating spiral wave shown to generate a pattern resembling TdP associated with a normal or long QT interval.73,74
Fig 8.
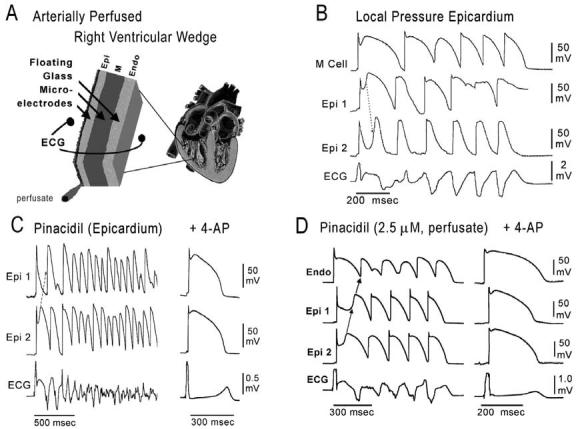
ECG and arrhythmias with typical features of the Brugada syndrome recorded from canine right ventricular wedge preparations (see inset, top left). A: Schematic of arterially perfused right ventricular wedge preparation. B: Pressure-induced phase 2 reentry and VT. Shown are transmembrane action potentials simultaneously recorded from two epicardial (Epi 1 and Epi 2) and one M region (M) site, together with a transmural ECG. Local application of pressure near Epi 2 results in loss of the action potential dome at that site but not at Epi 1 or M sites. The dome at Epi 1 then re-excites Epi 2, giving rise to a phase 2 reentrant extrasystole which triggers a short run of ventricular tachycardia. Note the ST segment elevation due to loss of the action potential dome in a segment of epicardium. C: Polymorphic VT/VF induced by local application of the potassium channel opener pinacidil (10 μM) to the epicardial surface of the wedge. Action potentials from two epicardial sites (Epi 1 and Epi 2) and a transmural ECG were simultaneously recorded. Loss of the dome at Epi 1 but not Epi 2 creates a marked dispersion of repolarization, giving rise to a phase 2 reentrant extrasystole. The extrasystolic beat then triggers a long episode of ventricular fibrillation (22 sec). Right panel: Addition of 4-AP (2 mM), a specific Ito blocker, to the perfusate restored the action potential dome at Epi 1, thus reducing dispersion of repolarization and suppressing all arrhythmic activity. BCL = 2000 msec. D: Phase 2 reentry gives rise to VT following addition of pinacidil (2.5 μM) to the coronary perfusate. Transmembrane action potentials from two epicardial sites (Epi 1 and Epi 2) and one endocardial site (Endo) as well as a transmural ECG were simultaneously recorded. Right panel: 4-AP (1 mM) markedly reduces the magnitude of the action potential notch in epicardium, thus restoring the action potential dome throughout the preparation and abolishing all arrhythmic activity. (Panel D is from ref. 72 with permission.)
Combined INa and ICa block has recently been shown to be more effective than INa inhibition alone in precipitating Brugada syndrome in the arterially perfused wedge preparation (Fig 9).75 High concentrations of terfenadine (5 μM) produce a potent block of INa and ICa, leading to accentuation of the epicardial action potential notch following acceleration of the rate from a BCL of 800 to 400 msec. The dramatic accentuation of the notch was due to the effect of the drug to depress phase 0, augment the magnitude of phase 1, and delay the appearance of the second upstroke. With continued rapid pacing, phase 1 became more accentuated, until all-or-none repolarization occurred at the end of phase 1 at some epicardial sites but not others, leading to the development of both epicardial (EDR) and transmural (TDR) dispersion of repolarization (Fig 9C). Propagation of the dome from the region where it was maintained to the region at which it was lost results in the development of local phase 2 reentry (Fig 9D). Figure 10 shows the ability of terfenadine-induced phase 2 reentry to generate an extrasystole, couplet, and polymorphic VT/VF. Figure 10D illustrates an example of programmed electrical stimulation to initiate VT/VF under similar conditions.
Fig 9.
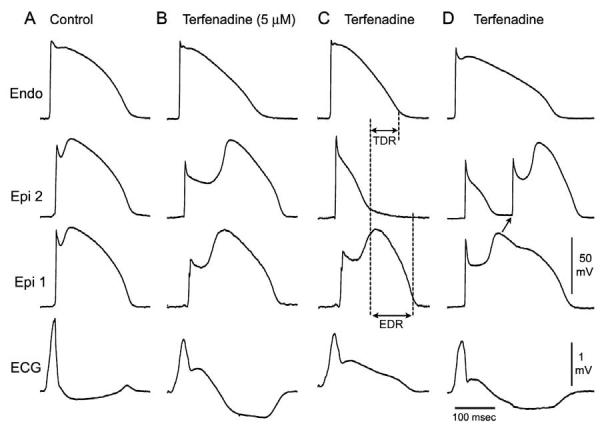
Terfenadine-induced ST segment elevation, T-wave inversion, transmural and endocardial dispersion of repolarization, and phase 2 reentry. Each panel shows transmembrane action potentials from one endocardial (top) and two epicardial sites together with a transmural ECG recorded from a canine arterially perfused right ventricular wedge preparation. A: Control (BCL 400 msec). B: Terfenadine (5 μM) accentuated the epicardial action potential notch creating a transmural voltage gradient that manifests as an ST segment elevation or exaggerated J-wave in the ECG. First beat recorded after changing from BCL 800 to BCL 400 msec. C: Continued pacing at BCL 400 msec results in all-or-none repolarization at the end of phase 1 at some epicardial sites but not others, creating a local epicardial dispersion of repolarization (EDR) as well as a transmural dispersion of repolarization (TDR). D: Phase 2 reentry occurs when the epicardial action potential dome propagates from a site where it is maintained to regions where it has been lost. (Modified from ref. 75 with permission.)
Fig 10.
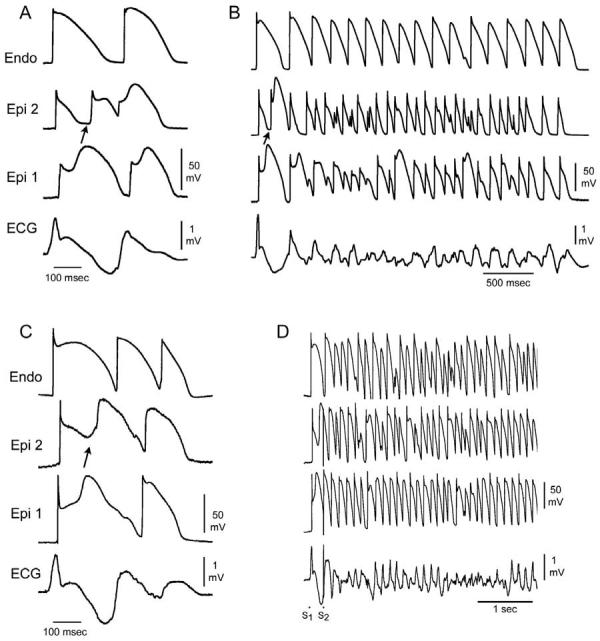
Spontaneous and programmed electrical stimulation-induced polymorphic VT in RV wedge preparations pretreated with terfenadine (5-10 μM). A: Phase 2 reentry in epicardium gives rise to a closely coupled extrasystole. B: Phase 2 reentrant extrasystole triggers a brief episode of polymorphic VT. C: Extrastimulus (S1-S2 = 250 msec) applied to epicardium triggers a polymorphic VT. (Modified from ref. 75 with permission.)
The electrocardiographic manifestations of the Brugada syndrome can be attributed to one of two basic mechanisms: (1) conduction delay in the right ventricular epicardial free wall in the region of the outflow tract (RVOT) or (2) premature repolarization of the right ventricular epicardial action potential secondary to loss of the action potential dome or a combination of the two. The cellular mechanisms thought to be responsible for the development of the Brugada phenotype via hypothesis 2 is schematically illustrated in Fig 11.76,77
Fig 11.
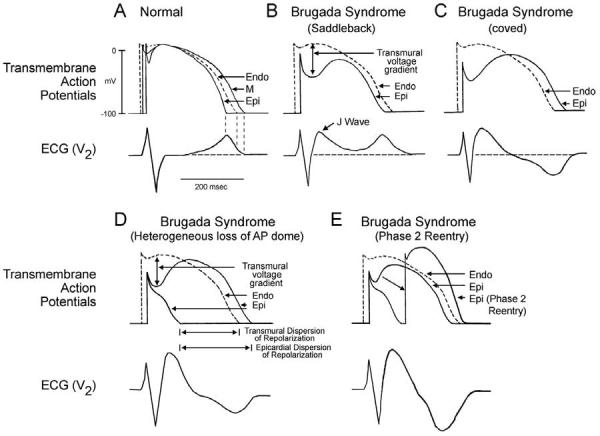
Schematic representation of right ventricular epicardial action potential changes proposed to underlie the electrocardiographic manifestation of the Brugada syndrome. (Modified from ref. 76 with permission.)
Under normal conditions, the ST segment is isoelectric because of the absence of transmural voltage gradients at the level of the action potential plateau (Fig 11A). Accentuation of the right ventricular notch under pathophysiologic conditions leads to exaggeration of transmural voltage gradients and thus to accentuation of the J-wave or to J-point elevation. When epicardial repolarization precedes repolarization of the cells in the M and endocardial regions, the T-wave remains positive. The result is a saddleback configuration of the repolarization waves (Fig 11B). Further accentuation of the notch may be accompanied by a prolongation of the epicardial action potential such that the direction of repolarization across the right ventricular wall and transmural voltage gradients are reversed, leading to the development of a coved-type ST segment elevation and inversion of the T-wave (Fig 11C), typically observed in the ECG of Brugada patients. A delay in epicardial activation may also contribute to inversion of the T-wave. The down-sloping ST segment elevation observed in the experimental wedge models often appears as an R′, suggesting that the appearance of a RBBB morphology in Brugada patients may be due at least in part to early repolarization of RV epicardium, rather than major impulse conduction block in the right bundle. Indeed a rigorous application of RBBB criteria reveals that a large majority of RBBB-like morphologies encountered in cases of Brugada syndrome do not fit the criteria for RBBB.78 Moreover, attempts by Miyazaki and coworkers to record delayed activation of the RV in Brugada patients met with failure.8 Although the typical Brugada morphology is present in Figs 11B and C, the substrate for reentry is not present. The arrhythmogenic substrate is thought to develop when a further shift in the balance of current leads to loss of the action potential dome at some epicardial sites but not others (Fig 11D). Loss of the action potential dome in epicardium but not endocardium results in the development of a marked transmural dispersion of repolarization and refractoriness responsible for the development of a vulnerable window during which a premature impulse or extrasystole can induce a reentrant arrhythmia. Conduction of the action potential dome from sites at which it is maintained to sites at which it is lost causes local re-excitation via a phase 2 reentry mechanism, leading to the development of a very closely coupled extrasystole, which captures the vulnerable window across the wall, thus triggering a circus movement reentry in the form of VT/VF (Fig 11E).61,72 The phase 2 reentrant beat fuses with the negative T-wave of the basic response. Because the extrasystole originates in epicardium, the QRS complex is largely composed of a Q-wave, which serves to accentuate the negative deflection of the inverted T-wave, giving the ECG a more symmetrical appearance. This morphology is often observed in the clinic preceding the onset of polymorphic VT.
Support for these hypotheses derives from experiments involving the arterially perfused right ventricular wedge preparation72 as well as from recent studies by Kurita et al in which monophasic action potential (MAP) electrodes were positioned on the epicardial and endocardial surfaces of the RVOT in patients with Brugada syndrome (Fig 12).2,79
Fig 12.
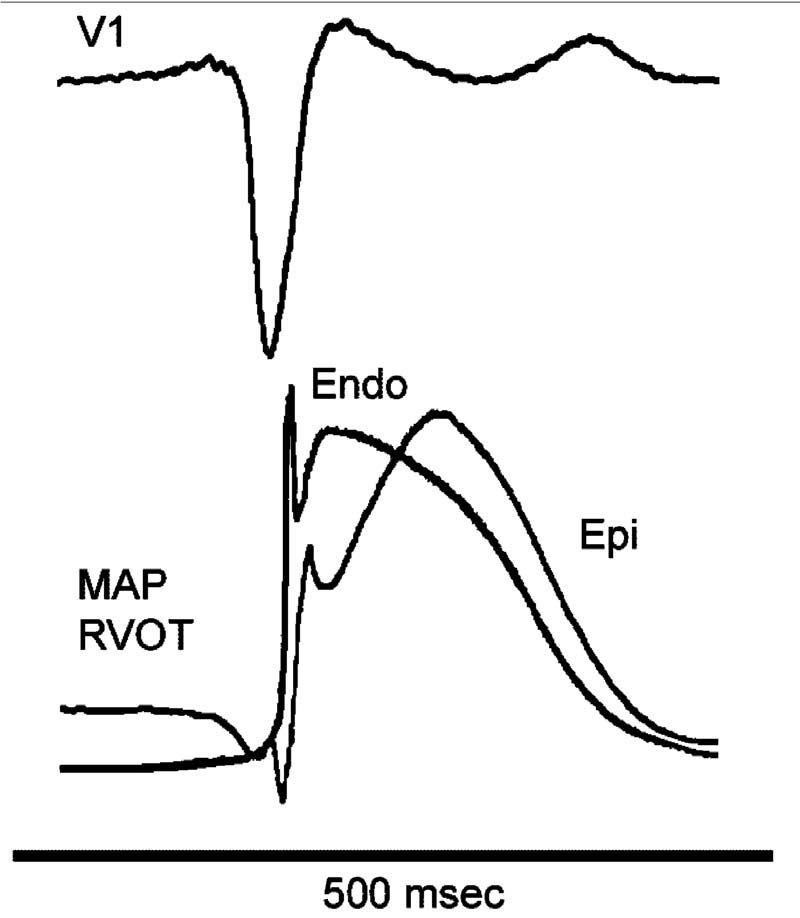
Monophasic action potential (MAP) recordings from endocardium (Endo) and epicardium (Epi) of the right ventricular outflow tract of a Brugada patient. A prominent action potential notch is apparent in the epicardial MAP, but not in the endocardial MAP, coincident with the appearance of the accentuated J-wave or ST segment elevation in the ECG (V1). (Modified from ref. 2 with permission.)
These characteristics of the action potential of ventricular epicardium suggest that activation forces, generated by the second upstroke of the right ventricular epicardial action potential and/or phase 2 reentry, may extend beyond the QRS in Brugada patients. Indeed, SAECG recordings have demonstrated late potentials in patients with the Brugada syndrome, especially in the anterior wall of the RVOT.35,80 The basis for these late potentials, commonly ascribed to delayed conduction within the ventricle, are largely unknown. Endocardial recordings have been unrevealing. Nagase and coworkers37 introduced a guide wire into the conus branch of the right coronary artery to record signals from the epicardial surface of the anterior wall of the RVOT in patients with Brugada syndrome. The unipolar recordings displayed delayed potentials which coincided with late potentials recorded in the SAECG, particularly after administration of class IC antiarrhythmic agents. The authors conclude that recordings from the conus branch of the right coronary artery can identify an “epicardial abnormality” in the RVOT which is accentuated in the presence of IC agents, thus uncovering part of the arrhythmogenic substrate responsible for VT/VF in Brugada syndrome, which may be related to the second upstroke or a concealed phase 2 reentrant beat. While late potentials are commonly regarded as being representative of delayed activation of the myocardium, in the case of the Brugada syndrome, other possibilities exist as discussed above. For example, the second upstroke of the epicardial action potential, thought to be greatly accentuated in Brugada syndrome,50 might be capable of generating late potentials when RVOT activation is otherwise normal. Moreover, the occurrence of phase 2 reentry, especially when concealed (ie, when it fails to trigger transmural reentry), may contribute to the generation of delayed unipolar and late SAECG potentials.
The rate dependence of the ECG sign may be helpful in discriminating between these two hypotheses. If the Brugada ECG sign is due to delayed conduction in the RVOT, acceleration of the rate would be expected to further aggravate conduction and thus accentuate the ST segment elevation and the RBBB morphology of the ECG. If, on the other hand, the Brugada sign is secondary to accentuation of the epicardial action potential notch, at some point leading to loss of the action potential dome, acceleration of the rate would be expected to normalize the ECG, by restoring the action potential dome and reducing the notch. This occurs because the transient outward current, which is at the heart of this mechanism, is slow to recover from inactivation and is less available at faster rates. The fact of the matter is that Brugada patients usually display a normalization of their ECG or no change when heart rate is increased, thus favoring the second hypothesis. Further evidence in support of this hypothesis derives from the recent of observations of Shimizu and coworkers.81 Using a unipolar catheter introduced into the great cardiac vein, they recorded unipolar activation recovery intervals (ARI), a measure of local action potential duration, from the epicardial surface of the RVOT in a 53-year-old Brugada patient. ARI in the RVOT was observed to abbreviate dramatically whenever the ST segment was elevated in V2 following a pause or the administration of a sodium channel blocker. Thus, the available data, both experimental and clinical, point to transmural voltage gradients that develop secondary to accentuation of the epicardial notch and loss of the action potential dome as being in large part responsible for the Brugada ECG signature.
Basis for the Greater Prevalence of the Brugada Phenotype in Males
Despite equal genetic transmission of the mutation between the sexes, the clinical phenotype is 8 to 10 times more prevalent in males than in females. The basis for this sex-related distinction was recently shown to be due to a more prominent Ito-mediated action potential notch in the RV epicardium of males vs. females82 (Figs 13 and 14). The more prominent Ito causes the end of phase 1 of the RV epicardial action potential to repolarize to more negative potentials in tissue and arterially perfused wedge preparations from males, facilitating loss of the action potential dome and the development of phase 2 reentry and polymorphic VT.
Fig 13.

Sex-based and interventricular differences in Ito. A: Mean I-V relationship for Ito recorded from RV epicardial cells isolated from hearts of male and female dogs. Inset, Representative Ito current traces and voltage protocol. Ito density was significantly greater in male vs. female RV epicardial cells. No sex differences were observed in LV. B: Transmembrane action potentials recorded from isolated canine RV epicardial male and female tissue slices. BCLs, 300, 500, 800, and 2000 ms. C: Rate dependence of phase 1 amplitude and voltage at end of phase 1 (V/phase 1, mV) in males (solid squares) vs. females (solid circles). (Modified from ref. 82 with permission.)
Fig 14.
Terfenadine induces Brugada phenotype more readily in male than female RV wedge preparations. Each panel shows action potentials recorded from two epicardial sites and one endocardial site, together with a transmural ECG. Control recordings were obtained at a BCL of 2000 msec, whereas terfenadine data were recorded at a BCL of 800 ms after a brief period of pacing at a BCL of 400 ms. A: Terfenadine (5 μM) induced, heterogeneous loss of action potential dome, ST-segment elevation, and phase 2 reentry (arrow) in a male RV wedge preparation. B: Terfenadine fails to induce Brugada phenotype in a female RV wedge preparation. C: Polymorphic VT triggered by spontaneous phase 2 reentry in a male preparation. D: Incidence of phase 2 reentry in male (6 of 7) vs. female (2 of 7) RV wedge preparations when perfused with 5 μM terfenadine for up to 2 hours. (Modified from ref. 82 with permission.)
Figure 15 summarizes the proposed cellular mechanism for the Brugada syndrome. The available data support the hypothesis that the Brugada syndrome results form amplification of heterogeneities intrinsic to the early phases of the action potential among the different transmural cell types. The amplification is secondary to a rebalancing of currents active during phase 1, including a decrease in INa or ICa or augmentation of any one of a number of outward currents. ST segment elevation similar to that observed in patients with the Brugada syndrome occurs as a consequence of the accentuation of the action potential notch, eventually leading to loss of the action potential dome in right ventricular epicardium, where Ito is most prominent. Loss of the dome gives rise to both a transmural as well as an epicardial dispersion of repolarization. The transmural dispersion is responsible for the development of ST segment elevation and the creation of a vulnerable window across the ventricular wall, whereas the epicardial dispersion gives to phase 2 reentry, which provides the extrasystole that captures the vulnerable window, thus precipitating VT/VF. The VT generated is usually polymorphic, resembling a very rapid form of Torsade de Pointes.
Fig 15.
Proposed mechanism for the Brugada syndrome. A shift in the balance of currents serves to amplify existing heterogeneities by causing loss of the action potential dome at some epicardial, but not endocardial sites. A vulnerable window develops as a result of the dispersion of repolarization and refractoriness within epicardium as well as across the wall. Epicardial dispersion leads to the development of phase 2 reentry, which provides the extrasystole that captures the vulnerable window and initiates VT/VF via a circus movement reentry mechanism. (Modified from ref. 77 with permission.)
Factors that Precipitate and Modulate the ECG and Arrhythmic Manifestations of the Brugada Syndrome
ST segment elevation in the Brugada syndrome can be very dynamic. The Brugada ECG is often concealed, but may be unmasked or modulated by sodium channel blockers, a febrile state, vagotonic agents, α-adrenergic agonists, β-adrenergic blockers, tricyclic or tetracyclic antidepressants, first-generation antihistaminics (dimenhydrinate), a combination of glucose and insulin, hyperkalemia, hypokalemia, hypercalcemia, and alcohol and cocaine toxicity (Fig 16).6-8,83-89 These agents may also induce acquired forms of the Brugada syndrome (Table 5). Until a definitive list of drugs to avoid in the Brugada syndrome is formulated, the list of agents in Table 5 may provide some guidance.
Fig 16.
Factors predisposing to the electrocardiographic and arrhythmic manifestations of the Brugada syndrome. (Modified from Nademanee et al 158 with permission.)
Acute ischemia or myocardial infarction due to vasospasm involving the RVOT mimics ST-segment elevation, similar to that in Brugada syndrome. This effect is secondary to the depression of ICa and the activation of IK-ATP during ischemia and suggests that patients with congenital and possibly acquired forms of Brugada syndrome may be at a higher risk for ischemia-related sudden cardiac death.90
VF and sudden death in the Brugada syndrome usually occur at rest and at night. Circadian variation of sympatho-vagal balance, hormones, and other metabolic factors are likely to contribute to this circadian pattern. Bradycardia, due to altered symaptho-vagal balance or other factors, may contribute to arrhythmia initiation.19,20,91 Abnormal 123I-MIBG uptake in 8 (17%) of the 17 Brugada syndrome patients but none in the control group was demonstrated by Wichter et al.92 There was segmental reduction of 123I-MIBG in the inferior and the septal left ventricular wall, indicating presynaptic sympathetic dysfunction. Of note, imaging of the right ventricle, particularly the RVOT, is difficult with this technique, so that insufficient information is available concerning sympathetic function in the regions known to harbor the arrhythmogenic substrate. Moreover, it remains unclear what role the reduced uptake function plays in the arrhythmogenesis of the Brugada syndrome. If indeed the RVOT is similarly affected, this defect may indeed alter the symaptho-vagal balance in favor of the development of an arrhythmogenic substrate.69,72
Hypokalemia has been implicated as a contributing cause for the high prevalence of SUDS in the Northeastern region of Thailand where potassium deficiency is endemic.89,93 Serum potassium in the Northeastern population is significantly lower than that of the population in Bangkok, which lies in the central part of Thailand where potassium is abundant in the food. A recent case report highlights the ability of hypokalemia to induce VF in a 60-year-old man who had asymptomatic Brugada syndrome, without a family history of sudden cardiac death.89 This patient was initially treated for asthma by steroids, which lowered serum potassium from 3.8 mmol/L on admission to 3.4 and 2.9 mmol/L on the 7th and 8th day of admission, respectively. Both were associated with unconsciousness. VF was documented during the last episode, which reverted spontaneously to sinus rhythm.
The Thai Ministry of Public Health Report (1990) found an association between a large meal of glutinous rice (“sticky rice”) or carbohydrates ingested on the night of death in SUNDS patients.93 Consistent with this observation, a recent study by Nogami et al found that glucose and insulin could unmask the Brugada ECG.88
Premature inactivation of the sodium channel in SCN5A mutations associated with Brugada syndrome has been shown to be accentuated at higher temperatures,53 suggesting that a febrile state may unmask Brugada syndrome. Indeed, several case reports have emerged recently demonstrating that febrile illness could reveal the Brugada ECG and precipitate VF.9,94-98 Anecdotal data point to hot baths as a possible precipitating factor. Of note, the Northeastern part of Thailand, where the Brugada syndrome is most prevalent, is known for its very hot climate. A study is underway to assess whether this extreme climate influences the prognosis of the disease.
Therapy of Congenital Brugada Syndrome
Despite impressive strides in the identification and characterization of Brugada syndrome over the past decade, progress relative to therapy has been less noteworthy. The various device and pharmacologic therapies tested clinically or suggested based on experimental evidence are listed in Table 7. Currently, an implantable cardioverter defibrillator (ICD) is the only proven effective treatment for the disease.99,100 In a multicenter trial of 690 patients with Brugada syndrome, in which 258 individuals received an ICD, efficacy of the device in reverting VF and preventing sudden cardiac death was 100%; appropriate shocks were delivered in 14, 20, 29, 38, and 52% of cases at 1, 2, 3, 4, and 5 years of follow-up, respectively (Fig 17). In the case of initially asymptomatic patients, appropriate ICD discharge was delivered in 4, 6, 9, 17, and 37% at 1, 2, 3, 4, and 5 years of follow-up, respectively.
TABLE 7.
Device and pharmacologic considerations for therapy in the Brugada syndrome
| Devices |
| √ ICD—only established effective therapy |
| ? Pacemaker |
| ? Ablation or cryosurgery |
| Pharmacologic |
| X Amiodarone—does not protect102 |
| X β-Blockers—does not protect102 |
| √ β-Adrenergic agonists—Isoproterenol8,10 |
| √ Phosphodiesterase inhibitors—cilostazol70 |
| X Class IC antiarrhythmics—Flecainide, Propafenone—contraindicated |
| Class IA antiarrhythmics |
| X Procainamide, Disopyramide—contraindicated |
| √ Quinidine72,104,105,108 |
| ? Tedisamil |
| √ Ito Blockers—cardioselective and ion channel specific |
Fig 17.
Kaplan-Meier curve of effectiveness of the ICD in 258 patients with electrocardiographic pattern of Brugada syndrome according to symptoms. (Modified from ref. 48 with permission.)
Current recommendations for ICD implantation are summarized as follows:
Symptomatic patients displaying the Type 1 Brugada ECG (either spontaneously or after sodium channel blockade) who present with aborted sudden death should receive an ICD without additional need for EPS. Similar patients presenting with related symptoms such as syncope, seizure, or noctural agonal respiration should also undergo ICD implantation after noncardiac causes of these symptoms have been carefully ruled out. EPS is recommended in symptomatic patients only for the assessment of supraventricular arrhythmia.
Asymptomatic patients displaying a Type 1 Brugada ECG (spontaneously or after sodium channel block) should undergo EPS if there is a family history of sudden cardiac death suspected to be due to Brugada syndrome. EPS may be justified when the family history is negative for sudden cardiac death if the Type 1 ECG occurs spontaneously. If inducible for ventricular arrhythmia, the patient should receive an ICD. Asymptomatic patients who have no family history and who develop a Type I ECG only after sodium channel blockade should be closely followed up.
As additional data become available, these recommendations will no doubt require further refinement.
ICD therapy is not an adequate solution for infants and young children or for patients residing in regions of the world where an ICD is unaffordable. Although arrhythmias and sudden cardiac death generally occur during sleep or at rest and have been associated with slow heart rates, a potential therapeutic role for cardiac pacing is largely unexplored. Data relative to a cryosurgical approach or the use of ablation therapy are limited to date. A recent report by Haissaguerre and coworkers101 indicates that focal radiofrequency ablation may be a potentially valuable tool in controlling arrhythmogenesis by focal ablation of the ventricular premature beats that trigger VT/VF in Brugada syndrome.
The pharmacologic approach to therapy, based on experimental data, has been tailored to a rebalancing of currents active during the early phases of the epicardial action potential in the right ventricle so as to reduce the magnitude of the action potential notch and/or restore the action potential dome (Table 7). Antiarrhythmic agents such as amiodarone and β-blockers have been shown to be ineffective.102 Class IC antiarrhythmic drugs (such as flecainide and propafenone) and class IA agents (such as procainamide and disopyramide) are contraindicated for reasons previously discussed. Other class IA agents, such as quinidine and tedisamil, however, may exert a therapeutic action because of their Ito blocking properties. Because the presence of a prominent transient outward current, Ito, in the right ventricle is at the heart of the mechanism underlying the Brugada syndrome, any agent that inhibits this current may be protective. Cardioselective and Ito-specific blockers are not currently available. The only agent on the market in the United States with significant Ito blocking properties is quinidine. It is for this reason that it was suggested several years ago that this agent may be of therapeutic value in the Brugada syndrome.103 Studies have shown quinidine to be effective in restoring the epicardial action potential dome, thus normalizing the ST segment and preventing phase 2 reentry and polymorphic VT in experimental models of the Brugada syndrome.72 Clinical evidence of the effectiveness of quinidine in normalizing ST-segment elevation in patients with Brugada syndrome has been reported (Fig 18),104,105 although clinical trials designed to assess the efficacy of this agent are not available. Agents that boost the L-type calcium current, such as isoproterenol, may be useful as well.50,72 Both types of agents (Ito blocker and agents that augment ICa) have been shown to be effective in normalizing ST-segment elevation in patients with Brugada syndrome and in controlling “electrical storms,” particularly in children.10,104-107 Other than the study by Belhassen and coworkers involving quinidine, none have as yet demonstrated long-term efficacy in the prevention of sudden cardiac death.104,108
Fig 18.
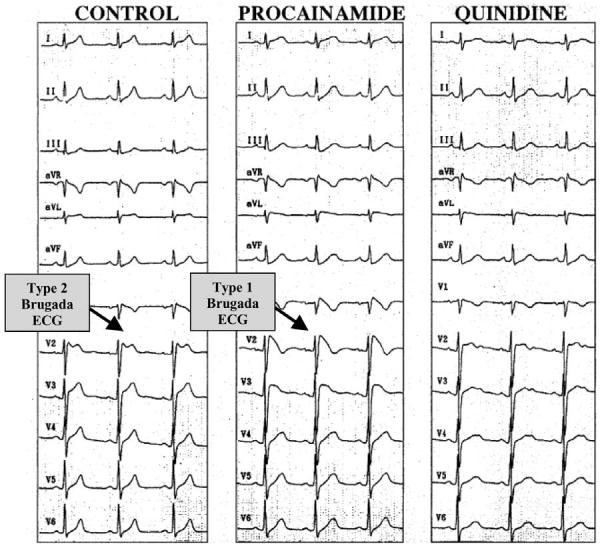
Twelve-lead electrocardiogram (ECG) tracings in an asymptomatic 26-year-old man with the Brugada syndrome. Left: Baseline: Type 2 ECG (not diagnostic) displaying a “saddleback-type” ST-segment elevation is observed in V2. Center: After intravenous administration of 750 mg procainamide, the Type 2 ECG is converted to the diagnostic Type 1 ECG consisting of a “coved-type” ST-segment elevation. Right: A few days after oral administration of quinidine bisulfate (1500 mg/day, serum quinidine level 2.6 mg/L), ST-segment elevation is attenuated displaying a nonspecific (neither Type 1-3 Brugada ECG) abnormal pattern in the right precordial leads. VF could be induced during control and procainamide Brugada ECG infusion, but not after quinidine. (From 104 with permission.)
The most recent addition to the pharmacological armamentarium is a phospodiesterase III inhibitor, cilostazol,70 which normalizes the ST segment most likely by augmenting calcium current (ICa) as well as by reducing Ito secondary to an increase in heart rate. Finally, an experimental antiarrhythmic agent, tedisamil, with potent actions to block Ito, among other outward currents, has been suggested as a therapeutic candidate.50 Tedisamil may be more potent than quinidine because it lacks the relatively strong inward current blocking actions of quinidine. The development of a cardioselective and Ito-specific blocker is important for the future to complement the limited therapeutic armamentarium currently available to combat this disease. Appropriate clinical trials are needed to establish the effectiveness of all of the above pharmacologic agents as well as the possible role of pacemakers.
M. M. Scheinman: Summary: We are indeed very much indebted to the authors for a superb, timely, and very current review of this disease. The authors are, in fact, the individuals most responsible for the major advances that have been recorded since the disease was first described in 1992. Several points are noteworthy; it was a keen clinician that recognized the importance of the Brugada ECG finding. All the wonderful genetic and pathophysiological findings grew out of the seminal clinical observation. This highlights the importance of the link between bedside and cellular physiologist. The work, beautifully summarized by the authors, suggests that we are currently at the stage for development of a large prospective NIH study to critically assess newer possible treatment modalities that might include drugs that block Ito (ie, quinidine, tedisimal) or enhance Ca++/Na+ current phosphodiesterase inhibitors, beta agonists.
One can envision trials using the high-risk cohort (as identified by the authors), where all subjects are treated with an ICD and only some receive drug therapy. Finding an effective drug would have enormous consequences in many countries where ICD therapy is not affordable.
Footnotes
Supported by Grants HL47678 and HL066169 from NHLBI (C.A., R.B.), American Heart Association (R.B., C.A.), and NYS and Florida Grand Lodges F.&A.M.
REFERENCES
- 1.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome: a multicenter report. J Am Coll Cardiol. 1992;20:1391–6. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 2.Antzelevitch C, Brugada P, Brugada J, et al. Brugada syndrome: a decade of progress. Circ Res. 2002;91:1114–8. doi: 10.1161/01.res.0000046046.53721.90. [DOI] [PubMed] [Google Scholar]
- 3.Wilde AA, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome. Eur Heart J. 2002;23:1648–54. doi: 10.1053/euhj.2002.3382. [DOI] [PubMed] [Google Scholar]
- 4.Wilde AA, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation. 2002;106:2514–9. doi: 10.1161/01.cir.0000034169.45752.4a. [DOI] [PubMed] [Google Scholar]
- 5.Brugada P, Brugada R, Antzelevitch C, Nademanee K, Towbin J, Brugada J. The Brugada Syndrome. In: Gussak I, Antzelevitch C, editors. Cardiac Repolarization. Bridging Basic and Clinical Sciences. Humana Press; Totowa, NJ: 2003. pp. 427–46. [Google Scholar]
- 6.Brugada P, Brugada J, Brugada R. Arrhythmia induction by antiarrhythmic drugs. PACE. 2000;23:291–2. doi: 10.1111/j.1540-8159.2000.tb06751.x. [DOI] [PubMed] [Google Scholar]
- 7.Brugada R, Brugada J, Antzelevitch C, et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101:510–5. doi: 10.1161/01.cir.101.5.510. [DOI] [PubMed] [Google Scholar]
- 8.Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol. 1996;27:1061–70. doi: 10.1016/0735-1097(95)00613-3. [DOI] [PubMed] [Google Scholar]
- 9.Antzelevitch C, Brugada R. Fever and the Brugada Syndrome. PACE. 2002;25:1637–9. doi: 10.1046/j.1460-9592.2002.01537.x. [DOI] [PubMed] [Google Scholar]
- 10.Shimizu W, Matsuo K, Takagi M, et al. Body surface distribution and response to drugs of ST segment elevation in Brugada syndrome: clinical implication of eighty-seven-lead body surface potential mapping and its application to twelve-lead electrocardiograms. J Cardiovasc Electrophysiol. 2000;11:396–404. doi: 10.1111/j.1540-8167.2000.tb00334.x. [DOI] [PubMed] [Google Scholar]
- 11.Sangwatanaroj S, Prechawat S, Sunsaneewitayakul B, Sitthisook S, Tosukhowong P, Tungsanga K. ew electrocardiographic leads and the procainamide test for the detection of the Brugada sign in sudden unexplained death syndrome survivors and their relatives. Eur Heart J. 2001;22:2290–6. doi: 10.1053/euhj.2001.2691. [DOI] [PubMed] [Google Scholar]
- 12.Alings M, Wilde A. “Brugada” syndrome: clinical data and suggested pathophysiological mechanism [see comments] Circulation. 1999;99:666–73. doi: 10.1161/01.cir.99.5.666. [DOI] [PubMed] [Google Scholar]
- 13.Bezzina C, Veldkamp MW, van Den Berg MP, et al. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–13. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 14.Priori SG, Napolitano C, Gasparini M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families [In Process Citation] Circulation. 2000;102:2509–15. doi: 10.1161/01.cir.102.20.2509. [DOI] [PubMed] [Google Scholar]
- 15.Pitzalis MV, Anaclerio M, Iacoviello M, et al. QT-interval prolongation in right precordial leads: an additional electrocardiographic hallmark of Brugada syndrome. J Am Coll Cardiol. 2003;42:1632–7. doi: 10.1016/j.jacc.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Smits JP, Eckardt L, Probst V, et al. Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J Am Coll Cardiol. 2002;40:350–6. doi: 10.1016/s0735-1097(02)01962-9. [DOI] [PubMed] [Google Scholar]
- 17.Morita H, Kusano-Fukushima K, Nagase S. Atrial fibrillation and atrial vulnerability in patients with Brugada syndrome. J Am Coll Cardiol. 2002;40:1437. doi: 10.1016/s0735-1097(02)02167-8. [DOI] [PubMed] [Google Scholar]
- 18.Takehara N, Makita N, Kawabe J, et al. A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J Intern Med. 2004;255:137–42. doi: 10.1046/j.0954-6820.2003.01247.x. [DOI] [PubMed] [Google Scholar]
- 19.Kasanuki H, Ohnishi S, Ohtuka M, et al. Idiopathic ventricular fibrillation induced with vagal activity in patients without obvious heart disease. Circulation. 1997;95:2277–85. doi: 10.1161/01.cir.95.9.2277. [DOI] [PubMed] [Google Scholar]
- 20.Proclemer A, Facchin D, Feruglio GA, Nucifora R. Recurrent ventricular fibrillation, right bundle-branch block and persistent ST segment elevation in V1-V3: a new arrhythmia syndrome? A clinical case report (see comments) G Ital Cardiol. 1993;23:1211–8. [PubMed] [Google Scholar]
- 21.Bjerregaard P, Gussak I, Kotar Sl, Gessler JE. Recurrent synocope in a patient with prominent J-wave. Am Heart J. 1994;127:1426–30. doi: 10.1016/0002-8703(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 22.Vatta M, Dumaine R, Varghese G, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11:337–45. doi: 10.1093/hmg/11.3.337. [DOI] [PubMed] [Google Scholar]
- 23.Sugai MA. Pathological study on sudden and unexpected death, especially on the cardiac death autopsied by medical examiners in Tokyo. Acta Pathol Jpn. 1959;9(Suppl):723–52. doi: 10.1111/j.1440-1827.1959.tb02963.x. [DOI] [PubMed] [Google Scholar]
- 24.Nademanee K, Veerakul G, Nimmannit S, et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation. 1997;96:2595–600. doi: 10.1161/01.cir.96.8.2595. [DOI] [PubMed] [Google Scholar]
- 25.Corrado D, Basso C, Buja G, Nava A, Rossi L, Thiene G. Right bundle branch block, right precordial ST-segment elevation, and sudden death in young people. Circulation. 2001;103:710–7. doi: 10.1161/01.cir.103.5.710. [DOI] [PubMed] [Google Scholar]
- 26.Ayerza MR, de Zutter M, Goethals M, Wellens F, Geelen P, Brugada P. Heart transplantation as last resort against Brugada syndrome. J Cardiovasc Electrophysiol. 2002;13:943–4. doi: 10.1046/j.1540-8167.2002.00943.x. [DOI] [PubMed] [Google Scholar]
- 27.Remme CA, Wever EFD, Wilde AAM, Derksen R, Hauer RNW. Diagnosis and long-term follow-up of Brugada syndrome in patients with idiopathic ventricular fibrillation. Eur Heart J. 2001;22:400–9. doi: 10.1053/euhj.2000.2366. [DOI] [PubMed] [Google Scholar]
- 28.Bezzina CR, Rook MB, Groenewegen WA, et al. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ Res. 2003;92:159–68. doi: 10.1161/01.res.0000052672.97759.36. [DOI] [PubMed] [Google Scholar]
- 29.Antzelevitch C. Molecular genetics of arrhythmias and cardiovascular conditions associated with arrhythmias. Invited Review—NASPE 25th Anniversary. J Cardiovasc Electrophysiol. 2003;14:1259–1272. doi: 10.1046/j.1540-8167.2003.03316.x. [DOI] [PubMed] [Google Scholar]
- 30.Weiss R, Barmada MM, Nguyen T, et al. Clinical and molecular heterogeneity in the Brugada syndrome. A novel gene locus on chromosome 3. Circulation. 2002;105:707–13. doi: 10.1161/hc0602.103618. [DOI] [PubMed] [Google Scholar]
- 31.Ahmed F, Li D, Karibe A, et al. Localization of a gene responsible for arrhythmogenic right ventricular dysplasia to chromosome 3p23. Circulation. 1998;98:2791–5. doi: 10.1161/01.cir.98.25.2791. [DOI] [PubMed] [Google Scholar]
- 32.Takagi M, Aihara N, Kuribayashi S, et al. Localized right ventricular morphological abnormalities detected by electron-beam computed tomography represent arrhythmogenic substrates in patients with the Brugada syndrome. Eur Heart J. 2001;22:1032–41. doi: 10.1053/euhj.2000.2424. [DOI] [PubMed] [Google Scholar]
- 33.Antzelevitch C. Brugada syndrome: historical perspectives and observations. Eur Heart J. 2002;23:676–8. doi: 10.1053/euhj.2001.3145. [DOI] [PubMed] [Google Scholar]
- 34.Antzelevitch C. Late potentials and the Brugada syndrome. J Am Coll Cardiol. 2002;39:1996–9. doi: 10.1016/s0735-1097(02)01887-9. [DOI] [PubMed] [Google Scholar]
- 35.Futterman LG, Lemberg L. Brugada. Am J Crit Care. 2001;10:360–4. [PubMed] [Google Scholar]
- 36.Fujiki A, Usui M, Nagasawa H, Mizumaki K, Hayashi H, Inoue H. ST segment elevation in the right precordial leads induced with class IC antiarrhythmic drugs: insight into the mechanism of Brugada syndrome. J Cardiovasc Electrophysiol. 1999;10:214–8. doi: 10.1111/j.1540-8167.1999.tb00662.x. [DOI] [PubMed] [Google Scholar]
- 37.Nagase S, Kusano KF, Morita H, et al. Epicardial electrogram at the right ventricular outflow tract in patients with Brugada syndrome—using epicardial lead. J Am Coll Cardiol. 2002;39:1992–5. doi: 10.1016/s0735-1097(02)01888-0. [DOI] [PubMed] [Google Scholar]
- 38.Shimizu W, Antzelevitch C, Suyama K, et al. Effect of sodium channel blockers on ST segment, QRS duration, and corrected QT interval in patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2000;11:1320–9. doi: 10.1046/j.1540-8167.2000.01320.x. [DOI] [PubMed] [Google Scholar]
- 39.Gurevitz O, Glikson M. Cardiac resynchronization therapy: a new frontier in the management of heart failure. Isr Med Assoc J. 2003;5:571–5. [PubMed] [Google Scholar]
- 40.Kalla H, Yan GX, Marinchak R. Ventricular fibrillation in a patient with prominent J (Osborn) waves and ST segment elevation in the inferior electrocardiographic leads: a Brugada syndrome variant? J Cardiovasc Electrophysiol. 2000;11:95–8. doi: 10.1111/j.1540-8167.2000.tb00743.x. [DOI] [PubMed] [Google Scholar]
- 41.Horigome H, Shigeta O, Kuga K, et al. Ventricular fibrillation during anesthesia in association with J waves in the left precordial leads in a child with coarctation of the aorta. J Electrocardiol. 2003;36:339–43. doi: 10.1016/s0022-0736(03)00079-7. [DOI] [PubMed] [Google Scholar]
- 42.Brugada J, Brugada R, Antzelevitch C, Towbin J, Nademanee K, Brugada P. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V(1) to V(3) Circulation. 2002;105:73–8. doi: 10.1161/hc0102.101354. [DOI] [PubMed] [Google Scholar]
- 43.Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105:1342–7. doi: 10.1161/hc1102.105288. [DOI] [PubMed] [Google Scholar]
- 44.Atarashi H, Ogawa S. For the idiopathic ventricular fibrillation investigators. New ECG criteria for high-risk Brugada syndrome. Circ J. 2003;67:8–10. doi: 10.1253/circj.67.8. [DOI] [PubMed] [Google Scholar]
- 45.Morita H, Takenaka-Morita S, Fukushima-Kusano K, et al. Risk stratification for asymptomatic patients with Brugada syndrome. Circ J. 2003;67:312–6. doi: 10.1253/circj.67.312. [DOI] [PubMed] [Google Scholar]
- 46.Kanda M, Shimizu W, Matsuo K, et al. Electrophysiologic characteristics and implications of induced ventricular fibrillation in symptomatic patients with Brugada syndrome. J Am Coll Cardiol. 2002;39:1799–805. doi: 10.1016/s0735-1097(02)01867-3. [DOI] [PubMed] [Google Scholar]
- 47.Eckardt L, Kirchhof P, Johna R, Haverkamp W, Breithardt G, Borggrefe M. Wolff-Parkinson-White syndrome associated with Brugada syndrome. PACE. 2001;24:1423–4. doi: 10.1046/j.1460-9592.2001.01423.x. [DOI] [PubMed] [Google Scholar]
- 48.Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation. 2003;108:3092–6. doi: 10.1161/01.CIR.0000104568.13957.4F. [DOI] [PubMed] [Google Scholar]
- 49.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanisms for idiopathic ventricular fibrillation. Nature. 1998;392:293–6. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 50.Antzelevitch C. The Brugada syndrome: ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol. 2001;12:268–72. doi: 10.1046/j.1540-8167.2001.00268.x. [DOI] [PubMed] [Google Scholar]
- 51.Balser JR. The cardiac sodium channel: gating function and molecular pharmacology. J Mol Cell Cardiol. 2001;33:599–613. doi: 10.1006/jmcc.2000.1346. [DOI] [PubMed] [Google Scholar]
- 52.Tan HL, Bezzina CR, Smits JP, Verkerk AO, Wilde AA. Genetic control of sodium channel function. Cardiovasc Res. 2003;57:961–73. doi: 10.1016/s0008-6363(02)00714-9. [DOI] [PubMed] [Google Scholar]
- 53.Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–9. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- 54.Schulze-Bahr E, Eckardt L, Breithardt G, Seidl K, Wichter T, Wolpert C, Borggrefe M, Haverkamp W. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Human Mut. 2003;21:651–2. doi: 10.1002/humu.9144. [DOI] [PubMed] [Google Scholar]
- 55.Antzelevitch C, Sicouri S, Litovsky SH, et al. Heterogeneity within the ventricular wall: electrophysiology and pharmacology of epicardial, endocardial and M cells. Circ Res. 1991;69:1427–49. doi: 10.1161/01.res.69.6.1427. [DOI] [PubMed] [Google Scholar]
- 56.Krishnan SC, Antzelevitch C. Sodium channel blockade produces opposite electrophysiologic effects in canine ventricular epicardium and endocardium. Circ Res. 1991;69:277–91. doi: 10.1161/01.res.69.2.277. [DOI] [PubMed] [Google Scholar]
- 57.Krishnan SC, Antzelevitch C. Flecainide-induced arrhythmia in canine ventricular epicardium: phase 2 reentry? Circulation. 1993;87:562–72. doi: 10.1161/01.cir.87.2.562. [DOI] [PubMed] [Google Scholar]
- 58.Litovsky SH, Antzelevitch C. Rate dependence of action potential duration and refractoriness in canine ventricular endocardium differs from that of epicardium: The role of the transient outward current. J Am Coll Cardiol. 1989;14:1053–66. doi: 10.1016/0735-1097(89)90490-7. [DOI] [PubMed] [Google Scholar]
- 59.Kilborn MJ, Fedida D. A study of the developmental changes in outward currents of rat ventricular myocytes. J Physiol (Lond) 1990;430:37–60. doi: 10.1113/jphysiol.1990.sp018280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Furukawa T, Kimura S, Cuevas J, Furukawa N, Bassett AL, Myerburg RJ. Role of cardiac ATP-regulated potassium channels in differential responses of endocardial and epicardial cells to ischemia. Circ Res. 1991;68:1693–702. doi: 10.1161/01.res.68.6.1693. [DOI] [PubMed] [Google Scholar]
- 61.Lukas A, Antzelevitch C. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. The antiarrhythmic effects of 4-aminopyridine. Cardiovasc Res. 1996;32:593–603. [PubMed] [Google Scholar]
- 62.Di Diego JM, Antzelevitch C. Pinacidil-induced electrical heterogeneity and extrasystolic activity in canine ventricular tissues: does activation of ATP-regulated potassium current promote phase 2 reentry? Circulation. 1993;88:1177–89. doi: 10.1161/01.cir.88.3.1177. [DOI] [PubMed] [Google Scholar]
- 63.Di Diego JM, Antzelevitch C. High [Ca2+]-induced electrical heterogeneity and extrasystolic activity in isolated canine ventricular epicardium: Phase 2 reentry. Circulation. 1994;89:1839–50. doi: 10.1161/01.cir.89.4.1839. [DOI] [PubMed] [Google Scholar]
- 64.Antzelevitch C, Sicouri S, Lukas A, et al. Clinical implications of electrical heterogeneity in the heart: The electrophysiology and pharmacology of epicardial, M and endocardial cells. In: Podrid PJ, Kowey PR, editors. Cardiac Arrhythmia: Mechanism, Diagnosis and Management. William & Wilkins; Baltimore, MD: 1995. pp. 88–107. [Google Scholar]
- 65.Aizawa Y, Tamura M, Chinushi M, et al. Idiopathic ventricular fibrillation and bradycardia-dependent intraverthcular block. Am Heart J. 1993;126:1473–4. doi: 10.1016/0002-8703(93)90550-s. [DOI] [PubMed] [Google Scholar]
- 66.Aizawa Y, Tamura M, Chinushi M, et al. An attempt at electrical catheter ablation of the arrhythmogenic area in idiopathic ventricular fibrillation. Am Heart J. 1992;123:257–60. doi: 10.1016/0002-8703(92)90786-u. [DOI] [PubMed] [Google Scholar]
- 67.Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93:372–9. doi: 10.1161/01.cir.93.2.372. [DOI] [PubMed] [Google Scholar]
- 68.Lukas A, Antzelevitch C. Differences in the electrophysiological response of canine ventricular epicardium and endocardium to ischemia: role of the transient outward current. Circulation. 1993;88:2903–15. doi: 10.1161/01.cir.88.6.2903. [DOI] [PubMed] [Google Scholar]
- 69.Litovsky SH, Antzelevitch C. Differences in the electrophysiological response of canine ventricular subendocardium and subepicardium to acetylcholine and isoproterenol. A direct effect of acetylcholine in ventricular myocardium. Circ Res. 1990;67:615–27. doi: 10.1161/01.res.67.3.615. [DOI] [PubMed] [Google Scholar]
- 70.Tsuchiya T, Ashikaga K, Honda T, Arita M. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 2002;13:698–701. doi: 10.1046/j.1540-8167.2002.00698.x. [DOI] [PubMed] [Google Scholar]
- 71.Di Diego JM, Sun ZQ, Antzelevitch C. Ito and action potential notch are smaller in left vs. right canine ventricular epicardium. Am J Physiol. 1996;271:H548–H561. doi: 10.1152/ajpheart.1996.271.2.H548. [DOI] [PubMed] [Google Scholar]
- 72.Yan GX, Antzelevitch C. Cellular basis for the Brugada Syndrome and other mechanisms of arrhythmogenesis associated with ST segment elevation. Circulation. 1999;100:1660–6. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 73.Pertsov AM, Davidenko JM, Salomonsz R, Baxter WT, Jalife J. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ Res. 1993;72:631–50. doi: 10.1161/01.res.72.3.631. [DOI] [PubMed] [Google Scholar]
- 74.Asano Y, Davidenko JM, Baxter WT, Gray RA, Jalife J. Optical mapping of drug-induced polymorphic arrhythmies and torsade de pointes in the isolated rabbit heart. J Am Coll Cardiol. 1997;29:831–42. doi: 10.1016/s0735-1097(96)00588-8. [DOI] [PubMed] [Google Scholar]
- 75.Fish JM, Antzelevitch C. Role of sodium and calcium channel block in unmasking the Brugada Syndrome. Heart Rhythm. 2004;1:210–7. doi: 10.1016/j.hrthm.2004.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Antzelevitch C. The Brugada syndrome: ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol. 2001;12:268–72. doi: 10.1046/j.1540-8167.2001.00268.x. [DOI] [PubMed] [Google Scholar]
- 77.Antzelevitch C. The Brugada Syndrome. Diagnostic criteria and cellular mechanisms. Eur Heart J. 2001;22:356–63. doi: 10.1053/euhj.2000.2461. [DOI] [PubMed] [Google Scholar]
- 78.Gussak I, Antzelevitch C, Bjerregaard P, Towbin JA, Chaitman BR. The Brugada syndrome: clinical, electrophysiological and genetic aspects. J Am Coll Cardiol. 1999;33:5–15. doi: 10.1016/s0735-1097(98)00528-2. [DOI] [PubMed] [Google Scholar]
- 79.Kurita T, Shimizu W, Inagaki M, et al. The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. J Am Coll Cardiol. 2002;40:330–4. doi: 10.1016/s0735-1097(02)01964-2. [DOI] [PubMed] [Google Scholar]
- 80.Fujiki A, Usui M, Nagasawa H, Mizumaki K, Hayashi H, Inoue H. ST segment elevation in the right precordial leads induced with class IC antiarrhythmic drugs: insight into the mechanism of Brugada syndrome. J Cardiovasc Electrophysiol. 1999;10:214–8. doi: 10.1111/j.1540-8167.1999.tb00662.x. [DOI] [PubMed] [Google Scholar]
- 81.Shimizu W, Aiba T, Kurita T, Kamakura S. Paradoxic abbreviation of repolarization in epicardium of the right ventricular outflow tract during augmentation of Brugada-type ST segment elevation. J Cardiovasc Electrophysiol. 2001;12:1418–21. doi: 10.1046/j.1540-8167.2001.01418.x. [DOI] [PubMed] [Google Scholar]
- 82.Di Diego JM, Cordeiro JM, Goodrow RJ, et al. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation. 2002;106:2004–11. doi: 10.1161/01.cir.0000032002.22105.7a. [DOI] [PubMed] [Google Scholar]
- 83.Babaliaros VC, Hurst JW. Tricyclic antidepressants and the Brugada syndrome: an example of Brugada waves appearing after the administration of desipramine. Clin Cardiol. 2002;25:395–8. doi: 10.1002/clc.4950250809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goldgran-Toledano D, Sideris G, Kevorkian JP. Overdose of cyclic antidepressants and the Brugada syndrome. N Engl J Med. 2002;346:1591–2. doi: 10.1056/NEJM200205163462020. [DOI] [PubMed] [Google Scholar]
- 85.Tada H, Sticherling C, Oral H, Morady F. Brugada syndrome mimicked by tricyclic antidepressant overdose. J Cardiovasc Electrophysiol. 2001;12:275. doi: 10.1046/j.1540-8167.2001.00275.x. [DOI] [PubMed] [Google Scholar]
- 86.Pastor A, Nunez A, Cantale C, Cosio FG. Asymptomatic brugada syndrome case unmasked during dimenhydrinate infusion. J Cardiovasc Electrophysiol. 2001;12:1192–4. doi: 10.1046/j.1540-8167.2001.01192.x. [DOI] [PubMed] [Google Scholar]
- 87.Ortega-Carnicer J, Bertos-Polo J, Gutierrez-Tirado C. Aborted sudden death, transient Brugada pattern, and wide QRS dysrrhythmias after massive cocaine ingestion. J Electrocardiol. 2001;34:345–9. doi: 10.1054/jelc.2001.26318. [DOI] [PubMed] [Google Scholar]
- 88.Nogami A, Nakao M, Kubota S, et al. Enhancement of J-ST-segment elevation by the glucose and insulin test in Brugada syndrome. PACE. 2003;26:332–7. doi: 10.1046/j.1460-9592.2003.00044.x. [DOI] [PubMed] [Google Scholar]
- 89.Araki T, Konno T, Itoh H, Ino H, Shimizu M. Brugada syndrome with ventricular tachycardia and fibrillation related to hypokalemia. Circ J. 2003;67:93–5. doi: 10.1253/circj.67.93. [DOI] [PubMed] [Google Scholar]
- 90.Noda T, Shimizu W, Taguchi A, et al. ST-segment elevation and ventricular fibrillation without coronary spasm by intracoronary injection of acetylcholine and/or ergonovine maleate in patients with Brugada syndrome. J Am Coll Cardiol. 2002;40:1841–7. doi: 10.1016/s0735-1097(02)02494-4. [DOI] [PubMed] [Google Scholar]
- 91.Mizumaki K, Fujiki A, Tsuneda T, et al. Vagal activity modulates spontaneous augmentation of ST elevation in daily life of patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2004;15:679–85. doi: 10.1046/j.1540-8167.2004.03601.x. [DOI] [PubMed] [Google Scholar]
- 92.Wichter T, Matheja P, Eckardt L, et al. Cardiac autonomic dysfunction in Brugada syndrome. Circulation. 2002;105:702–6. doi: 10.1161/hc0602.103677. [DOI] [PubMed] [Google Scholar]
- 93.Nimmannit S, Malasit P, Chaovakul V, Susaengrat W, Vasuvattakul S, Nilwarangkur S. Pathogenesis of sudden unexplained nocturnal death (lai tai) and endemic distal renal tubular acidosis. Lancet. 1991;338:930–2. doi: 10.1016/0140-6736(91)91786-t. [DOI] [PubMed] [Google Scholar]
- 94.Gonzalez Rebollo G, Madrid H, Carcia A, Garcia de Casto A, Moro AM. Recurrent ventricular fibrillation during a febrile illness in a patient with the Brugada Syndrome. Rev Esp Cardiol. 2000;53:755–7. doi: 10.1016/s0300-8932(00)75151-7. [DOI] [PubMed] [Google Scholar]
- 95.Madle A, Kratochvil Z, Polivkova A. The Brugada syndrome. Vnitr Lek. 2002;48:255–8. [PubMed] [Google Scholar]
- 96.Saura D, Garcia-Alberola A, Carrillo P, Pascual D, Martinez-Sanchez J, Valdes M. Brugada-like electrocardiographic pattern induced by fever. PACE. 2002;25:856–9. doi: 10.1046/j.1460-9592.2002.t01-1-00856.x. [DOI] [PubMed] [Google Scholar]
- 97.Porres JM, Brugada J, Urbistondo V, Garcia F, Reviejo K, Marco P. Fever unmasking the Brugada syndrome. PACE. 2002;25:1646–8. doi: 10.1046/j.1460-9592.2002.01646.x. [DOI] [PubMed] [Google Scholar]
- 98.Kum L, Fung JWH, Chan WWL, Chan GK, Chan YS, Sanderson JE. Brugada syndrome unmasked by febrile illness. PACE. 2002;25:1660–1. doi: 10.1046/j.1460-9592.2002.01660.x. [DOI] [PubMed] [Google Scholar]
- 99.Brugada J, Brugada R, Brugada P. Pharmacological and device approach to therapy of inherited cardiac diseases associated with cardiac arrhythmias and sudden death. J Electrocardiol. 2000;33(Suppl):41–7. doi: 10.1054/jelc.2000.20322. [DOI] [PubMed] [Google Scholar]
- 100.Brugada P, Brugada R, Brugada J, Geelen P. Use of the prophylactic implantable cardioverter defibrillator for patients with normal hearts. Am J Cardiol. 1999;83:98D–100D. doi: 10.1016/s0002-9149(98)01009-1. [DOI] [PubMed] [Google Scholar]
- 101.Haissaguerre M, Extramiana F, Hocini M, et al. Mapping and ablation of ventricular fibrillation associated with long-QT and Brugada syndromes. Circulation. 2003;108:925–8. doi: 10.1161/01.CIR.0000088781.99943.95. [DOI] [PubMed] [Google Scholar]
- 102.Brugada J, Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3. A marker for sudden death in patients without demonstrable structural heart disease. Circulation. 1998;97:457–60. doi: 10.1161/01.cir.97.5.457. [DOI] [PubMed] [Google Scholar]
- 103.Antzelevitch C, Brugada P, Brugada J, Brugada R, Nademanee K, Towbin JA. The Brugada Syndrome. Futura Publishing Company, Inc.; Armonk, NY: 1999. p. 1. [Google Scholar]
- 104.Belhassen B, Viskin S, Antzelevitch C. The Brugada Syndrome: is ICD the only therapeutic option? PACE. 2002;25:1634–40. doi: 10.1046/j.1460-9592.2002.01634.x. [DOI] [PubMed] [Google Scholar]
- 105.Alings M, Dekker L, Sadee A, Wilde A. Quinidine induced electrocardiographic normalization in two patients with Brugada syndrome. PACE. 2001;24:1420–2. doi: 10.1046/j.1460-9592.2001.01420.x. [DOI] [PubMed] [Google Scholar]
- 106.Suzuki H, Torigoe K, Numata O, Yazaki S. Infant case with a malignant form of Brugada syndrome. J Cardiovasc Electrophysiol. 2000;11:1277–80. doi: 10.1046/j.1540-8167.2000.01277.x. [DOI] [PubMed] [Google Scholar]
- 107.Tanaka H, Kinoshita O, Uchikawa S, et al. Successful prevention of recurrent ventricular fibrillation by intravenous isoproterenol in a patient with Brugada syndrome. PACE. 2001;24:1293–4. doi: 10.1046/j.1460-9592.2001.01293.x. [DOI] [PubMed] [Google Scholar]
- 108.Belhassen B, Viskin S, Fish R, Glick A, Setbon I, Eldar M. Effects of electro-physiologic-guided therapy with Class IA antiarrhythmic drugs on the long-term outcome of patients with idiopathic ventricular fibrillation with or without the Brugada syndrome. J Cardiovasc Electrophysiol. 1999;10:1301–12. doi: 10.1111/j.1540-8167.1999.tb00183.x. [DOI] [PubMed] [Google Scholar]
- 109.Wang K, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med. 2003;349:2128–35. doi: 10.1056/NEJMra022580. [DOI] [PubMed] [Google Scholar]
- 110.Myers GB. Other QRS-T patterns that may be mistaken for myocardial infarction. IV. Alterations in blood potassium; myocardial ischemia; subepicardial myocarditis; distortion associated with arrhythmias. Circulation. 1950;2:75–93. doi: 10.1161/01.cir.2.1.75. [DOI] [PubMed] [Google Scholar]
- 111.Abbott JA, Cheitlin MD. The nonspecific camel-hump sign. JAMA. 1976;235:413–4. [PubMed] [Google Scholar]
- 112.Hersch C. Electrocardiographic changes in head injuries. Circulation. 1961;23:853–60. doi: 10.1161/01.cir.23.6.853. [DOI] [PubMed] [Google Scholar]
- 113.Perloff JK, Henze E, Schelbert HR. Alterations in regional myocardial metabolism, perfusion, and wall motion in Duchenne muscular dystrophy studied by radionuclide imaging. Circulation. 1984;69:33–42. doi: 10.1161/01.cir.69.1.33. [DOI] [PubMed] [Google Scholar]
- 114.Read DH, Harrington DD. Experimentally induced thiamine deficiency in beagle dogs: clinical observations. Am J Vet Res. 1981;42:984–91. [PubMed] [Google Scholar]
- 115.Merrill JP, Levine HD, Somerville W, Smith S. Clinical recognition and treatment of acute potassium intoxication. Ann Intern Med. 1950;33:797–830. doi: 10.7326/0003-4819-33-4-797. [DOI] [PubMed] [Google Scholar]
- 116.Ortega-Carnicer J, Benezet J, Ruiz-Lorenzo F, Alcazar R. Transient Brugada-type electrocardiographic abnormalities in renal failure reversed by dialysis. Resuscitation. 2002;55:215–9. doi: 10.1016/s0300-9572(02)00210-1. [DOI] [PubMed] [Google Scholar]
- 117.Douglas PS, Carmichael KA, Palevsky PM. Extreme hypercalcemia and electrocardiographic changes. Am J Cardiol. 1984;54:674–5. doi: 10.1016/0002-9149(84)90274-1. [DOI] [PubMed] [Google Scholar]
- 118.Sridharan MR, Horan LG. Electrocardiographic J wave of hypercalcemia. Am J Cardiol. 1984;54:672–3. doi: 10.1016/0002-9149(84)90273-x. [DOI] [PubMed] [Google Scholar]
- 119.Corrado D, Nava A, Buja G, et al. Familial cardiomyopathy underlies syndrome of right bundle branch block. ST segment elevation and sudden death. J Am Coll Cardiol. 1996;27:443–8. doi: 10.1016/0735-1097(95)00485-8. [DOI] [PubMed] [Google Scholar]
- 120.Osborn JJ. Experimental hypothermia: respiratory and blood pH changes in relation to cardiac function. Am J Physiol. 1953;175:389–98. doi: 10.1152/ajplegacy.1953.175.3.389. [DOI] [PubMed] [Google Scholar]
- 121.Noda T, Shimizu W, Tanaka K, Chayama K. Prominent J wave and ST segment elevation: serial electrocardiographic changes in accidental hypothermia. J Cardiovasc Electrophysiol. 2003;14:223. doi: 10.1046/j.1540-8167.2003.02384.x. [DOI] [PubMed] [Google Scholar]
- 122.Tarin N, Farre J, Rubio JM, Tunon J, Castro-Dorticos J. Brugada-like electrocardiographic pattern in a patient with a mediastinal tumor. PACE. 1999;22:1264–6. doi: 10.1111/j.1540-8159.1999.tb00613.x. [DOI] [PubMed] [Google Scholar]
- 123.Tomcsanyi J, Simor T, Papp L. Images in cardiology. Haemopericardium and Brugada-like ECG pattern in rheumatoid arthritis. Heart. 2002;87:234. doi: 10.1136/heart.87.3.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Krishnan SC, Josephson ME. ST segment elevation induced by class IC antiarrhythmic agents: underlying electrophysiologic mechanisms and insights into drug-induced proarrhythmia. J Cardiovasc Electrophysiol. 1998;9:1167–72. doi: 10.1111/j.1540-8167.1998.tb00088.x. [DOI] [PubMed] [Google Scholar]
- 125.Fujiki A, Usui M, Nagasawa H, Mizumaki K, Hayashi H, Inoue H. ST segment elevation in the right precordial leads induced with class IC antiarrhythmic drugs: insight into the mechanism of Brugada syndrome. J Cardiovasc Electrophysiol. 1999;10:214–8. doi: 10.1111/j.1540-8167.1999.tb00662.x. [DOI] [PubMed] [Google Scholar]
- 126.Gasparini M, Priori SG, Mantica M, et al. Flecainide test in Brugada syndrome: a reproducible but risky tool. PACE. 2003;26:338–41. doi: 10.1046/j.1460-9592.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- 127.Takenaka S, Emori T, Koyama S, Morita H, Fukushima K, Ohe T. Asymptomatic form of Brugada syndrome. PACE. 1999;22:1261–3. doi: 10.1111/j.1540-8159.1999.tb00612.x. [DOI] [PubMed] [Google Scholar]
- 128.Matana A, Goldner V, Stanic K, Mavric Z, Zaputovic L, Matana Z. Unmasking effect of propafenone on the concealed form of the Brugada phenomenon. PACE. 2000;23:416–8. doi: 10.1111/j.1540-8159.2000.tb06774.x. [DOI] [PubMed] [Google Scholar]
- 129.Rolf S, Bruns HJ, Wichter T, et al. The ajmaline challenge in Brugada syndrome: diagnostic impact, safety, and recommended protocol. Eur Heart J. 2003;24:1104–12. doi: 10.1016/s0195-668x(03)00195-7. [DOI] [PubMed] [Google Scholar]
- 130.Tada H, Nogami A, Shimizu W, et al. ST segment and T wave alternans in a patient with Brugada syndrome. PACE. 2000;23:413–5. doi: 10.1111/j.1540-8159.2000.tb06773.x. [DOI] [PubMed] [Google Scholar]
- 131.Matsuo K, Shimizu W, Kurita T, Inagaki M, Aihara N, Kamakura S. Dynamic changes of 12-lead electrocardiograms in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 1998;9:508–12. doi: 10.1111/j.1540-8167.1998.tb01843.x. [DOI] [PubMed] [Google Scholar]
- 132.Bolognesi R, Tsialtas D, Vasini P, Conti M, Manca C. Abnormal ventricular repolarization mimicking myocardial infarction after heterocyclic antidepressant overdose. Am J Cardiol. 1997;79:242–5. doi: 10.1016/s0002-9149(96)00727-8. [DOI] [PubMed] [Google Scholar]
- 133.Rouleau F, Asfar P, Boulet S, et al. Transient ST segment elevation in right precordial leads induced by psychotropic drugs: relationship to the Brugada syndrome. J Cardiovasc Electrophysiol. 2001;12:61–5. doi: 10.1046/j.1540-8167.2001.00061.x. [DOI] [PubMed] [Google Scholar]
- 134.Antzelevitch C. The Brugada syndrome. J Cardiovasc Electrophysiol. 1998;9:513–6. doi: 10.1111/j.1540-8167.1998.tb01844.x. [DOI] [PubMed] [Google Scholar]
- 135.Pastor A, Nunez A, Cantale C, Cosio FG. Asymptomatic Brugada syndrome case unmasked during dimenhydrinate infusion. J Cardiovasc Electrophysiol. 2001;12:1192–4. doi: 10.1046/j.1540-8167.2001.01192.x. [DOI] [PubMed] [Google Scholar]
- 136.Littmann L, Monroe MH, Svenson RH. Brugada-type electrocardiographic pattern induced by cocaine. Mayo Clin Proc. 2000;75:845–9. doi: 10.4065/75.8.845. [DOI] [PubMed] [Google Scholar]
- 137.Vatta M, Dumaine R, Antzelevitch C, et al. Novel mutations in domain I of SCN5A cause Brugada syndrome. Mol Genet Metab. 2002;75:317–24. doi: 10.1016/S1096-7192(02)00006-9. [DOI] [PubMed] [Google Scholar]
- 138.Wan X, Chen S, Sadeghpour A, Wang Q, Kirsch GE. Accelerated inactivation in a mutant Na(+) channel associated with idiopathic ventricular fibrillation. Am J Physiol Heart Circ Physiol. 2001;280:H354–H360. doi: 10.1152/ajpheart.2001.280.1.H354. [DOI] [PubMed] [Google Scholar]
- 139.Mok NS, Priori SG, Napolitano C, Chan NY, Chahine M, Baroudi G. A newly characterized SCN5A mutation underlying Brugada syndrome unmasked by hyperthermia. J Cardiovasc Electrophysiol. 2003;14:407–11. doi: 10.1046/j.1540-8167.2003.02379.x. [DOI] [PubMed] [Google Scholar]
- 140.Potet F, Mabo P, Le Coq G, et al. Novel brugada SCN5A mutation leading to ST segment elevation in the inferior or the right precordial leads. J Cardiovasc Electrophysiol. 2003;14:200–3. doi: 10.1046/j.1540-8167.2003.02382.x. [DOI] [PubMed] [Google Scholar]
- 141.Baroudi G, Acharfi S, Larouche C, Chahine M. Expression and intracellular localization of an SCN5A double mutant R1232W/T1620M implicated in Brugada Syndrome. Circ Res. 2002;90:E11–E16. [PubMed] [Google Scholar]
- 142.Wan X, Wang Q, Kirsch GE. Functional suppression of sodium channels by beta(1)-subunits as a molecular mechanism of idiopathic ventricular fibrillation. J Mol Cell Cardiol. 2000;32:1873–84. doi: 10.1006/jmcc.2000.1223. [DOI] [PubMed] [Google Scholar]
- 143.Kyndt F, Probst V, Potet F, et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation. 2001;104:3081–6. doi: 10.1161/hc5001.100834. [DOI] [PubMed] [Google Scholar]
- 144.Deschenes I, Baroudi G, Berthet M, et al. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc Res. 2000;46:55–65. doi: 10.1016/s0008-6363(00)00006-7. [DOI] [PubMed] [Google Scholar]
- 145.Baroudi G, Pouliot V, Denjoy I, Guicheney P, Shrier A, Chahine M. Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G) Circ Res. 2001;88:E78–E83. doi: 10.1161/hh1201.093270. [DOI] [PubMed] [Google Scholar]
- 146.Rook MB, Alshinawi CB, Groenewegen WA, et al. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc Res. 1999;44:507–17. doi: 10.1016/s0008-6363(99)00350-8. [DOI] [PubMed] [Google Scholar]
- 147.Shirai N, Makita N, Sasaki K, et al. A mutant cardiac sodium channel with multiple biophysical defects associated with overlapping clinical features of Brugada syndrome and cardiac conduction disease. Cardiovasc Res. 2002;53:348–54. doi: 10.1016/s0008-6363(01)00494-1. [DOI] [PubMed] [Google Scholar]
- 148.Baroudi G, Carbonneau E, Pouliot V, Chahine M. SCN5A mutation (T1620M) causing Brugada syndrome exhibits different phenotypes when expressed in Xenopus oocytes and mammalian cells. FEBS Lett. 2000;467:12–6. doi: 10.1016/s0014-5793(00)01099-1. [DOI] [PubMed] [Google Scholar]
- 149.Wang DW, Makita N, Kitabatake A, Balser JR, George AL., Jr. Enhanced Na(+) channel intermediate inactivation in Brugada syndrome. Circ Res. 2000;87:E37–E43. doi: 10.1161/01.res.87.8.e37. [DOI] [PubMed] [Google Scholar]
- 150.Kiehn J, Karle C, Thomas D, Yao X, Brachmann J, Kubler W. HERG potassium channel activation is shifted by phorbol esters via protein kinase A-dependent pathways. J Biol Chem. 1998;273:25285–91. doi: 10.1074/jbc.273.39.25285. [DOI] [PubMed] [Google Scholar]
- 151.Akai J, Makita N, Sakurada H, et al. A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 2000;479:29–34. doi: 10.1016/s0014-5793(00)01875-5. [DOI] [PubMed] [Google Scholar]
- 152.Veldkamp MW, Viswanathan PC, Bezzina C, Baartscheer A, Wilde AA, Balser JR. Two distinct congenital arrhythmias evoked by a multidysfunctional Na(+) channel. Circ Res. 2000;86:E91–E97. doi: 10.1161/01.res.86.9.e91. [DOI] [PubMed] [Google Scholar]
- 153.Viswanathan PC, Bezzina CR, George AL, Jr, Roden DM, Wilde AA, Balser JR. Gating-dependent mechanisms for flecainide action in SCN5A-linked arrhythmia syndromes. Circulation. 2001;104:1200–5. doi: 10.1161/hc3501.093797. [DOI] [PubMed] [Google Scholar]
- 154.Baroudi G, Chahine M. Biophysical phenotypes of SCN5A mutations causing long QT and Brugada syndromes. FEBS Lett. 2000;487:224–8. doi: 10.1016/s0014-5793(00)02360-7. [DOI] [PubMed] [Google Scholar]
- 155.Rivolta I, Abriel H, Tateyama M, et al. Inherited Brugada and LQT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. J Biol Chem. 2001:30623–30. doi: 10.1074/jbc.M104471200. [DOI] [PubMed] [Google Scholar]
- 156.Tan HL, Kupershmidt S, Zhang R, et al. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442–7. doi: 10.1038/415442a. [DOI] [PubMed] [Google Scholar]
- 157.Tan HL. Biophysical analysis of mutant sodium channels in Brugada syndrome. In: Antzelevitch C, Brugada P, editors. Brugada Syndrome. From Bench to Bedside. Blackwell Futura; Malden, MA: 2005. pp. 26–4. [Google Scholar]
- 158.Nademanee K, Veerakul G, Schwab M. Predisposing factors in the Brugada syndrome. In: Antzelevitch C, Brugada P, editors. Brugada Syndrome. From Bench to Bedside. Blackwell Futura; Malden, MA: 2005. pp. 157–65. [Google Scholar]
- 159.Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Riera ARP, Tan H, Shimizu W, SchultzeBahr E, Wilde A. Brugada syndrome: overview. In: Antzelevitch C, editor. The Brugada Syndrome. From Bench to Bedside. Blackwell Futura; Malden, MA: 2004. pp. l–22. [Google Scholar]