

Abstract
The cannabinoid CB1 receptor has been shown to be critically involved in the extinction of fear memory. Systemic injection of a CB1 receptor antagonist prior to extinction training blocked extinction. Conversely, administration of the cannabinoid uptake inhibitor AM404 facilitated extinction in a dose-dependent manner. Here we show that bilateral infusion of CB1 receptor agonists into the amygdala after memory reactivation blocked reconsolidation of fear memory measured with fear-potentiated startle. The effect was dose-dependent and could be blocked by AM251, a specific CB1 receptor antagonist. In contrast, the effect of CB1 agonists on reconsolidation was no longer seen if memory reactivation was omitted. Concomitant with block of reconsolidation, CB1 agonist-treated animals did not exhibit shock-induced reinstatement or spontaneous recovery of fear. The absence of recovery was not attributable to permanent damage to the amygdala in WIN-treated rats, nor did the effect result from alteration of baseline startle or shock reactivity. These results suggest that CB1 agonists could impair fear memory via blocking reconsolidation.
Synthetic and endogenous cannabinoids have profound effects on the central neurons. They inhibited pain (Pertwee 2001) and reduced neuronal damage in models of ischemia and traumatic brain injury (Panikashvili et al. 2001). They impaired memory in animals, particularly in hippocampus-dependent tasks such as an eight-arm radial maze, spatial alteration in a T-maze, and delayed matching/non-matching to a position task with lever presentation (Lichtman et al. 1995; Davis et al. 2002). On the other hand, SR141716A, a specific antagonist of the cannabinoid CB1 receptor, blocked the disruptive effects of cannabinoids on rate and accuracy of responding (Brodkin and Moerschbaecher 1997). Cannabinoids produce marked alterations in behavior and mood in animals and humans. Administration of a CB1 antagonist elicited an anxiety-like response (Navarro et al. 1997), whereas active inhibitors of fatty acid amide hydrolase (FAAH), which catalyzes endogenous cannabinoid anadamide hydrolysis, induced anxiolytic effects in rats (Kathuria et al. 2003).
Pavlovian fear conditioning is a behavioral procedure in which a cue (conditioned stimulus, CS) comes to induce a fear response when it is repeatedly paired with a noxious stimulus, often a foot-shock (unconditioned stimulus, US). Fear conditioning is not only a sensitive measure of anticipatory fear or anxiety but is also a leading behavioral paradigm for studying the neural mechanisms through which emotional memory is formed and stored (Davis 2000; LeDoux 2000). Extinction, on the other hand, refers to gradual disappearance of the previously acquired responses if animals are exposed only to the cue without pairing with a shock (Rescorla 2001; Myers and Davis 2002). Recently, endocannabinoids were demonstrated to be critically involved in the extinction of fear memory because mutant mice lacking CB1 receptors were specifically impaired in extinction (Marsicano et al. 2002).
Many observations in animal studies, including spontaneous recovery with time (Bouton and Peck 1989), reinstatement after unpaired US presentations (Rescorla and Heth 1975), and renewal with context change (Bouton and King 1983), indicate that extinction is a new inhibitory learning, which leaves the original memory intact (Quirk et al. 2000; Herry and Garcia 2002; Myers and Davis 2002; Maren and Quirk 2004). It has been shown that treatment of rats with an inhibitor of cannabinoid reuptake, AM404, enhanced extinction (Chhatwal et al. 2005). However, animals that had received AM404 during extinction training exhibited less reinstatement effect. It is possible that extinction seen following AM404 treatment was more robust and less susceptible to subsequent US reinstatement. Alternatively, it may suggest the possibility of additional mechanisms. Following retrieval, memory became labile for a period before being reconsolidated and re-stored. Thus, in theory, memory would not return after a block of reconsolidation (Duvarci and Nader 2004). Extinction training usually consisted of CS-alone trials that induced memory retrieval. Therefore, it is reasonable to speculate that CB1 receptor agonists may act on the reconsolidation of fear memory.
Results
On day 1, rats were conditioned with 10 light–shock pairings. On day 2, they were infused with vehicle or a CB1 receptor agonist, WIN55212–2 (WIN, 1 or 11 μg per side), bilaterally into the amygdala within 1 h after a retention test (Test 1). Memory was assessed 24 h after Test 1 (Test 2). Figure 1A shows that infusion of WIN resulted in an impairment of fear memory. Startle potentiations were 171.4% ± 8.3% (n = 6) in vehicle controls, 99.0% ± 13.6% (1 μg per side, n = 5), and 46.0% ± 7.7% (11 μg per side, n = 10) in WIN-treated animals. The ANOVA for startle scores showed a significant effect for group (F(2,18) = 48.17, P < 0.001), and post hoc t-tests showed that the two WIN groups differed from the vehicle group (P < 0.001). Furthermore, less startle reflex occurred in the high-dose group than in the low-dose group (P < 0.01), indicating a dose-dependent effect. The infusion cannula tip locations are shown in Figure 1B. Only rats with cannula tips at or within the boundaries of LA and BLA were included in the data analysis.
Figure 1.

CB1 receptor agonists block reconsolidation of fear memory. (A) Rats were infused with vehicle (n = 6), 1 μg of WIN (n = 5), or 11 μg of WIN (n = 10) within 1 h after the test, and memory retention was assessed 24 h later. ***P < 0.001 vs. vehicle. (B) Cannula tip placements from rats infused with vehicle (●), 1 μg of WIN (▴), or 11 μg of WIN (★) in the experiments shown in A. (C) Dose–response relationship of HU210 on reconsolidation. *P < 0.05, ***P < 0.001 vs. vehicle. (D) Cannula tip placements from rats infused with vehicle (●), 1 μg of HU210 (▴), or 10 μg of HU210 (★) in the experiments shown in C.
A similar result was obtained with another CB1 agonist, HU210. Post-test infusion of HU210 significantly attenuated startle reflex. Startle potentiations were 170.5% ± 14.1% (n = 6) in vehicle controls, 106.4% ± 19.9% (1 μg per side, n = 5, P < 0.05 vs. vehicle), and 61.1% ± 15.2% (10 μg per side, n = 6, P < 0.001 vs. vehicle) in HU210-treated animals (Fig. 1C). Cannula tip placements are shown in Figure 1D.
AM251 is a selective CB1 antagonist. To ensure that the memory-impairing effect of WIN was mediated by the CB1 receptor, we determined whether AM251 could reverse the effects of WIN and HU210. AM251 (20 μg per side) and WIN (11 μg per side) were sequentially infused into the amygdala with an interval of 20–25 min. As shown in Figure 2, AM251 blocked the effects of WIN and HU210 (10 μg per side) such that there was no difference in the amount of startle amplitude between the vehicle and WIN/AM251 groups (t(12) = 0.18, P = 0.86) and between the vehicle and HU210/AM251 groups (t(7) = 0.68, P = 0.52). As a control, vehicle and AM251 also were sequentially infused into the amygdala to investigate the effect of AM251 on reconsolidation. The result showed that there was no difference between the vehicle and veh/AM251 groups (t(8) = 0.32, P = 0.75), suggesting that AM251 by itself did not affect reconsolidation and that concentrations of endocannabinoids were below threshold during the retention test to activate CB1 receptors.
Figure 2.

Block of the effect of CB1 agonists on reconsolidation by AM251. AM251 (20 μg per side) was administered 20–25 min before WIN (11 μg per side) or HU210 (10 μg per side). There was no difference in the amount of startle amplitude between the vehicle and WIN/AM251 groups (t(12) = 0.18, P = 0.86) and between the vehicle and HU210/AM251 groups (t(7) = 0.68, P = 0.52). AM251 and vehicle were also infused into the amygdala, and there was no difference between the vehicle and veh/AM251 groups (t(8) = 0.32, P = 0.75).
We repeated the experiments to determine the effects of WIN on post-reactivation of short-term memory (PR-STM) at 4 h and long-term memory (PR-LTM) at 24 h after Test 1. An ANOVA comparing the drug group across trials (PR-STM and PR-LTM) demonstrated a significant interaction (F(3,20) = 11.94, P < 0.001). Newman-Keuls post hoc analysis revealed that the WIN group was significantly different from the vehicle group both in the PR-STM (P < 0.05) and PR-LTM (P < 0.001) (Fig. 3). Taken together, these results indicate that CB1 receptor agonists impair fear memory when given shortly after memory reactivation.
Figure 3.

Effects of post–Test 1 infusion of WIN on STM and LTM. Rats were infused with vehicle or 11 μg of WIN within 1 h after the test, and STM was assessed at 4 h and followed by LTM at 24 h after administration of WIN. *P < 0.05, ***P < 0.001 vs. vehicle. Cannula tip placements from rats infused with vehicle (○) or WIN (●).
To determine whether the observed impairment of fear memory required memory reactivation, we omitted Test 1. Conditioned rats were infused with WIN, HU210, or vehicle in the absence of Test 1. Memory retention was assessed 24 h after drug application. Figure 4 shows that neither WIN (11 μg per side) nor HU210 (10 μg per side) had an effect on the startle reflex. Furthermore, WIN still failed to induce extinction even though the dose was increased to 33 μg per side. These results suggest that the effects of WIN and HU210 require memory reactivation as demonstrated by the lack of amnesia when Test 1 is omitted.
Figure 4.

Requirement of memory retrieval for the action of CB1 agonists. (A) There was no difference in startle reflex between vehicle- and WIN- or HU210-treated rats when Test 1 was omitted. (B) Cannula tip placements from rats infused with vehicle (▴), 11 μg WIN (◽), 33 μg WIN (◼), or HU210 (★).
To examine the possibility that WIN might damage the amygdala neurons, we performed a histological analysis. Figure 5A shows that there was no evidence of increased gliosis or cell loss in vehicle- or WIN-treated rats. We further determined whether WIN induced cell apoptosis by staining neurons with Hoechst 33,342. WIN or vehicle was infused into the amygdala, and 24 h later apoptotic features including dense chromatin condensation and nuclear pyknosis were examined with a fluorescence microscope. There was no difference in abnormal nuclei-positive cells between vehicle- and WIN-treated animals (Fig. 5B).
Figure 5.

WIN55212–2 did not lesion the amygdala. (A) Representative photomicrographs show amygdala slices from rats infused with DMSO (left) or WIN (right). There was no evidence of increased cell loss or gliosis in the amygdala in the DMSO or WIN-treated animals. Bar, 0.5 mm. (B) WIN (11 μg/side) or vehicle were infused into the amygdala, and 24 h later morphological studies were conducted by Hoechst 33,342 staining. Bar, 10 μm.
We assessed whether WIN-treated rats exhibited reinstatement of fear memory. Rats were trained according to our previous reconsolidation paradigm and then tested for memory recovery by application of a reminder shock (Fig. 6A). Vehicle control rats were divided into two groups with or without exposing to CS-alone trials that led to extinction. An ANOVA on Test 1, PR-LTM, and reinstatement showed a significant interaction with drug treatment (F(5,33) = 24.12, P < 0.0001). Post hoc comparisons revealed that Test 1 scores were the same for the vehicle and WIN groups (P > 0.05). However, WIN rats demonstrated less startle reflex than controls on both PR-LTM (P < 0.001) and reinstatement (P < 0.001). In contrast, subsequent exposure of vehicle extinction rats to 10 foot-shocks reinstated the startle. Furthermore, there was no increase in the startle amplitude of WIN-treated animals after the reminder shock (t(6) = 1.21, P = 0.27). To rule out the possibility that the lack of recovery was attributable to WIN-induced damage to the amygdala, five out of seven WIN-treated rats were retrained. Figure 6B shows that startle reflex in all five WIN-treated rats was significantly increased to levels (183.1 ± 13.9, t(4) = 5.98, P < 0.005 vs. reinstatement) comparable with control animals on Test 1. This result suggests that the lack of reinstatement is not attributable to the inability of animals to learn.
Figure 6.

Retardation of reinstatement of fear memory by CB1 receptor agonists. (A) Behavioral procedure used for the experiments shown in B. (B) WIN (11 μg per side) or HU210 (10 μg per side) were infused into the amygdala bilaterally within 1 h after Test 1, which blocked reconsolidation. Amnesia resulting from CB1 agonist infusions did not show reinstatement with unconditioned foot-shocks. After retraining, the levels of startle potentiation in the WIN or HU210 rats were comparable with their Test 1. Vehicle extinction animals were trained and then exposed to three sessions of 10 CS-alone trials that led to extinction. Subsequent exposure of these rats to 10 foot-shocks reinstated the startle. ***P < 0.001 vs. vehicle. (C) Cannula tip placements from rats infused with vehicle (●), vehicle extinction (○), WIN (▴), or HU210 (◼) in the experiments shown in B.
Similar experiments were performed with HU210 (10 μg per side). ANOVA analysis on Test 1, PR-LTM, and reinstatement showed a significant interaction with drug treatment (F(5,27) = 14.14, P < 0.0001). Post hoc comparisons revealed that Test 1 scores were the same for both groups (P > 0.05), whereas the HU210 rats demonstrated less startle reflex than controls on both PR-LTM (P < 0.001) and reinstatement (P < 0.001). In addition, there was no increase in the startle amplitude of HU210-treated animals after a reminder shock (t(4) = 0.57, P = 0.60). 5 d later, these HU210-treated rats were retrained and, as shown in Figure 6B, the level of startle potentiation was increased to 151.7% ± 18.3% (t(4) = 5.50, P < 0.01 vs. reinstatement).
We examined whether the memory would recover spontaneously from reactivation amnesia in WIN-treated rats. Animals were trained according to our previous reconsolidation paradigm and then tested for memory recovery 7 d after training. To match the levels of startle in the WIN group, vehicle control rats were given 30 trials of CS-alone extinction training ∼30 min after Test 1. As shown in Figure 7B, testing animals 7 d after training revealed a recovery of startle in vehicle controls. In contrast, the conditioned responses of the WIN (11 μg per side) and HU210 (10 μg per side) groups were significantly less than vehicle controls 7 d after training (WIN: t(10) = 2.40, P < 0.05; HU210: t(10) = 2.95, P < 0.02), indicating an inhibition of spontaneous recovery by CB1 agonists.
Figure 7.

Retardation of spontaneous recovery by CB1 receptor agonists. (A) Behavioral procedure used in the experiment shown in B. (B) Animals were trained and then tested the next day. WIN (11 μg per side) or HU210 (10 μg per side) were infused into the amygdala bilaterally within 1 h after the test. Recovery of memory was assessed 7 d after training. Vehicle control rats were given extinction training to match the levels of startle in WIN group. ***P < 0.001 vs. vehicle. (C) Cannula tip placements from rats infused with vehicle (●), WIN (▴), or HU210 (◼) in the experiments shown in B.
We assessed whether WIN produced an analgesic effect and affected baseline anxiety by measuring the shock reactivity and baseline startle, respectively, according to the methods described by Chhatwal et al. (2005). A separate group of conditioned rats was given an intra-amygdala infusion of WIN (n = 5) and 30 min later was presented with three shocks and 42 startle stimuli identical to those used in the above studies (0.6-mA, 0.5-sec foot-shocks, 95-dB startle stimuli). 3 d later, the same rats were returned to the startle box, injected with vehicle, and similarly tested. Figure 8 shows that there was no difference in shock sensitivity (P = 0.32) or baseline startle amplitude (P = 0.67) in rats given WIN or vehicle. Thus, intra-amygdala administration of WIN has no effect on pain sensitivity or baseline startle amplitude.
Figure 8.
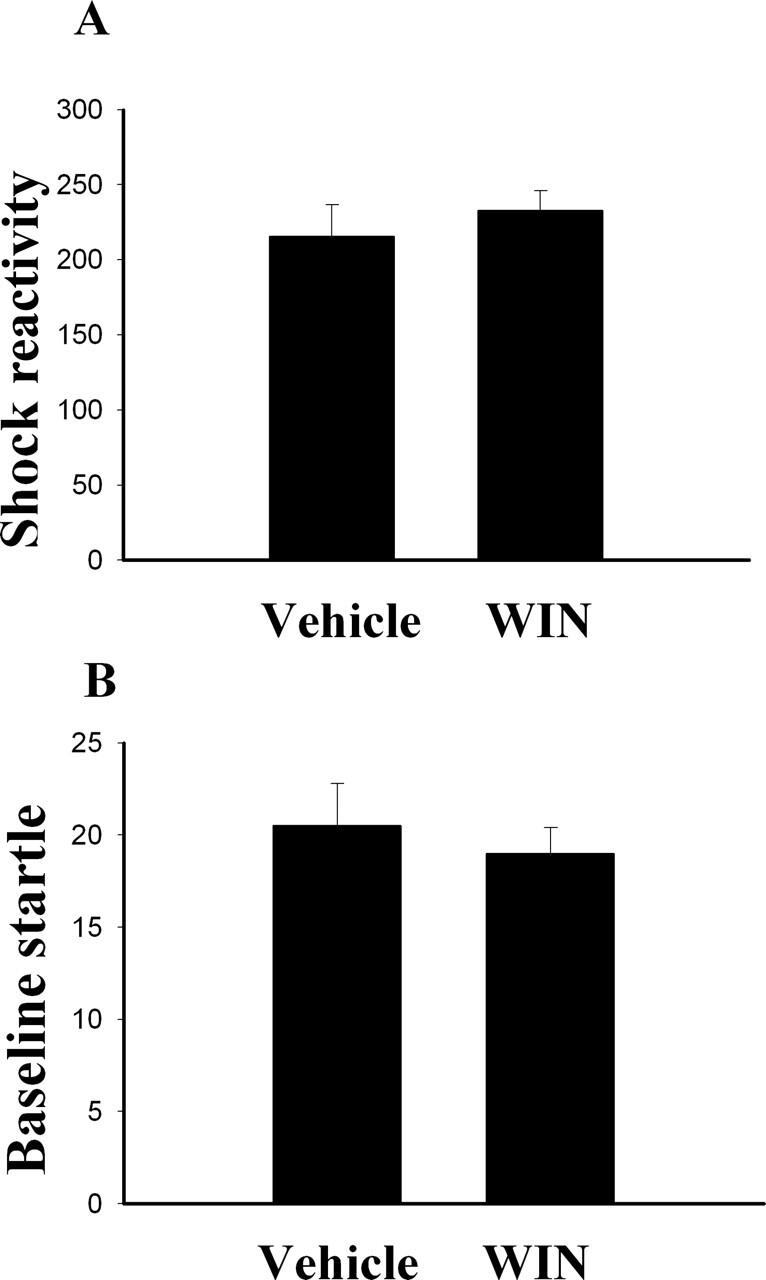
Effect of WIN on shock reactivity and baseline anxiety. Conditioned rats received an intra-amygdala infusion of WIN (11 μg/side, n = 5) and 30 min later were presented with three shocks and 42 startle stimuli (0.6-mA, 0.5-sec shocks, 95-dB noise-burst startle). 3 d later, the same rats were returned to the startle box, injected with vehicle, and similarly tested. (A) Shock reactivity represents the average response to three foot-shocks. (B) Baseline startle amplitude represents the average response to 42 startle stimuli.
Discussion
In the present study, we have shown that post-test infusion of WIN or HU210 into the amygdala significantly impaired fear memory in a dose-dependent manner. The effects of WIN or HU210 could be reversed by the selective CB1 receptor antagonist and were no longer seen if the test was omitted. Re-exposing WIN-treated rats to the US failed to reinstate learned fear. In addition, the WIN-treated rats did not show spontaneous recovery. Finally, administration of CB1 agonists at the dose used in this study did not damage the amygdala neurons, induce apoptosis, or produce an obvious analgesic effect. Taken together, these results suggest that intra-amygdala infusion of CB1 receptor agonists could impair fear memory via an effect on reconsolidation.
Memory testing caused memory reactivation and initiated two potentially dissociable but opposite processes: reconsolidation and extinction (Nader et al. 2000; Myers and Davis 2002; Nader 2003; Suzuki et al. 2004). We have demonstrated that activation of the CB1 receptor in the amygdala impaired fear memory when CB1 agonists were administered immediately after test, but were not effective when administered without a test. In addition, no evidence of reinstatement and spontaneous recovery was found in WIN-treated animals. Based on the notion that original memory became labile and would not return after a specific block of reconsolidation (Duvarci and Nader 2004), reactivation-induced amnesia by CB1 agonists could be attributable to the block of reconsolidation. Extinction of conditioned fear in general was considered to be an inhibitory learning that prevented the expression of intact association rather than erasing it. If a memory deficit induced by CB1 agonists after memory reactivation was attributable to enhanced extinction, then re-exposing animals to the US prior to the test would restore its representation and reinstate the learned responses. In addition, testing animals at different time points after extinction should reveal a recovery of retention. A previous study by Chhatwal et al. (2005) has shown that systemic injection of a CB1 receptor antagonist prior to extinction training blocked extinction. Conversely, administration of the cannabinoid uptake inhibitor AM404 facilitated extinction in a dose-dependent manner. The difference between their results and ours is not clear, but could be due to different training protocols applied (extinction vs. memory testing) or the route of drug administration (systemic vs. intra-amygdala administration). Activation of CB1 receptors could facilitate extinction on one hand and block reconsolidation on the other.
Reinstatement and spontaneous recovery are signs of preservation of the original memory after extinction training. Theoretically, they could be used to judge whether a manipulation facilitates extinction as opposed to blocking reconsolidation. However, it should be cautioned that under certain circumstances if extinguishment of memory was caused by the erasure of original memory, then reinstatement and spontaneous recovery are not valid to differentiate between the facilitation of extinction and blocking of reconsolidation.
It is noted that intra-amygdala injection of a CB1 agonist immediately after the test impaired both PR-STM and PR-LTM, suggesting that CB1 agonists block a fast cascade of events necessary for memory reconsolidation. It has been shown that PKA phosphorylation of S845 in GluR1 increased the peak open probability (Banke et al. 2000) of AMPA receptors as well as the surface reinsertion of GluR1 (Ehlers 2000). Furthermore, fear memory formation required the coupling of GluR1 and PKA by A-kinase anchoring proteins (AKAPs) through synapse-associated protein 97 kDa (SAP97) in the lateral amygdala (Moita et al. 2002). Thus, it is likely that activation of CB1 receptors negatively regulates adenylyl cyclase (Howlett et al. 1986; Bidaut-Russell et al. 1990), PKA, and phosphorylation of AMPA receptors, resulting in the retardation of formation and maintenance of STM. In this context, it has been shown recently that, using a low-intensity training protocol (1.3-mA US foot-shock), activation of PKA in the amygdala enhanced reconsolidation. In contrast, inhibition of PKA impaired reconsolidation when a high-intensity training protocol (2.0-mA US foot-shock) was applied (Tronson et al. 2006).
In summary, retrieval of memory would put it into a new vulnerable phase so that a reconsolidation blockade could lead to erasure of memory, not by inhibiting the expression of memory as extinction training did. Here, we have demonstrated that activation of CB1 receptors blocked reconsolidation, and rats given CB1 agonists immediately after a memory test failed to exhibit reinstatement and spontaneous recovery. Thus, CB1 agonists could be useful for the treatment of patients with post-traumatic stress disorders (PTSD) because the drug-treated patients may be less likely to relapse after a stressful experience.
Materials and Methods
Surgery
Rats anesthetized with sodium pentobarbital (50 mg/kg, i.p.) were mounted on a stereotaxic apparatus, and two cannulae made of 22-gauge stainless steel tubing were implanted bilaterally into the LA or BLA. The coordinates were AP −2.3 mm, ML ±4.5 mm, DV −7.0 mm according to Paxinos and Watson (1986). Only rats with cannula tips within the boundaries of LA and BLA were included in the data analysis. Rats were monitored and handled daily and were given 7 d to recover. WIN55212–2, HU210, and AM251 were obtained from Tocris Cookson Ltd. The drugs were dissolved in DMSO (50%) and administered bilaterally in a volume of 1 μL at a rate of 0.1 μL/min.
Behavioral apparatus and procedures
Rats were trained and tested in a stabilimeter device. A piezoelectric device mounted below the stabilimeter detects and transduces the motion of the cylinder produced by the whole body startle response of the rat (San Diego Instrument). The whole set-up was enclosed in a ventilated, sound-attenuating cabinet (length 38 cm, width 38 cm, height 55 cm). The acoustic startle stimulus was a 50-ms white noise at the intensity of 95 dB. The visual CS was a 3.7-sec light produced by an 8W fluorescent bulb attached to the back of the stabilimeter. The US was a 0.6-mA foot-shock with a duration of 0.5 sec.
Acclimation
On three consecutive days, rats were placed in the startle test boxes for 10 min and returned to their home cages.
Matching
On two consecutive days, rats were placed in the startle box and 3 min later presented with 10 startle stimuli at 2-min intertrial intervals (ITI). On the basis of their mean startle amplitudes in the second of these two sessions, rats were matched into groups with similar response levels.
Training
Rats were placed in the startle boxes and received 10 light–foot-shock pairings with an ITI of 2 min.
Test
24 h after training, rats were tested for fear-potentiated startle. This involved 10 startle-eliciting noise bursts presented alone (noise-alone trial) and 10 noise bursts presented 3.2 sec after onset of the 3.7-sec light (light–noise trials). The two trial types were presented in a balanced mixed order (ITI, 30 sec). The percentage of fear-potentiated startle was computed as follows: [(startle amplitude on CS-noise minus noise-alone trials) / (noise-alone trials)] × 100.
Reconsolidation
Rats were trained and memory was tested 24 h later (Test 1). Rats were infused with WIN55212–2, HU210, or vehicle within 1 h after termination of Test 1. A post-reactivation short-term memory (PR-STM) test was performed 4 h later, followed by a PR-LTM test 24 h after Test 1.
Reinstatement
Animals were trained according to the reconsolidation paradigm, returned to the testing chamber 24 h later, and presented with 10 foot-shocks. Animals underwent a test for memory reinstatement 24 h after foot-shock. 5 d later, rats were retrained with 10 light–foot-shock pairings, and the following day they were tested for the LTM of the retrained memory. A group of vehicle control rats was exposed to 30 trials of CS-alone extinction training to match the degree of startle reflex in WIN-treated animals.
Shock reactivity and baseline startle measurement
A group of conditioned rats was injected with WIN bilaterally into the amygdala, placed in the training box, and presented with three unpaired foot-shocks and 42 startle stimuli (0.6-mA, 0.5-sec shocks, 95-dB noise-burst startle). The same group of rats was returned to the same startle box 3 d later, injected with vehicle, and presented with identical foot-shocks and startle stimuli.
Histology
At the end of experiments, animals received an overdose of pentobarbital (100mg/kg), and the brains were removed from the skull and fixed in buffered 4% paraformaldehyde (pH 7.4) for 48 h. Brains were sectioned with a sliding MicroSlicer (DTK-1000, Ted Pella Inc.), and sections (40-μm thickness) were stained for Nissl bodies and DNA dye Hoechst 33,342 (bis-benzimide, Sigma). Nuclei were visualized using a fluorescence microscope.
Data analysis
Data were analyzed with ANOVA. A single-factor ANOVA and post hoc comparisons were used to analyze the dose-dependent effect of WIN55212–2 in blocking reconsolidation and the difference between the effect of drugs on STM and LTM. An unpaired t-test was used to analyze differences of startle reflex between the drug-treated and vehicle control groups. A paired t-test was used to analyze the difference in startle amplitude before and after a reminder shock in drug-treated rats (reinstatement experiments). All values in the text and figure legends are mean ±SEM.
Acknowledgments
This study was supported by the National Health Research Institutes (NHRI-EX92-9202NI) and the National Science Council (NSC94-2752-B-006-001-PAE).
Footnotes
Article published online before print. Article and publication date are at http://www.learnmem.org/cgi/doi/10.1101/lm.217006
References
- Banke T.G., Bowie D., Lee H.K., Huganir R.L., Schousboe A., Traynelis S.F. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidaut-Russell M., Devane W.A., Howlett A.C. Cannabinoid receptors and regulation of cyclic AMP accumulation in the rat brain. J. Neurochem. 1990;55:21–26. doi: 10.1111/j.1471-4159.1990.tb08815.x. [DOI] [PubMed] [Google Scholar]
- Bouton M.E., King D.A. Contextual control of the extinction of conditioned fear: Tests for the associative value of the context. J. Exp. Psychol. Anim. Behav. Process. 1983;9:248–265. [PubMed] [Google Scholar]
- Bouton M.E., Peck C.A. Spontaneous recovery in cross-motivational transfer (counterconditioning). Anim. Learn. Behav. 1989;20:313–321. [Google Scholar]
- Brodkin J., Moerschbaecher J.M. SR141716A antagonizes the disruptive effects of cannabinoid ligands on learning in rats. J. Pharmacol. Exp. Ther. 1997;282:1526–1532. [PubMed] [Google Scholar]
- Chhatwal J., Davis M., Maguschak K.A., Ressler K.J. Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology. 2005;30:516–524. doi: 10.1038/sj.npp.1300655. [DOI] [PubMed] [Google Scholar]
- Davis M.2000. The role of the amygdala in conditioned and unconditioned fear and anxiety. In The amygdala: A functional analysis (ed. J.P. Aggleton) pp. 213–287. Oxford University Press; New York [Google Scholar]
- Davis S.N., Pertwee R.G., Riedel G. Functions of cannabinoid receptors in the hippocampus. Neuropharmacology. 2002;42:993–1007. doi: 10.1016/s0028-3908(02)00060-6. [DOI] [PubMed] [Google Scholar]
- Duvarci S., Nader K. Characterization of fear memory reconsolidation. J. Neurosci. 2004;24:9269–9275. doi: 10.1523/JNEUROSCI.2971-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers M.D. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Herry C., Garcia R. Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J. Neurosci. 2002;22:577–583. doi: 10.1523/JNEUROSCI.22-02-00577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett A.C., Qualy J.M., Khachatrian L.L. Involvement of Gi in the inhibition of adenylyl cyclase by cannabimimetic drugs. Mol. Pharmacol. 1986;29:161–165. [PubMed] [Google Scholar]
- Kathuria S., Gaetani S., Fegley D., Valino F., Duranti A., Tontini A., Mor M., Tarzia F.G., La Rana G., Calignano A., et al. Modulation of anxiety through blockade of anadamide hydrolysis. Nat. Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- LeDoux J.E. Emotion circuits in the brain. Annu. Rev. Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Lichtman A.H., Dimen K.R., Martin B.R. Systemic or intrahippocampal cannabinoid administration impairs spatial memory in rats. Psychopharmacology. 1995;119:282–290. doi: 10.1007/BF02246292. [DOI] [PubMed] [Google Scholar]
- Maren S., Quirk G.J. Neuronal signalling of fear memory. Nat. Rev. Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- Marsicano G., Wotjak C.T., Azad S.C., Bisogno T., Rammes G., Cascio M.G., Hermann H., Tang J., Hofmann C., Zieglgansberger W., et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Moita M.A., Lamprecht R., Nader K., LeDoux J.L. A-kinase anchoring proteins in amygdala are involved in auditory fear memory. Nat. Neurosci. 2002;5:837–838. doi: 10.1038/nn901. [DOI] [PubMed] [Google Scholar]
- Myers K.M., Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- Nader K. Memory traces unbound. Trends Neurosci. 2003;26:65–72. doi: 10.1016/S0166-2236(02)00042-5. [DOI] [PubMed] [Google Scholar]
- Nader K., Schafe G.E., LeDoux J.E. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Navarro M., Hernandez E., Munoz R.M., del Arco I., Villanua M., Carrera M.R.A. Acute administration of the CB1 cannabinoid receptor antagonist SR141716A induces anxiety-like responses in the rat. Neuroreport. 1997;8:491–496. doi: 10.1097/00001756-199701200-00023. [DOI] [PubMed] [Google Scholar]
- Panikashvili D., Simeonidou C., Ben-Shabat S., Hanus L., Breuer A., Mechoulam R., Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- Paxinos G., Watson C. The rat brain in stereotaxic coordinates. Academic Press; New York.: 1986. [DOI] [PubMed] [Google Scholar]
- Pertwee R.G. Cannabinoid receptors and pain. Prog. Neurobiol. 2001;63:569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- Quirk G.J., Russo G.K., Barron J.L., Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J. Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla R.A.2001. Experimental extinction. In Handbook of contemporary learning theories (eds. R.R. Mowrer and S. Klein), pp. 119–154. Erlbaum; Mahwah, NJ [Google Scholar]
- Rescorla R.A., Heth C.D. Reinstatement of fear to an extinguished conditioned stimulus. J. Exp. Psychol. Anim. Behav. Process. 1975;1:88–96. [PubMed] [Google Scholar]
- Suzuki A., Josselyn S.A., Frankland P.W., Masushige S., Silva A.J., Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J. Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson N.C., Wiseman S.L., Olausson P., Taylor J.R. Bidirectional behavioral plasticity of memory reconsolidation depends on amygdalar protein kinase A. Nat. Neurosci. 2006;9:167–169. doi: 10.1038/nn1628. [DOI] [PubMed] [Google Scholar]