

Abstract
Long-term memory formation is regulated by many distinct molecular mechanisms that control gene expression. An emerging model for effecting a stable, coordinated pattern of gene transcription involves epigenetic tagging through modifications of histones or DNA. In this study, we investigated the regulation of histone phosphorylation in the hippocampus by the ERK/MAPK (extracellular signal-regulated kinase/mitogen-activated protein kinase) pathway. We found that activation of ERK/MAPK in vitro significantly increased histone H3 phosphorylation in hippocampal area CA1. Furthermore, we found that contextual fear conditioning in vivo leads to a rapid time-dependent increase in histone H3 phosphorylation in area CA1. This increase paralleled the time course of contextual fear-dependent activation of ERK, and was inhibited in vivo by a latent inhibition paradigm as well as by injection of an N-methyl-d-aspartic acid receptor (NMDA-R) antagonist. Finally, injection of an inhibitor of MEK (MAP kinase/ERK kinase), the unique dual-specificity kinase upstream of ERK, blocked the increase in histone H3 phosphorylation seen after contextual fear conditioning. These results demonstrate that changes in histone phosphorylation in the hippocampus are regulated by ERK/MAPK following a behavioral fear conditioning paradigm.
Long-term memory formation is a complex process involving biochemical signaling cascades that lead to a change in gene expression in neurons. In mammalian associative memory tasks, activation of the ERK/MAPK (extracellular signal-regulated kinase/mitogen-activated protein kinase) cascade in the hippocampus is necessary for consolidation of the memory (Atkins et al. 1998). This is accomplished typically by activation of the NMDA (N-methyl-d-aspartic acid) subtype of glutamate receptors, leading to an increase in intracellular Ca2+ (Fanselow et al. 1994). Ca2+ activates Ca2+-sensitive protein kinase C (PKC) and adenylyl cyclase/protein kinase A (PKA), triggering a series of events that eventually converge upon ERK (Adams and Sweatt 2002). Once activated, ERK translocates into the nucleus to coordinate and effect changes in gene expression (Davis et al. 2000). ERK is known to regulate transcription factors such as CREB (Cre-binding protein) and Elk-1 (Sweatt 2001), which help initiate transcription of memory-associated genes that contain their respective regulatory elements.
There is growing evidence that memory formation also utilizes epigenetic mechanisms that modify the structure of chromatin (Swank and Sweatt 2001; Guan et al. 2002; Alarcon et al. 2004; Korzus et al. 2004; Wood et al. 2005; for review, see Levenson and Sweatt 2005). Epigenetic modifications can serve as enduring changes to the epigenome that help drive stable changes in gene expression (Rakyan et al. 2001), which in turn manifest as a long-lasting change in behavior. Mechanistically, this is accomplished by direct modification of DNA or post-translational modification of histone proteins, including methylation, acetylation, and phosphorylation (Berger 2002). These modifications exert their effects either by physical remodeling of chromatin structure or by further recruitment of signaling complexes that drive or repress transcription. In addition, epigenetic marks themselves constitute a form of cellular memory. Cellular storage and propagation of information is critical in processes such as mitosis and differentiation, during which patterns of gene expression are preserved and transmitted as unique traits of a particular cell (Ehrenhofer-Murray 2004). Although adult neurons are terminally differentiated and no longer divide, recent studies indicate that epigenetic mechanisms may come into play to subserve information storage in the adult nervous system as well (Guan et al. 2002; Alarcon et al. 2004; Korzus et al. 2004; Levenson et al. 2004b; Wood et al. 2005).
Regulation of histone modifications has been observed in neurons with a variety of physiological stimuli. Exposure of animals to light pulses resulted in transient changes in histone phosphorylation in neurons of the suprachiasmatic nucleus (Crosio et al. 2000). Neuronal stimulation by several neurotransmitter signaling pathways induced changes in histone phosphorylation and acetylation, and also resulted in immediate-early gene (IEG) expression (Crosio et al. 2003). Also, boosting histone acetylation levels in the hippocampus was found to enhance long-term potentiation, a physiological correlate of learning (Levenson et al. 2004b), and disruption of the CREB-binding protein CBP, a known transcriptional activator and histone acetyltransferase, was found to impair both memory consolidation and long-term potentiation in the hippocampus (Alarcon et al. 2004; Korzus et al. 2004). All of these studies suggest that chromatin is a highly dynamic structure in the nervous system that is modified in response to various environmental signals.
Histone phosphorylation has been implicated as a target of the ERK/MAPK pathway. The kinases MSK1 and MSK2 (mitogen- and stress-activated protein kinase), which are downstream of ERK, mediate the mitogen- and stress-induced phosphorylation of histone H3 in cell culture systems in vitro (Soloaga et al. 2003). Levels of histone H3 phosphorylation were significantly diminished in knockout cells for MSK1 and MSK2, and this deficiency was partially rescued by transfection of GFP-tagged MSK2 (Soloaga et al. 2003). Oncogene-transformed fibroblasts, in which the Ras-MAPK pathway is constitutively active, show an increase in histone H1 and H3 phosphorylation and have relaxed chromatin structure (Dunn et al. 2005). Also, when activated by ERK, MSK phosphorylates histone H3 and is required for transcription of nuclear orphan receptors in cultured fibroblasts (Darragh et al. 2005). Interestingly, the same phosphorylation site on histone H3 is used as a marker for dividing chromosomes in mitosis (Hendzel et al. 1997), indicating that a single histone modification may have disparate functions within the cell depending on its context (Jenuwein and Allis 2001).
Given the importance of ERK in mammalian associative learning and the importance of histone phosphorylation in regulating gene expression, we investigated whether ERK regulates histone phosphorylation in CA1 hippocampal neurons during long-term memory formation. We report that activation of ERK in hippocampal slices in vitro, using activators of PKA and PKC, resulted in a significant increase in histone H3 phosphorylation. We then turned to the behaving animal to assess histone H3 phosphorylation with contextual fear conditioning, a robust model of associative learning. H3 phosphorylation was regulated by this paradigm in a time-dependent manner, peaking at 1 h after training before returning to baseline. This effect also required the activation of NMDA receptors and was blocked by a latent inhibition training paradigm. Finally, the increases in histone H3 phosphorylation in the fear-conditioned animal were reduced by inhibition of MAP kinase/ERK kinase (MEK), the kinase upstream of ERK, after training. These results demonstrate that phosphorylation of histone H3 at serine 10 (Ser10) is regulated by an ERK-dependent process in long-term memory.
Results
Histone phosphorylation is regulated by ERK activation in vitro
We first investigated whether activation of ERK in vitro was capable of regulating histone phosphorylation. We used phorbol 12,13-diacetate (PDA, 3 μM) and forskolin (FSK, 50 μM), drugs that activate protein kinase C (PKC) and protein kinase A (PKA) pathways, respectively, in acute hippocampal slices. The phosphodiesterase inhibitor Ro20-1724 (100 μM) was added along with FSK to prevent breakdown of cyclic AMP (cAMP). When slices were treated with PDA or FSK plus Ro20-1724 for 1 h, phosphorylation of ERK2 was significantly increased in area CA1 (Fig. 1A). Pre-incubation of slices with U0126 (20 μM), a selective inhibitor of MEK, for 10 min followed by PDA or FSK blocked the increase in ERK2 phosphorylation (Fig. 1A). This indicates that PDA and FSK activated ERK through MEK. Levels of total ERK protein, as measured by the p44/42 MAPK antibody, did not change with any treatment (Fig. 1A).
Figure 1.
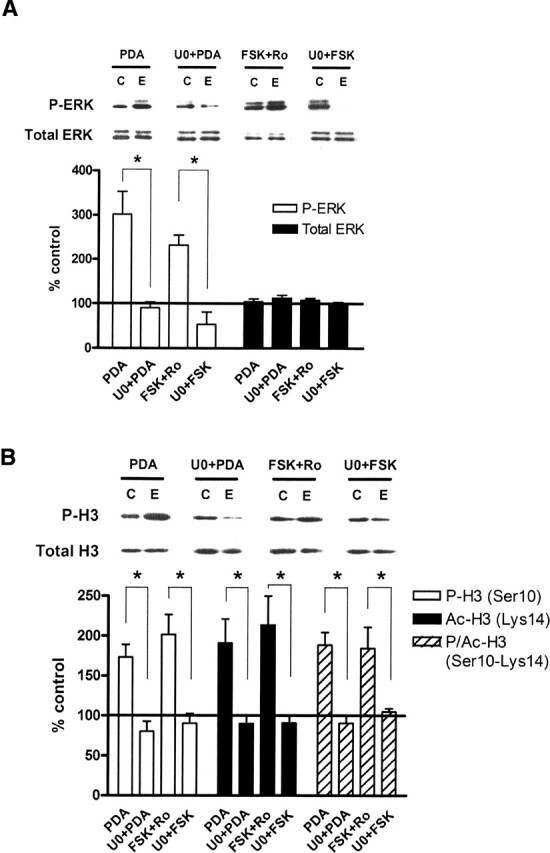
Activation of ERK regulates histone H3 phosphorylation, acetylation, and phospho-acetylation in vitro. (A) Quantification of immunoblot densities for phospho-ERK and total ERK. Treatment of hippocampal slices with PDA (3 μM, n = 9) or FSK (50 μM with 100 μM Ro20-1724, n = 9) for 1 h significantly increased phosphorylation of ERK2 in area CA1. Pre-incubation of slices with U0126 (20 μM, n = 5) for 10 min followed by PDA or FSK blocked the increase in ERK2 phosphorylation. Total ERK protein was unchanged in all treatments. Representative immunoblots for P-ERK and total ERK are shown for each treatment condition. Control (C) samples appear on the left, and experimental (E) samples appear on the right. (B) Quantification of immunoblot densities for phospho-histone H3, acetyl-histone H3, and phospho-acetyl-histone H3. Treatment of hippocampal slices with PDA (3 μM, n = 7) or FSK (50 μM with 100 μM Ro20-1724, n = 6) for 1 h significantly increased phosphorylation, acetylation, and phospho-acetylation of histone H3 in area CA1. Pre-incubation of slices with U0126 (20 μM, n = 5) for 10 min followed by PDA or FSK blocked these changes. Total histone H3 protein was unchanged. Representative immunoblots for P-H3 and total H3 are shown for each treatment condition. Control (C) samples appear on the left, and experimental (E) samples appear on the right. All drug-treated slices were compared with vehicle-treated controls. Error bars indicate standard error of the mean. Asterisks denote significant differences (P < 0.05) as determined by Tukey’s multiple comparison test.
We then asked whether histone H3 phosphorylation was regulated by ERK through PKC- and PKA-mediated pathways. Both PDA and FSK treatment led to significant increases in histone H3 phosphorylation at Ser10 in area CA1 (Fig. 1B). In addition, histone H3 acetylation at Lys14 was significantly regulated, confirming previous results (Levenson et al. 2004b), and phospho-acetylation at both Ser10 and Lys14 simultaneously also increased (Fig. 1B). Pre-incubation of slices with U0126 blocked the increases in phosphorylation, as well as acetylation and phospho-acetylation (Fig. 1B). These results demonstrate that regulation of these changes in histone H3 occurs through the ERK signaling pathway via the upstream kinase MEK. Interestingly, the blockade of histone phosphorylation by U0126 suggests that neither PKC nor PKA directly phosphorylates histone H3 in the hippocampus. Total histone H3 protein was unchanged in any of the drug treatments (Fig. 1B). Together these results indicate that histone H3 phosphorylation is regulated by the ERK/MAPK signaling pathway in hippocampal area CA1 in vitro.
Histone phosphorylation is regulated by contextual fear conditioning in vivo
Given that ERK regulates histone H3 phosphorylation in vitro, we next determined if contextual fear conditioning, a robust model of associative learning and long-term memory formation, was capable of regulating changes in histone phosphorylation in vivo. Contextual fear conditioning leads to formation of a transcription-dependent long-term fear memory 24 h after the training period (Atkins et al. 1998). We first confirmed that 24 h following training, freezing behavior was significantly elevated in animals receiving the fear conditioning paradigm compared with baseline naive control animals (Fig. 2A). Latent inhibition, in which the animals became habituated to the context for 2 h, resulted in significantly less freezing than the shocked animals (Fig. 2A). In addition, the increase in freezing was dependent upon NMDA receptor activation, which is required for long-term memory formation (Fanselow et al. 1994). Injection of the NMDA receptor (NMDA-R) antagonist MK801 prior to training resulted in significantly less freezing than injection of saline (Fig. 2A).
Figure 2.
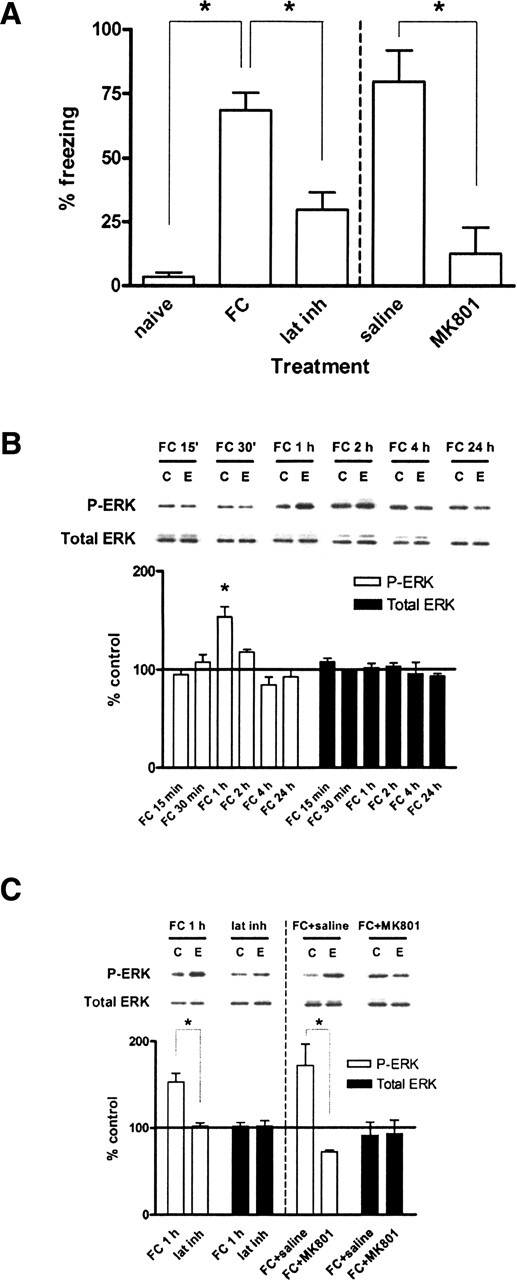
Contextual fear conditioning leads to formation of long-term fear memory and regulates ERK phosphorylation in vivo. (A) Quantification of freezing behavior 24 h following the training period. Animals trained under the contextual fear conditioning paradigm (FC, n = 12) displayed significantly greater freezing than either naive animals (naive, n = 4) or animals trained under the latent inhibition paradigm (lat inh, n = 12). Naive animals had not been previously exposed to either the training chamber or shocks. Animals injected with MK801 (300 μg/kg, n = 4) displayed significantly less freezing than animals injected with saline (0.9% NaCl, 1.25 mL/kg, n = 5). (B) Quantification of immunoblot densities for phospho-ERK and total ERK at different time points following contextual fear conditioning. ERK2 phosphorylation in area CA1 was significantly increased at 1 h after training (FC 1 h, n = 11) before returning to baseline and remaining even after 24 h. Total ERK was unchanged. Representative immunoblots for P-ERK and total ERK are shown for each time point. Control (C) samples appear on the left and experimental (E) samples appear on the right. (C) Quantification of immunoblot densities for phospho-ERK and total ERK. The latent inhibition paradigm (lat inh, n = 6) significantly reduced ERK2 phosphorylation in area CA1 compared with fear conditioning (FC 1 h, n = 11). Injection of animals with MK801 (300 μg/kg) prior to fear conditioning (FC + MK801, n = 3) significantly reduced ERK2 phosphorylation in area CA1 compared with injection with saline (0.9% NaCl, 1.25 mL/kg) prior to fear conditioning (FC + saline, n = 3). Total ERK was unchanged. Representative immunoblots for P-ERK and total ERK are shown for each condition. Control (C) samples appear on the left and experimental (E) samples appear on the right. Error bars indicate standard error of the mean. Asterisks denote significant differences (P < 0.05) as determined by Tukey’s multiple comparison test.
To determine the temporal dynamics of ERK phosphorylation in response to long-term memory formation in the hippocampus, we performed a series of time course experiments. ERK2 phosphorylation in hippocampal area CA1 peaked 1 h following training before returning to baseline (Fig. 2B). Levels of total ERK were unchanged (Fig. 2B). When ERK2 phosphorylation levels were measured following latent inhibition, they were not significantly different from baseline (Fig. 2C). Also, ERK2 phosphorylation was dependent upon NMDA receptor activation, since MK801 injections blocked this increase in area CA1 compared with saline injections when measured 1 h after fear conditioning (Fig. 2C). These observations indicate that fear conditioning leads to NMDA receptor-dependent ERK activation in the hippocampus, as previously reported (Atkins et al. 1998). Furthermore, these data indicate that the ERK activation is not simply a direct consequence of the shocks, as animals in the latent inhibition paradigm receive exactly the same shocks as the fear conditioned animals. Again, levels of total ERK protein were unchanged with any condition (Fig. 2C).
Having shown that our behavioral paradigm leads to long-term contextual fear memory and regulates ERK in a time-dependent manner, we then investigated whether contextual fear conditioning also leads to changes in histone phosphorylation. We found that in area CA1, there was a time-dependent increase in histone H3 phosphorylation at Ser10 which peaked at 1 h after training (Fig. 3A). This coincided with acetylation (Lys14) and phospho-acetylation (Ser10–Lys14) of histone H3, all of which peaked 1 h after training (Fig. 3A). In contrast, there was no significant difference in phosphorylation of histone H3 when measured following latent inhibition (Fig. 3B). Latent inhibition also blocked acetylation and phospho-acetylation of histone H3 (Fig. 3B). In addition, NMDA receptor activation was required for these changes, since MK801 injections blocked the increase in histone H3 phosphorylation, acetylation, and phospho-acetylation in fear-conditioned animals compared with saline-injected controls (Fig. 3B). There was no change in total histone H3 protein in all treatments (Fig. 3A,B). Taken together, these data demonstrate that in the behaving animal, contextual fear conditioning regulates histone H3 phosphorylation in area CA1, and this regulation requires both proper timing of the presentation of novel context plus shocks and the activation of NMDA receptors.
Figure 3.
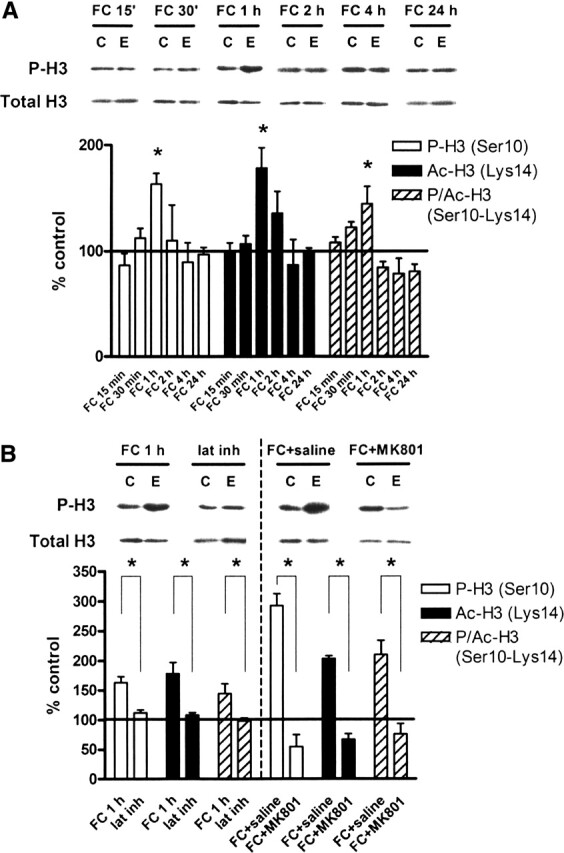
Contextual fear conditioning regulates histone H3 phosphorylation, acetylation, and phospho-acetylation in vivo. (A) Quantification of immunoblot densities for phospho-histone H3, acetyl-histone H3, and phospho-acetyl-histone H3 at different time points following contextual fear conditioning. Histone H3 phosphorylation, acetylation, and phospho-acetylation in area CA1 were significantly increased at 1 h after training (FC 1 h, n = 8) before returning to baseline and remaining even after 24 h. Total histone H3 was unchanged. Representative immunoblots for P-H3 and total H3 are shown for each time point. Control (C) samples appear on the left, and experimental (E) samples appear on the right. (B) Quantification of immunoblot densities for phospho-histone H3, acetyl-histone H3, and phospho-acetyl-histone H3. The latent inhibition paradigm (lat inh, n = 4) significantly reduced histone H3 phosphorylation, acetylation, and phospho-acetylation in area CA1 compared with fear conditioning (FC 1 h, n = 8). Injection of animals with MK801 (300 μg/kg) prior to fear conditioning (FC + MK801, n = 3) significantly reduced histone H3 phosphorylation, acetylation, and phospho-acetylation in area CA1 compared with injection with saline (0.9% NaCl, 1.25 mL/kg) prior to fear conditioning (FC + saline, n = 3). Total histone H3 was unchanged. Representative immunoblots for P-H3 and total H3 are shown for each condition. Control (C) samples appear on the left, and experimental (E) samples appear on the right. Error bars indicate standard error of the mean. Asterisks denote significant differences (P < 0.05) as determined by Tukey’s multiple comparison test.
Activation of MEK is important for regulation of histone phosphorylation by contextual fear conditioning
In a final set of experiments, we investigated whether the activation of MEK, the kinase immediately upstream of ERK, contributes to the fear conditioning-induced changes in histone phosphorylation. We used SL327, a selective inhibitor of MEK that crosses the blood-brain barrier (Atkins et al. 1998). Injections were performed immediately after training to avoid any drug-induced alteration of the animal’s behavioral state during training. We found that SL327-injected animals exhibited significantly less freezing 24 h after receiving shocks compared with dimethyl sulfoxide (DMSO)-injected controls (Fig. 4A). This confirms previous studies suggesting that ERK activation via MEK is necessary for the formation of contextual fear memory (Atkins et al. 1998).
Figure 4.

MEK signaling through ERK contributes to long-term fear memory and contextual fear conditioning-induced changes in histone H3. (A) Quantification of freezing behavior 24 h following the training period. Animals that were fear conditioned and injected with DMSO (2.99 mL/kg, n = 4) displayed significantly greater freezing than either naive animals (naive, n = 4) or animals that were fear-conditioned and injected with SL327 (100 mg/kg, n = 7). Naive animals had not been previously exposed to either the training chamber or shocks. (B) Quantification of immunoblot densities for phospho-ERK and total ERK. Injection of animals with SL327 (100 mg/kg) following fear conditioning (FC + SL327, n = 9) significantly reduced ERK2 phosphorylation in area CA1 compared with injection with DMSO (2.99 mL/kg) following fear conditioning (FC + DMSO, n = 3). Total ERK was unchanged. Representative immunoblots for P-ERK and total ERK are shown for each condition. Control (C) samples appear on the left and experimental (E) samples appear on the right. (C) Quantification of immunoblot densities for phospho-histone H3, acetyl-histone H3, and phospho-acetyl-histone H3. Injection of animals with SL327 (100 mg/kg) following fear conditioning (FC + SL327, n = 3) significantly reduced histone H3 phosphorylation, acetylation, and phospho-acetylation in area CA1 compared with injection with DMSO (2.99 mL/kg) following fear conditioning (FC + DMSO, n = 3). Total histone H3 was unchanged. Representative immunoblots for P-H3 and total H3 are shown for each condition. Control (C) samples appear on the left, and experimental (E) samples appear on the right. Error bars indicate standard error of the mean. Asterisks denote significant differences (P < 0.05) as determined by Tukey’s multiple comparison test.
To confirm that SL327 inhibits hippocampal ERK activation normally seen with fear conditioning, we measured ERK2 phosphorylation following training plus either DMSO or SL327 injection. One hour after training and DMSO injection, ERK2 phosphorylation levels in area CA1 were significantly higher than baseline (Fig. 4B) and comparable with animals receiving training alone with no injection (see Fig. 2B). However, 1 h after training and SL327 injection, ERK2 phosphorylation was reduced to less than 15% of baseline controls (Fig. 4B). This confirmed that ERK activation induced by contextual fear conditioning occurs through the upstream kinase MEK. Total ERK was unchanged (Fig. 4B).
Lastly, we investigated whether MEK activation was important for changes in histone phosphorylation with fear conditioning. One hour after training animals and injecting DMSO, we found a significant increase in histone H3 phosphorylation at Ser10 in area CA1 (Fig. 4C), comparable with trained animals without injection (see Fig. 3A). There were similar increases in histone H3 acetylation (Lys14) and phospho-acetylation (Ser10–Lys14) (Fig. 4C). However, animals that were trained and received SL327 injection showed a marked reduction in histone H3 phosphorylation, acetylation, and phospho-acetylation (Fig. 4C). Total histone H3 protein was unchanged (Fig. 4C). These results support the hypothesis that MEK signaling through ERK contributes to the increase in histone H3 phosphorylation observed with contextual fear conditioning.
Discussion
Our results demonstrate that histone phosphorylation is regulated in the hippocampus during induction and consolidation of long-term memory, probably serving as an epigenetic mark for these events. This is remarkable for several reasons. Histone phosphorylation may be part of an epigenetic “histone code” for memory formation. Multiple post-translational histone modifications may be integrated together, combinatorially driving gene expression patterns by remodeling the structure of chromatin or by recruiting signaling complexes (Jenuwein and Allis 2001). Thus there is a mapping of histone alteration states onto subsets of genes that are transcribed as a result of those changes. With contextual fear conditioning, we observe that histone H3 phosphorylation, acetylation, and phospho-acetylation in the hippocampus peak 1 h following training. The timing of these changes corresponds to the period when rapid hippocampal gene induction is seen (Levenson et al. 2004a). We hypothesize that phosphorylation and acetylation of histone H3 are part of a histone code that is interpreted as a pattern of gene transcription specific to fear memory. Furthermore, while the observed molecular events at 1 h are important for the behavioral expression of fear memory at 24 h, the changes rapidly return to baseline after 1 h. This leaves the possibility of a separate histone code for the maintenance and expression phases of long-term memory in addition to induction. Different patterns or types of histone modifications may operate to drive longer-lasting changes in gene expression that are maintained at the 24-h time point and beyond. This would presumably occur at the actual sites of long-term memory storage, such as the cortex, and therefore be distinct from that occurring in the hippocampus. Further work will be needed to determine these additional patterns of histone regulation in other brain regions at later time points.
Our results implicating ERK in histone phosphorylation and acetylation also support the idea that chromatin acts as a signal integration platform (Schreiber and Bernstein 2002). Each of the core histones of the nucleosome contains N-terminal tails upon which the majority of histone modifications take place (Peterson and Laniel 2004). Each modification may be targeted by separate, independent signaling pathways that converge upon reaching the nucleus. We have shown that phosphorylation and acetylation of histone H3 are both targeted by the ERK/MAPK signaling pathway in hippocampal neurons; however, studies in cultured embryonic fibroblasts have also implicated the p38 MAPK and SAPK2 (stress-activated protein kinase 2) pathways in targeting histone phosphorylation and acetylation (Thomson et al. 1999). It is possible that both mitogen-activated and stress-activated pathways target histones independently, and their effects on transcriptional output are additive or redundant. It remains to be seen precisely what other signaling pathways may be involved and how their relative contributions in histone regulation are integrated to produce a coordinated transcriptional output.
Although histone modifications at specific residues have been associated with transcriptional activation or repression, the interplay between modifications is complex and poorly understood. Some studies posit a correlation between histone phosphorylation and acetylation, leading to the proposal that they are synergistic and coupled (Cheung et al. 2000). Gcn5, a transcriptional activator that contains intrinsic HAT activity (Brownell et al. 1996), has been shown to bind preferentially to phospho-Ser10 on histone H3 in vitro (Lo et al. 2000). Thus phosphorylation may be a prerequisite for nearby histone residues to become acetylated through the binding of Gcn5. Other studies have found that histone phosphorylation and acetylation are in fact not coupled but rather occur independently and only transiently in combination (Thomson et al. 2001). We observed that phosphorylation, acetylation, and phospho-acetylation of histone H3 in the hippocampus are correlated and occur with similar kinetics following conditioned fear training. While this suggests that the two modifications may be coupled in long-term memory formation, further research will be necessary to determine whether phosphorylation predisposes to acetylation or if phosphorylation and acetylation are truly independent events.
In order for ERK to effect a change in histone phosphorylation, it must translocate from the cytoplasm into the nucleus, where it is known to regulate transcription factor activity (Sweatt 2001). Several additional questions remain regarding the precise molecular mechanisms at play. What is the identity of the actual histone kinase? Given our findings, it is unlikely that either PKC or PKA are viable candidates. As we have stated, the blockade of histone phosphorylation by U0126 suggests that neither PKC nor PKA directly phosphorylates histone H3 in the hippocampus but instead does so through MEK and ERK. However, besides ERK itself, other potential kinases include ribosomal S6 kinase 2 (RSK2) and MSK1/2 downstream from ERK, which have been implicated in studies involving mitogenic stimulation of cultured fibroblasts (Sassone-Corsi et al. 1999; Thomson et al. 1999). The Aurora family of kinases, which phosphorylate histones in mitotic cells (Giet and Glover 2001), have yet to be characterized in neurons. It is also unknown how the histone kinase finds its target(s) in the vast solenoid structure that makes up chromatin. The kinase may phosphorylate histones indiscriminately as part of a general metaplastic signal or bind to specific target sequences or nuclear factors to affect transcription of a unique subset of genes. MSK1, one of the candidate histone kinases, is required for CREB phosphorylation in vitro (Arthur and Cohen 2000), suggesting that histones nearby CREB-driven genes may be specifically phosphorylated as a consequence of CREB binding by MSK.
We have demonstrated in this study that hippocampal memory-associated signal transduction pathways regulate histone phosphorylation, acetylation, and phospho-acetylation both in vitro and in the behaving animal. While there are still many complexities to be explored, we report that these findings suggest a role for histone phosphorylation in the induction of long-term memory. As our understanding of chromatin function and epigenetic tagging increases, we will hopefully be able to investigate further the importance of different histone modifications and delineate their role in memory formation in more molecular detail.
Materials and Methods
Hippocampal slice preparation
All experiments were performed in compliance with the Baylor College of Medicine Institutional Animal Care and Use Committee as well as national regulations and policies. Adult male Sprague-Dawley rats (80–100 g) were used in all experiments. Animals were sacrificed by decapitation, and the brains were removed and immersed in ice-cold cutting solution (CS) (100 mM sucrose, 60 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 28 mM NaHCO3, 0.5 mM CaCl2, 7 mM MgCl2, 5 mM glucose, 0.6 mM ascorbate). The caudal portion of the brain containing hippocampus and entorhinal cortex was isolated, and 400 μm transverse slices were prepared using a Vibratome (The Vibratome Company) and stored in ice-cold CS. The hippocampus was isolated from cortical tissue in all slices, and all hippocampi were equilibrated in 50% CS and 50% artificial cerebrospinal fluid (ACSF) (125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 2 mM CaCl2, 1 mM MgCl2, 25 mM glucose) for 45 min at room temperature. Slices were removed and further incubated in 100% ACSF for 45 min at room temperature, then 100% ACSF for 1 h at 32°C. Drugs or vehicle were then added to the slices for 1 h before area CA1 was dissected. All solutions were continuously bubbled with 95% O2/5% CO2.
Fear conditioning
Animals were housed under a 12-h light/12-h dark schedule and given access to rodent chow and water ad libitum. Animals were transported to the laboratory and allowed to acclimate for at least 30 min prior to fear conditioning. Animals were then placed in the fear conditioning apparatus and allowed to explore for 2 min before receiving an electric shock (1 sec, 0.5 mA). The 2-min/1 shock pairing was repeated an additional two times, for a total of three shocks. The animal was left in the chamber for a final minute before being removed. Latent inhibition was performed in the same manner, except that the animal was placed in the apparatus for 2 h before receiving the 2-min/1 shock pairings. For MK801 injection experiments, animals received saline (0.9% NaCl, 1.25 mL/kg) or MK801 (300 μg/kg) injections intraperitoneally 1 h prior to being placed in the chamber. For SL327 injection experiments, animals received DMSO (2.99 mL/kg) or SL327 (100 mg/kg) injections intraperitoneally immediately after being removed from the training chamber. For all behavior experiments, freezing was measured 24 h after training by observing the animals once every 10 sec for 7 min (length of training period). For histone extraction experiments, animals were sacrificed at various time points following the training period, and brains were removed and immersed in oxygenated ice-cold CS before area CA1 was dissected.
Area CA1 dissection
Hippocampal slices were removed following drug or vehicle treatment and immersed in ice-cold CS. For both slices and whole brains, area CA1 of the hippocampus was isolated using a dissecting microscope. Tissue from two animals was pooled for each experimental sample. Area CA1 was placed in 4 mL of ice-cold homogenization buffer (250 mM sucrose, 50 mM Tris-HCl pH 7.5, 25 mM KCl, 0.5 mM phenylmethylsulfonyl fluoride, 0.9 mM sodium butyrate, 1.0 mM sodium orthovanadate, 1% Sigma protease inhibitor cocktail).
Histone extraction
Samples were homogenized for 12 strokes at 4000 rpm using Teflon grinders. All steps were performed on ice, and all centrifugations were done at 4°C. Homogenized samples were centrifuged at 7700g for 1 min. The supernatant was stored at −80°C as the cytoplasmic fraction. The pellet containing the nuclear fraction was resuspended in 1 mL of 0.4 N H2SO4 for 30 min to extract histones. Samples were centrifuged at 14,000g for 10 min. The supernatant was transferred to a fresh tube, and 250 μL trichloroacetic acid with 4 mg/mL deoxycholic acid was added to precipitate proteins. The precipitate was incubated on ice for 30 min, then centrifuged at 14,000g for 30 min. The supernatant was discarded, and the protein pellet was washed with 1 mL acidified acetone (0.1% HCl) and 1 mL pure acetone for 5 min each, with centrifugation at 14,000g for 5 min after each wash. The final protein pellet was resuspended in 10 mM Tris pH 8.0 and stored at −80°C.
Western blotting
Protein concentrations were measured using commercially available reagents (protein assay concentrate, Bio-Rad Laboratories). Four times sample buffer (final concentration 6.25 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 1.25% 2-mercaptoethanol, 0.1% bromophenol blue) was added to each sample. One microgram of protein from each sample was loaded and run on a 4% acrylamide stacking gel and 20% acrylamide resolving gel. Proteins were transferred to polyvinylidene difluoride membranes which were processed for immunoblotting. Membranes were first blocked in 3% bovine serum albumin in Tris-buffered saline with Tween (TTBS) (150 mM NaCl, 20 mM Tris-HCl pH 7.5, 0.05% Tween-20) for 45 min at room temperature, then incubated with primary antibody overnight at 4°C. Primary antibodies and dilutions used were p44/42 MAPK (1:1000, Cell Signaling), phospho-p44/42 MAPK (1:1000, Cell Signaling), anti-histone H3 (1:500, Upstate Biotechnology), anti-phospho-histone H3 (Ser10, 1:500, Upstate Biotechnology), anti-acetyl-histone H3 (Lys14, 1:1000, Upstate Biotechnology), and anti-phospho-acetyl-histone H3 (Ser10-Lys14, 1:500, Upstate Biotechnology). Subsequently membranes were washed with TTBS and incubated with secondary antibody for 2.5 h at room temperature. Secondary antibody used was horseradish peroxidase-conjugated goat anti-rabbit IgG heavy and light chain (Jackson ImmunoResearch). Finally, membranes were washed with TTBS and 1 × TBS, and then immunolabeled by chemiluminescence (ECL, Amersham Biosciences). Luminescence was recorded onto BioMax MR films (Kodak Scientific Imaging Systems). ImageJ (National Institutes of Health) was used to quantify integrated densities of each band on film.
Statistical analysis
Western blot immunodensities were analyzed using a one-way ANOVA followed by Tukey’s multiple comparison test. Comparisons in freezing behavior were analyzed using a one-way ANOVA followed by Tukey’s multiple comparison test. Significant differences were denoted by P < 0.05.
Acknowledgments
This work was supported by grants from the National Institutes of Health (MH57014, NS37444, NS13546) and the American Health Assistance Foundation to J.D.S. W.B.C. was supported in part by grants from the C. Thomas Caskey Scholarship Fund and the Chinese American Doctors Association of Houston.
Footnotes
Article and publication are at http://www.learnmem.org/cgi/doi/10.1101/lm.152906
References
- Adams J.P., Sweatt J.D. Molecular psychology: Roles for the ERK MAP kinase cascade in memory. Annu. Rev. Pharmacol. Toxicol. 2002;42:135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- Alarcon J.M., Malleret G., Touzani K., Vronskaya S., Ishii S., Kandel E.R., Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: A model for the cognitive deficit in Rubenstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Arthur J.S., Cohen P. MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett. 2000;482:44–48. doi: 10.1016/s0014-5793(00)02031-7. [DOI] [PubMed] [Google Scholar]
- Atkins C.M., Selcher J.C., Petraitis J.J., Trzaskos J.M., Sweatt J.D. The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Berger S.L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002;12:142–148. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- Brownell J.E., Zhou J., Ranalli T., Kobayashi R., Edmondson D.G., Roth S.Y., Allis C.D. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- Cheung P., Tanner K.G., Cheung W.L., Sassone-Corsi P., Denu J.M., Allis C.D. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell. 2000;5:905–915. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]
- Crosio C., Cermakian N., Allis C.D., Sassone-Corsi P. Light induces chromatin modification in cells of the mammalian circadian clock. Nat. Neurosci. 2000;3:1241–1247. doi: 10.1038/81767. [DOI] [PubMed] [Google Scholar]
- Crosio C., Heitz E., Allis C.D., Borrelli E., Sassone-Corsi P. Chromatin remodeling and neuronal response: Multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J. Cell Sci. 2003;116:4905–4914. doi: 10.1242/jcs.00804. [DOI] [PubMed] [Google Scholar]
- Darragh J., Soloaga A., Beardmore V.A., Wingate A.D., Wiggin G.R., Peggie M., Arthur J.S. MSKs are required for the transcription of the nuclear orphan receptors Nur77, Nurr1 and Nor1 downstream of MAPK signalling. Biochem. J. 2005;390:749–759. doi: 10.1042/BJ20050196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S., Vanhoutte P., Pages C., Caboche J., Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J. Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn K.L., Espino P.S., Drobic B., He S., Davie J.R. The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochem. Cell Biol. 2005;83:1–14. doi: 10.1139/o04-121. [DOI] [PubMed] [Google Scholar]
- Ehrenhofer-Murray A.E. Chromatin dynamics at DNA replication, transcription and repair. Eur. J. Biochem. 2004;271:2335–2349. doi: 10.1111/j.1432-1033.2004.04162.x. [DOI] [PubMed] [Google Scholar]
- Fanselow M.S., Kim J.J., Yipp J., De Oca B. Differential effects of the N-methyl-d-aspartate antagonist DL-2-amino-5-phosphonovalerate on acquisition of fear of auditory and contextual cues. Behav. Neurosci. 1994;108:235–240. doi: 10.1037//0735-7044.108.2.235. [DOI] [PubMed] [Google Scholar]
- Giet R., Glover D.M. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J. Cell Biol. 2001;152:669–682. doi: 10.1083/jcb.152.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z., Guistetto M., Lomvardas S., Kim J.H., Miniaci M.C., Schwartz J.H., Thanos D., Kandel E.R. Integration of long-term-memory-related synaptic plasticity involves bi-directional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Hendzel M.J., Wei Y., Mancini M.A., Van Hooser A., Ranalli T., Brinkley B.R., Bazett-Jones D.P., Allis C.D. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- Jenuwein T., Allis C.D. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Korzus E., Rosenfeld M.G., Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson J.M., Sweatt J.D. Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Levenson J.M., Choi S., Lee S.Y., Cao Y.A., Ahn H.J., Worley K.C., Pizzi M., Liou H.C., Sweatt J.D. A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-rel. J. Neurosci. 2004a;24:3933–3943. doi: 10.1523/JNEUROSCI.5646-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson J.M., O’Riordan K.J., Brown K.D., Trinh M.A., Molfese D.L., Sweatt J.D. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004b;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Lo W.S., Trievel R.C., Rojas J.R., Duggan L., Hsu J.Y., Allis C.D., Marmorstein R., Berger S.L. Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol. Cell. 2000;5:917–926. doi: 10.1016/s1097-2765(00)80257-9. [DOI] [PubMed] [Google Scholar]
- Peterson C.L., Laniel M.A. Histones and histone modifications. Curr. Biol. 2004;14:R546–R551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Rakyan V.K., Preis J., Morgan H.D., Whitelaw E. The marks, mechanisms and memory of epigenetic states in mammals. Biochem. J. 2001;356:1–10. doi: 10.1042/0264-6021:3560001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassone-Corsi P., Mizzen C.A., Cheung P., Crosio C., Monaco L., Jacquot S., Hanauer A., Allis C.D. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science. 1999;285:886–891. doi: 10.1126/science.285.5429.886. [DOI] [PubMed] [Google Scholar]
- Schreiber S.L., Bernstein B.E. Signaling network model of chromatin. Cell. 2002;111:771–778. doi: 10.1016/s0092-8674(02)01196-0. [DOI] [PubMed] [Google Scholar]
- Soloaga A., Thomson S., Wiggin G.R., Rampersaud N., Dyson M.H., Hazzalin C.A., Mahadevan L.C., Arthur J.S. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swank M.W., Sweatt J.D. Increased histone acetyltransferase and lysine acetyltransferase activity and biphasic activation of the ERK/RSK cascade in insular cortex during novel taste learning. J. Neurosci. 2001;21:3383–3391. doi: 10.1523/JNEUROSCI.21-10-03383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt J.D. The neuronal MAP kinase cascade: A biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- Thomson S., Clayton A.L., Hazzalin C.A., Rose S., Barratt M.J., Mahadevan L.C. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 1999;18:4779–4793. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson S., Clayton A.L., Mahadevan L.C. Independent dynamic regulation of histone phosphorylation and acetylation during immediate-early gene induction. Mol. Cell. 2001;8:1231–1241. doi: 10.1016/s1097-2765(01)00404-x. [DOI] [PubMed] [Google Scholar]
- Wood M.A., Kaplan M.P., Park A., Blanchard E.J., Oliveira A.M., Lombardi T.L., Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn. Mem. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]