

Abstract
PURPOSE. To identify pathogenic mutations in the guanylate cyclase-activating protein 1 (GCAP1) and GCAP2 genes and to characterize the biochemical effect of mutation on guanylate cyclase (GC) stimulation.
METHODS. The GCAP1 and GCAP2 genes were screened by direct sequencing for mutations in 216 patients and 421 patients, respectively, with various hereditary retinal diseases. A mutation in GCAP1 segregating with autosomal dominant cone degeneration was further evaluated biochemically by employing recombinant proteins, immunoblotting, Ca2+-dependent stimulation of GC, fluorescence emission spectra, and limited proteolysis in the absence and presence of Ca2+.
RESULTS. A novel GCAP1 mutation, I143NT (substitution of Ile at codon 143 by Asn and Thr), affecting the EF4 Ca2+-binding loop, was identified in a heterozygote father and son with autosomal dominant cone degeneration. Both patients had much greater loss of cone function versus rod function; previous histopathologic evaluation of the father's eyes at autopsy (age 75 years) showed no foveal cones but a few, scattered cones remaining in the peripheral retina. Biochemical analysis showed that the GCAP1-I143NT mutant adopted a conformation susceptible to proteolysis, and the mutant inhibited GC only partially at high Ca2+ concentrations. Individual patients with atypical or recessive retinitis pigmentosa (RP) had additional heterozygous GCAP1-T114I and GCAP2 gene changes (V85M and F150C) of unknown pathogenicity.
CONCLUSIONS. A novel GCAP1 mutation, I143NT, caused a form of autosomal dominant cone degeneration that destroys foveal cones by mid-life but spares some cones in the peripheral retina up to 75 years. Properties of the GCAP1-I143NT mutant protein suggested that it is incompletely inactivated by high Ca2+ concentrations as should occur with dark adaptation. The continued activity of the mutant GCAP1 likely results in higher-than-normal scotopic cGMP levels which may, in turn, account for the progressive loss of cones.
Rod and cone phototransduction cascades rapidly activate a cGMP phosphodiesterase when rhodopsin is photoactivated. The final steps of the phototransduction cascade are a reduction of cytoplasmic cGMP and the closure of cGMP-gated cation channels in the plasma membrane.1 To recover from a photoresponse, cGMP must be replenished by photoreceptor guanylate cyclases (GCs). When the level of cGMP (1-10 μM) is replenished, cGMP-gated channels reopen, and dark-adapted Ca2+ levels (600-1000 nM) are restored. Activity of the photoreceptor GC is regulated via retina-specific guanylate cyclaseactivating proteins, or GCAPs. In the dark, when the intracellular Ca2+ level is relatively high due to its continuous influx through open channels, Ca2+-bound GCAPs are inactive. In the light, the closed channels result in reduced Ca2+ levels, thereby allowing the Ca2+ ions to dissociate from the GCAPs. Ca2+-free GCAPs are then able to stimulate GC, resulting in an acceleration of cGMP synthesis. Disruption of these events by mutations in genes encoding PDE subunits,2-4 GC1,5,6 GCAP1,7-9 and the subunits of the rod and cone cGMP-gated channels,10-15 cause photoreceptor degenerations such as retinitis pigmentosa, Leber congenital amaurosis, cone dystrophy, or congenital stationary night blindness.16,17
Multiple GCAP gene duplications have taken place during evolution. Up to eight GCAPs (GCAP1 to -8) have been identified in teleosts.18 In the mammalian retina, only three GCAP isoforms (GCAP1 to -3) are expressed.19 In humans, GCAP1 and GCAP2 are encoded by a tail-to-tail gene array on chromosome 6p20 while the GCAP3 gene is located on chromosome 3q.21 All three GCAPs are expressed in rods and cones,19 but mutations in only the GCAP1 gene have been associated with human retinal disease, namely, autosomal dominant cone dystrophy (adCD). Cone-specific degenerations due to GCAP1 defects are consistent with the high levels of GCAP1 in the outer segments of this cell type. One reported mutation (Y99C) results in a Tyr to Cys change in EF3.22,23 Another mutation, E155G,9 replacing the last acidic residue of the EF hand with a neutral residue, is predicted to affect Ca2+ coordination at EF4. Acidic Glu/Asp residues, occupying the first and last positions of the loop, are required for effective Ca2+coordination.24 E155 is 100% conserved in all GCAPs sequenced so far, suggesting that this residue is essential. A third reportedly pathogenic mutation (GCAP1-P50L) found in one family with cone-rod dystrophy25 causes disease by an entirely different mechanism26 or may be a nonpathogenic rare variant. No disease-causing mutations have yet been reported in the human GCAP227 and GCAP3 genes.
This contribution describes our recent searches for mutations in the GCAP1 and GCAP2 genes among patients with hereditary retinal diseases. Analysis of the GCAP1 gene was focused on patients whose disease included cone dysfunction or degeneration as a prominent feature. Analysis of the GCAP2 gene included patients with rod and cone dysfunction or degeneration, since this gene is thought to be expressed in both photoreceptor cell types.28,29
Materials and Methods
Ascertainment of Patients and Controls
This study conformed to the tenets of the Declaration of Helsinki and was approved by Internal Review Boards at the Massachusetts Eye and Ear Infirmary and Harvard Medical School. Almost all patients were recruited from the Berman-Gund Laboratory and were diagnosed with a hereditary retinal disease after an eye examination that included an electroretinogram (ERG). Patients with cone-rod degeneration had a substantially more severe reduction in cone versus rod ERG amplitudes. Patients with retinitis pigmentosa had more severe reduction in rod versus cone ERG amplitudes or had comparable reductions in rod and cone amplitudes. Patients with achromatopsia had absent color vision and severely reduced cone ERGs and normal to slightly subnormal rod ERGs from early childhood. Patients with cone degeneration had reduced central visual acuity, a reduction in cone ERGs, and normal or slightly subnormal rod ERGs. Patients with cone dysfunction had some but not all clinical features diagnostic of cone degeneration or achromatopsia. Control individuals had no symptoms of hereditary retinal disease and had no known blood relatives with such diseases.
ERGs were recorded after maximal dilation of the pupil and at least 40 minutes of dark-adaptation.30-32 Blood samples were obtained from the patients. Relatives of some patients were also recruited for this study and donated blood samples. Control blood samples were also obtained from individuals without symptoms of or family history of retinal disease. Leukocyte DNA was purified using standard procedures.
Mutation Screening
The open reading frames of the GCAP1 and GCAP2 genes, including their flanking intron splice sites, were amplified using the polymerase chain reaction and the primers listed in Table 1. The DNA sequences were obtained by direct DNA sequencing using a dye-terminator cycle sequencing kit (version 3.1; Applied Biosystems, Foster City, CA) and an automated sequencer (ABI model 3100; Applied Biosystems). The numbering of the codons of the cDNA of GCAP1 and GCAP2 in this article are according to GenBank accession numbers L36859 and NM-002098, respectively20,33; NT-007592 was used for the numbering of the genomic sequence for both genes (GCAP1 and GCAP2 genes are separated by 5420 bp).
TABLE 1.
PCR Primers Used in this Study
| Primer | Exon | Primer Sequence | |
|---|---|---|---|
| GCAP1 | |||
| 4949 | 1S | 5′-GGACTCAGGCCTGTGAGAGA | |
| 4950 | 1A | 5′-CCAGCTGGTCAGGCTTCC | |
| 4951 | 2-4S | 5′-CGGAGCCTTGGGTTATGAT | |
| 4952 | 2-4A | 5′-GCGAGCTAAGCCTCTGAGTT | |
| 5094 | 2R* | 5′-CCTCAGTCCCACATCCTGTC | |
| 5095 | 3F* | 5′-TCTACTCCTCACTGCTCTTG | |
| 5097 | 4F* | 5′-ACGTGGGCTCTGTCCCTGCC | |
| GCAP2 | |||
| 5098 | 1S | 5′-AGTCACCCAGGAGATGATGG | |
| 5099 | 1A | 5′-ACGGAGTGGAAAGAAGAGCA | |
| 5100 | 2S | 5′-GGCCACATGGATTAAAAGGA | |
| 5101 | 2A | 5′-CACAGGAAACACACTCAGAATGA | |
| 5102 | 3S | 5′-GTGATCCTAAGACGCACAGC | |
| 5103 | 3A | 5′-TTCTCCACAGCCTTCTCACA | |
| 5104 | 4S | 5′-TGAGGTCTGAGACTGCAGGA | |
| 5105 | 4A | 5′-GAAGTCAACACCAGGGGAAGA |
Exons terminating in an “S” are in the sense direction; those with “A” are in the antisense direction.
These primers were used for sequencing the PCR product amplified by 4951 and 4952.
Expression Constructs of Mutant GCAPs
The mutations were introduced into a human GCAP1 plasmid (hGCAP1) by site-directed mutagenesis,33 and the mutant GCAPs were expressed in insect cells. Briefly, the EcoRI-digested insert of hGCAP1 was cloned into pFastBac-1, and transformed into XL1-Gold Escherichia coli strain. The orientation of the resulting plasmid was confirmed with Pst1 digests and the mutations were verified by DNA sequencing with the primers 5′-GTTGGCTACGTATACTCCGG and 5′-GTAAAACCTCTACAAATGTGG. Mutagenesis to generate GCAP1-I143NT was carried out in this plasmid using the sense primer 5′-CAGTGTTCTCCAAGAATACCGACGTCAACGGGGAT and the anti-sense primer 5′-ATCCCCGTTGACGTCGGTATTCTTGGAGAACACTG. Mutagenesis to generate GCAP1-E155G9 as an internal control was carried out with the sense primer 5′-AACTCTCCCTGGAAGGGTTTATAGAGGGCGT and the anti-sense primer 5′-ACGCCCTCTATAAACCCTTCCAGGGAGAGTT. Wild-type human GCAP1, GCAP1-E155G, and GCAP1-I143NT pFastBac plasmids were transformed into DH10Bac E. coli to generate the recombinant bacmid DNAs (Bac-to-Bac Baculovirus Expression System, Invitrogen, Carlsbad, CA). The bacmids were transfected into High five insect cells (Invitrogen) to produce recombinant baculovirus and proteins.
SDS PAGE and Western Blot Analysis
The expression of GCAP1 proteins was confirmed with UW101 antibody using SDS PAGE and Western blot analysis as described.34 All purification procedures were performed at 4°C. After homogenization, cell suspensions were centrifuged at 100,000g for 20 minutes. Again, all purification procedures were carried out at 4°C. Supernatants were used as a source of recombinant proteins for protein purification. GCAPs were purified using monoclonal antibody (G2) coupled to CNBr-activated Sepharose resin (∼5 mg IgG/1 mL gel) as described previously.35 Purified proteins were dialyzed overnight against 10 mM Bis-Tris Propane (1,3-bis(tris[hydroxymethyl]amino) propane (pH 7.5), containing 100 mM NaCl at 4°C. The purity of the proteins was estimated by SDS-PAGE and Coomassie staining. SDS-PAGE in the presence and absence of Ca2+, and limited proteolysis of purified GCAP1 and GCAP1 mutants, were carried out as described previously.36
GC Activity Assay
Washed rod outer segment (ROS) membranes37 were prepared from fresh bovine retinas reconstituted with recombinant GCAPs and assayed as described.38 [Ca2+] was calculated using the computer program Chelator 1.0039 and adjusted to higher concentrations by increasing the amount of CaCl2. All assays were repeated at least twice.
Fluorescence Measurements
Fluorescence measurements of GCAP1 and its mutants were carried out on a spectrofluorimeter (LS 50B; Perkin-Elmer, Boston, MA) using a 1 × 1-cm quartz cuvette. Emission spectra were recorded with excitation at 280 nm at 5 nm slit widths. Spectra were determined in 50 mM HEPES (pH 7.8), containing 60 mM KCl, 20 mM NaCl, 1 mM EGTA, 1 mM dithiothreitol, and 4.6×10-8 to 2.4×10-6 M CaCl2.
Results and Discussion
GCAP1 Gene Mutations
The GCAP1 gene was analyzed in 216 patients with cone degeneration, cone-rod degeneration, achromatopsia, or other hereditary retinal disease in which cones degenerate or are dysfunctional (Table 2). Five DNA sequence changes were found in a total of six patients (Table 3). One of the changes, I143NT (c.428delTinsACAC), affected a residue in the α-helix immediately flanking the Ca2+-binding site (EF4). This change was found in an index patient with autosomal dominant cone degeneration, and it was not found among 93 control individuals. The index patient IV-1 (family #3761) and his affected son V-1 were both heterozygous for this change; the one unaffected relative (a daughter, V-2) who was analyzed did not carry the change (Fig. 1).
TABLE 2.
Numbers and Diagnoses of Patients Screened
| Diagnosis | GCAP1 | GCAP2 |
|---|---|---|
| Cone-rod degeneration | 82 | 0 |
| Atypical RP | 50 | 56 |
| Cone dysfunction | 37 | 36 |
| Achromatopsia | 35 | 35 |
| Cone degeneration | 12 | 11 |
| Dominant RP | 0 | 190 |
| Recessive RP | 0 | 93 |
| Total | 216 | 421 |
TABLE 3.
Sequence Variants Identified in GCAP1 Gene
| Protein | Nucleotide | Patient | Diagnosis |
|---|---|---|---|
| None | c.-26A>G | 065-001 | Achromatopsia |
| T114I | c.341C>T | 199-005 | Atypical RP |
| I143NT | c.428delTinsACAC | 195-001 | Cone degeneration |
| None | g.IVS3+171delG | 065-011* | Achromatopsia |
| D189D | c.577C>T | 111-007 | Achromatopsia |
| D189D | c.577C>T | 270-003 | Macular degeneration |
This patient was later shown to be a homozygote with the pathogenic mutation T383fs in the CNGB3 gene encoding β-subunit of the cone cGMP-gated channel (data not shown).
FIGURE 1.
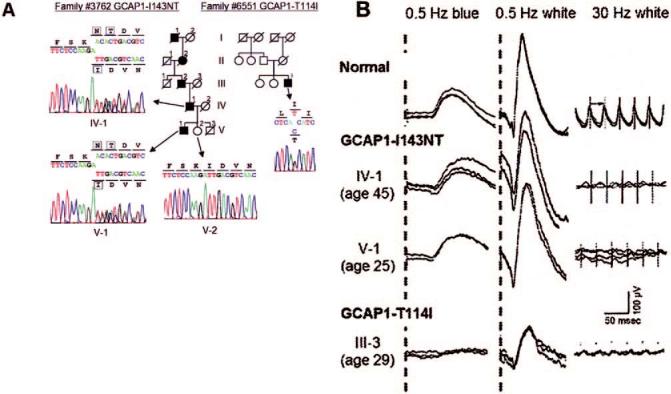
Analyses of family #3762 with GCAP1-I143NT and family 6551 with GCAP1-T114I. (A) Sequences of two novel missense changes in GCAP1 and schematic pedigrees of the corresponding families. Filled symbols: affected individuals; open symbols: unaffected individuals. Arrows point to the sequence results of relevant family members. Boxes denote residues at the mutation site. (B) Dark-adapted, full-field ERGs from a normal control individual (top), two patients with GCAP1-I143NT (middle), and a patient with GCAP1-T114I (bottom). The patients with GCAP1-I143NT are individuals IV-1 and V-1 of family #3762; the patient with GCAP1-T114I is individual III-3 of family #6551. Going from left to right, the columns show rod responses to 0.5-Hz blue flashes (1.2 log ft/L, λmax = 440 nm), rod-plus-cone responses to 0.5-Hz white flashes (3.8 log ft/L), and cone responses to 30-Hz white flashes (3.8 log ft/L). For all three columns, the scale bar (right column above the bottom traces) indicates 100 μV vertically and 50 ms horizontally. For each test condition, two to four consecutive sweeps are shown to illustrate reproducibility.
A missense change, T114I (c.341C>T), was identified in another index patient, also a heterozygote, with an atypical form of retinal degeneration (Fig. 1A). The patient had no known affected relatives, and the few living unaffected relatives could not be located. Thus we could not determine whether they also carried this change. At age 29 years, the patient had rod ERG amplitudes reduced approximately 70%-75% below the lower limit of normal, and cone ERG amplitudes reduced only approximately 65% below the lower limit of normal (Fig. 1B). This reduction in rod and cone ERG amplitudes was very different from that found in previously reported patients with GCAP1 mutations and was different from our patient with the GCAP1-I143NT mutation.
Three other novel GCAP1 changes were found in one or two patients each, all heterozygotes, and were interpreted as likely nonpathogenic rare variants: an A-to-G transition in the 5′-untranslated region, a 1-bp deletion in intron 3, and an isocoding change in codon D189 (Table 3). None of these three changes is predicted to affect the amino acid sequence of the encoded protein. None of these three changes, and neither the I143NT nor the T114I change, appears to create or destroy any splice donor or acceptor site, based on analysis with splice-site prediction software (available at http://www.fruitfly.org/seq_tools/splice.html).40
Ocular Findings of Patients with GCAP1-I143NT
The ocular findings and ERGs of the affected father and son from this family with the GCAP1-I143NT mutation have been previously reported.41 Although the son at age 15 years had visual acuity of 20/20 in both eyes, his cone function was severely compromised, as indicated by his absent color vision (i.e., his inability to arrange the Farnsworth D-15 panel or to correctly identify Hardy-Rand-Rittler (HRR) color plates). The father at age 45 had photophobia, visual acuity of 20/300 in both eyes, absent color vision, and small regions of retinal atrophy in the center of the macula of both eyes. ERGs of both the father at age 45 and the son at age 25 showed minimal if any 30-Hz cone ERG responses and intact 0.5-Hz rod ERG responses to blue or white light (Fig. 1B).41 Early receptor potential amplitudes after a bleaching light recovered at a rate slower than normal and corresponded to the normal rate of rhodopsin regeneration, indicating little to no cone function.42,43 The eyes of the father, obtained at autopsy at age 75, revealed the absence of rod and cone photoreceptors in the central macula; some degenerating photoreceptor inner and outer segments were seen in the parafoveal region.44 In the periphery, the rod photoreceptors were intact and normal in number, and a few intact cone photoreceptors were also present.44
GCAP2 Gene Mutations
The GCAP2 gene was analyzed in 190 patients with autosomal dominant retinitis pigmentosa, 93 with autosomal recessive retinitis pigmentosa, and 138 patients with various forms of retinal degeneration/dysfunction (Table 2). Five rare variants were identified in a total of six heterozygous patients (Table 4). Two of them were missense changes, both affecting residues in α-helices flanking the Ca2+-binding domain. A V85M mutation was found in a heterozygote patient with atypical retinal degeneration with no family history. An F150C change was found in a heterozygous patient with recessive RP who was the offspring of a consanguineous marriage. No likely mutation affecting the other allele was identified in either patient. The remaining three rare variants were isocoding or intron changes predicted not to affect the encoded protein (Table 4). Six DNA sequence changes were found that were interpreted as non-pathogenic polymorphisms because they were found in >1% of alleles (Table 4). One of the polymorphisms is a previously reported27 missense alteration (E155D) that affects the last residue in the highly conserved Ca2+-binding domain EF3. However, this mutation was previously also found in a normal control.27 Splice-site prediction software found no evidence that any of the GCAP2 changes created or destroyed RNA splice sites.
TABLE 4.
Sequence Variants Identified in GCAP2 Gene
| Rare Variants | |||
|---|---|---|---|
| Protein | Nucleotide | Patient | Diagnosis |
| V85M | c.253G>A | 038-114 | Atypical RP |
| N106N | c.318T>C | 001-473 | Dominant RP |
| None | g.IVS3+22delA | 115-018 | Cone dysfunction |
| None | g.IVS3+22delA | 001-051 | Dominant RP |
| None | g.IVS3+65G>A | 001-245 | Dominant RP |
| F150C | c.449T>G | 003-059 | Recessive RP |
| Polymorphisms | |||
| Protein | Nucleotide | Allele Frequency* (hom/het/hom) | |
| Y57Y | c.171T>C | 56/206/159 | |
| None | g.IVS2+81C>T | 299/83/8 | |
| E155D† | c.465G>T | 410/11/0 | |
| None | g.IVS2-74 G>A | 9/96/280 | |
| None | g.IVS3+8G>A | 402/15/0 | |
| None† | g.IVS3-30A>G | 389/11/0 | |
Some patients were not included in the analysis of some poly-morphisms.
Reported in Reference 27.
Biochemical Analysis of the GCAP1-I143NT Mutant
Although the GCAP1-I143NT mutation cosegregated with cone degeneration in family #3761, the available family members were few in number and the cosegregation of the mutation with the disease could have been by chance. Further evidence for the pathogenicity of this mutation, however, came from analyses of the ability of the mutant recombinant protein to stimulate GC as a function of [Ca2+].
The residue Ile143 precedes the EF4 loop in GCAP1 (Fig. 2A). This position, typically occupied by a hydrophobic residue, is invariant in the EF4 hand motifs of GCAP1 (Fig. 2A), and in the closely related GCAP2 and GCAP3 sequences (not shown). To test the presumed importance of this residue for Ca2+ binding, human GCAP1, the mutant GCAP1-I143NT, and the mutant GCAP1-E155G which had earlier been shown to be associated with cone dystrophy,9 were expressed in insect cells. When the affinity-purified proteins were analyzed by SDS PAGE (Fig. 2C) in the presence of high Ca2+ (1 mM) and in the absence of Ca2+ (1 mM EGTA), normal and mutant GCAP1s exhibited a change in mobility (“Ca2+-shift”). This mobility shift is typical for calmodulin-like Ca2+-binding proteins and indicated a large rearrangement of the 3D structure from an open form (Ca2+-free, the slower moving active form) to a more compact Ca2+-bound conformation (the faster moving inactive form). The Ca2+-shift was somewhat smaller in the mutant proteins, consistent with impaired Ca2+ binding to EF4.
FIGURE 2.
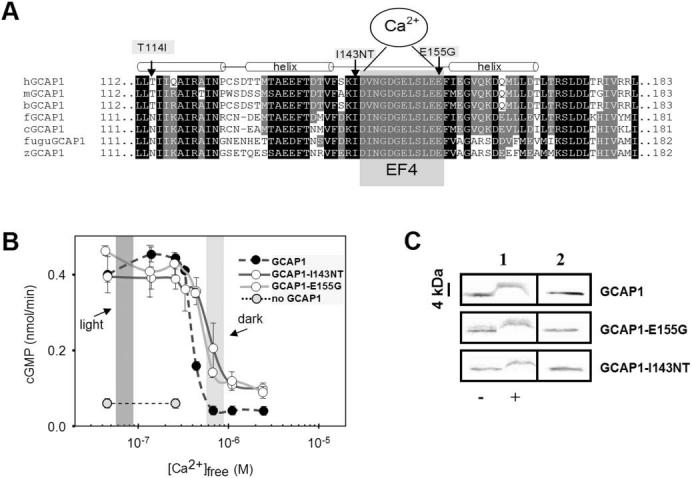
Amino acid sequence of vertebrate GCAP1s around EF4 and biochemical analysis of mutant GCAPs. (A) Sequence of vertebrate GCAP1s around the Ca2+-binding loop EF4. EF4 is highlighted by gray shading, surrounded by two α-helical structures (approximately three helical turns). The mutations changing the hydrophobic residue Ile143 and the acidic residue E155 are identified by arrows. A third mutation found in an independent family (Fig. 1B) is situated in the C-terminal helix of EF3. It is unclear whether this mutation causes retinal disease. (B) Stimulation of GC activity in ROS membranes by normal and mutant GCAPs as a function of Ca2+. The dark-shaded area indicates low [Ca2+]free expected in the light-adapted photoreceptors; the light-shaded area reflects high [Ca2+]free expected in the dark-adapted photoreceptors. Assays were carried out with addition of 3 μM GCAPs and were repeated at least three times. Error bars: SD for GC activity stimulated by GCAPs. (C) Stained SDS gel of GCAP1, GCAP1-I143NT, and GCAP1-E155G in the presence or absence of Ca2+ (column 1);-, absence of EGTA, +, presence of EGTA. Column 2 is a control immunoblot for GCAPs in water. GCAPs display a shift in mobility in the absence of bound Ca2+. The antibody used is UW14.21
Persistent Stimulation of Photoreceptor GC by GCAP1-I143NT
We next assayed the degree to which mutant GCAP1-I143NT stimulated GC as a function of [Ca2+]. We expected incomplete inactivation of GC1 at the high Ca2+ levels found in dark-adapted photoreceptors, similar to what has been reported for GCAP1-Y99C22,23 and GCAP1-E155G.9 As shown in Figure 2B, GCAP1-I143NT was active at low Ca2+ (<200 nM), as was wild-type GCAP1, but the mutant remained active (30%-50%) even at high dark [Ca2+]free-levels (∼700 nM) when wild-type GCAP1 was inhibited. Similar incomplete suppression of GC activity was previously found with a GCAP1-Y99C mutation affecting EF3.23 The mutant GCAP1-E155G exhibited similar incomplete suppression of GC activity, suggesting that in both mutants, as evidenced by the rightward shift in the Ca2+-titration curve, EF4 binds Ca2+ with an impaired affinity. This conclusion is consistent with our previous finding that the GCAP1-E155D36 mutant, which is not able to bind Ca2+ to this loop, showed activity even at high, nonphysiological Ca2+ levels (50 μM [Ca2+]free). Mutagenesis experiments showed that when both EF3 and EF4 of GCAP1 are defective and allow no Ca2+ binding, GCAP1 was converted into a Ca2+-insensitive GC stimulator (Fig. 6B in Ref.36). Taken together, the findings indicate that Ca2+ binding by EF3 and EF4 is critical for inactivation of GCAP1, and dissociation of Ca2+ from these sites converts GCAP1 from the inactive to an active state. Mutations affecting only one of these sites will partially impair GC inhibition, leading to persistent stimulation in dark-adapted photoreceptors.
Fluorescence Emission Spectra of GCAP1 Mutants
A sensitive assay for changes in Ca2+-binding is to measure intrinsic Trp fluorescence. Trp21 in the N-terminal domain, Trp51 between EF1 and EF4, and Trp94 near EF3 can be used to monitor changes in conformation when excited with UV light45 (Fig. 3). An increase of Ca2+ bound to GCAP1 caused a decrease in fluorescence intensity with a minimum occurring at 200-300 nM [Ca2+]free (Fig. 3A). Further increases in Ca2+ levels reversed this trend and caused an increase in fluorescence intensity (Fig. 3A). These changes in the fluorescence correlated with a structural rearrangement of GCAP1 and a transition from an activator to an inhibitor of photoreceptor GC. However, when the GCAP1 mutants were analyzed, we noticed a significantly modified dependence of fluorescence intensity on Ca2+ levels: high Ca2+ levels did not increase the fluorescence of the mutant proteins (Figs. 3B and 3C) apart from a small shoulder at 300-600 nM [Ca2+]free. Since Trp residues were not present only in the Ca2+-binding sites, the fluorescence data provided general information about structural changes in GCAP1, unrelated to specific regions such as EF4.
FIGURE 3.
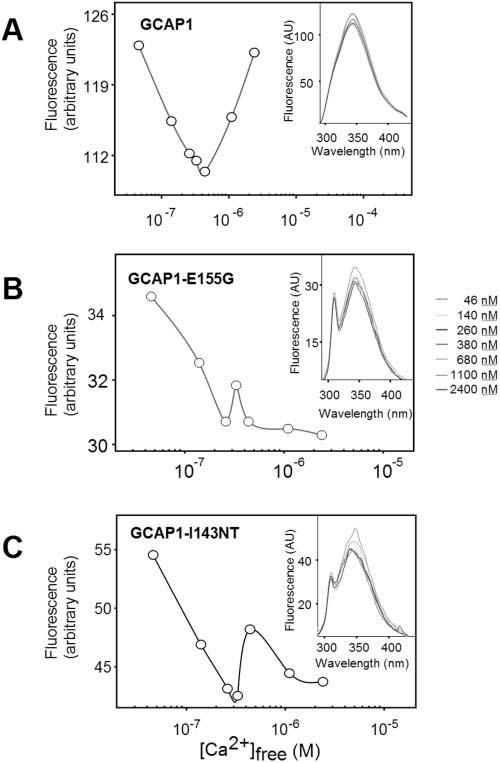
Ca2+-induced conformational changes in GCAP1 and its mutants observed by fluorescence spectroscopy. Changes in fluorescence at λex = 290 nm and λem = 343 nm as a function of [Ca2+]. Insets, fluorescence emission spectra of GCAPs using excitation at 280 nm from 4.6 × 10-8 M to 2.4 × 10-6 M Ca2+. (A) Recombinant human GCAP1; (B) GCAP1-E155G; (C) GCAP1-I143NT.
GCAP1 Structure and the Consequence of I143NT Mutation
The four EF hands of GCAP1 are arranged in pairs that gave rise to N- and C-terminal domains which form a relatively compact structure (Fig. 4). Three Ca2+ ions are bound to GCAP1 at EF2, EF3, and EF4, as predicted on the basis of its amino acid sequence and by numerous site-directed mutagenesis experiments.23,36,38,45-47 EF1 does not bind Ca2+ because the binding loop is distorted by a Pro at the fourth position of the 12 residue loop. In addition, the preceding residue (Cys) is not suitable for the coordination of Ca2+ ions. Modeling of EF4 of GCAP1, based on the recoverin crystal structure,48 and the structure of GCAP2, revealed through nuclear magnetic resonance techniques,49 suggests that substitution of Ile143, positioned at the N-terminal end of EF4, by two polar residues will change the orientation of the N-terminal α-helix, distorting the loop conformation that is essential for Ca2+ binding, and decreasing the affinity for Ca2+ (Figs. 4A and 4B). Changes in these positions among other Ca2+-binding proteins have also been shown to impair Ca2+ coordination.24 The effect of the mutation on the structure of GCAP1 can be monitored by subjecting GCAP1 and GCAP1-I143NT to limited proteolysis (Fig. 4C). In the absence of Ca2+, GCAP1 assumed an open conformation and was readily digested by trypsin (last lane, both gels). In the presence of Ca2+, GCAP1 assumed a compact structure, and proteolysis was restricted. Under the same conditions, GCAP1-I143NT, in which Ca2+-binding is impeded at EF4, was more susceptible to proteolysis.
FIGURE 4.
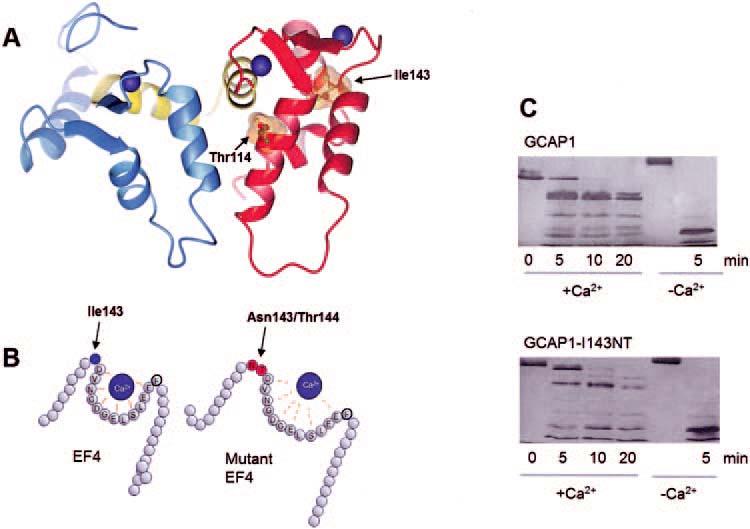
Model of GCAP1 and distortion of EF4 by insertion of two residues. (A) The model of GCAP1 is based on the NMR structure of GCAP2 (as deposited in the Protein Data Bank, PDB ID# 1JBA).50 The N-terminal domain is in blue, the C-terminal domain is in red, and the central helix is in yellow. Ca2+ ions are shown in blue, and the side chains of the residues Ile143 and Thr114 are shown with the van der Waals spheres. (B) A cartoon of EF4 depicting how the replacement of hydrophobic Ile143 by two polar residues may lower affinity for Ca2+.(C) Limited proteolysis of GCAP1 and GCAP1-I143NT by trypsin. The digestion was carried out at 30°C at a ratio of GCAP1/trypsin 300:1, and the digest was analyzed by SDS-PAGE at 0, 5, 10, and 15 minutes; +Ca2+ represents 2 μM [Ca2+] and -Ca2+ indicates 30 nM [Ca2+]. Note that already after 5 minutes of digestion, the high molecular weight components are more extensively digested in GCAP1-I143NT.
Thr114 (Table 3), a residue which was mutated to Ile in family 6551 (Fig. 1A), was located on the C-terminal helix of EF3 (Fig. 4). Substitution at this conserved region among GCAPs18,19 may also affect coordination of Ca2+ to this loop, consistent with the GCAP1-Y99C mutant affecting Ca2+ binding in EF3, or it may modify the interaction with GCs. Since the patient with the GCAP1-T114I changes had no family history of retinal disease and since his retinal disease was very different from that found in other patients with GCAP1 mutations, this mutant was not further biochemically characterized.
Conclusions
In summary, GCAP1-I143NT stimulated photoreceptor GC effectively at low [Ca2+]free, to a degree comparable to wild-type GCAP1 and the GCAP1-E155G mutant. However, the Ca2+-sensitivity of the GCAP1 mutants was markedly altered, causing persistent stimulation of GC when exposed to high Ca2+ levels such as those present in dark-adapted photoreceptors. This activity is consistent with the dominant phenotype produced by the mutant alleles causing cone degeneration. Persistent stimulation of GC by the mutant proteins is predicted to lead to elevated levels of cGMP in the dark-adapted retina, which in turn causes a higher percentage of cGMP-gated channels in the plasma membrane to be open. It is likely that the actual cGMP level in the dark-adapted photoreceptors will be a balance between the excessive production of cGMP by GC stimulated by the mutant GCAP1 and hydrolysis of cGMP through the basal activity of PDE. The altered physiological cGMP levels may be subtle and thus cause a relatively slow retinal degeneration. The reason for the mostly cone-specific degeneration in response to this physiological defect may be because GCAP1 may be more active in cones than rods, or, alternatively, it may reflect other differences in cGMP metabolism between rods and cones.
It remains unexplained why some cones in the peripheral retina44 can survive for over 70 years, while rods and cones in the fovea and perifovea degenerate. Since the abnormality of photoreceptor physiology in the affected patients would be expected to occur mainly in darkness or dim light (persistent GC stimulation in the dark), it is doubtful that reducing light exposure (such as by wearing sunglasses) would slow their rate of progressive visual loss. Whether reducing exposure to darkness (such as by keeping lights on at night while sleeping) would be helpful is uncertain. Animal models with homologous dominant GCAP1 mutations might be helpful to address this uncertainty.
Acknowledgments
The authors thank Sławomit Filipek for the preparation of the structural model shown in Figure 4.
Footnotes
Supported by Grants EY09339, EY08123, EY00169, and EY08683 from the National Institutes of Health; a grant from Research to Prevent Blindness, Inc. (RPB) to the Departments of Ophthalmology at the University of Washington and the University of Utah; a grant from the Macular Vision Research Foundation; the Foundation Fighting Blindness, Owings Mills, MD; and a grant from the E.K. Bishop Foundation (KP).
Disclosure: K.M. Nishiguchi, None[b]; I. Sokal, None; L. Yang, None; N. Roychowdhury, None; K. Palczewski, None; E.L. Berson, None; T.P. Dryja, None; W. Baehr, None
References
- 1.Polans A, Baehr W, Palczewski K. Turned on by Ca2+ The physiology and pathology of Ca2+ binding proteins in the retina. Trends Neurosci. 1996;19:547–554. doi: 10.1016/s0166-2236(96)10059-x. [DOI] [PubMed] [Google Scholar]
- 2.Huang SH, Pittler SJ, Huang XH, Oliveira L, Berson EL, Dryja TP. Autosomal recessive retinitis pigmentosa caused by mutations in the β-subunit of rod cGMP phosphodiesterase. Nature Genet. 1995;11:468–471. doi: 10.1038/ng1295-468. [DOI] [PubMed] [Google Scholar]
- 3.McLaughlin ME, Sandberg MA, Berson EL, Dryja TP. Recessive mutations in the gene encoding the β-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nature Genet. 1993;4:130–134. doi: 10.1038/ng0693-130. [DOI] [PubMed] [Google Scholar]
- 4.Gal A, Orth U, Baehr W, Schwinger E, Rosenberg T. Heterozygous missense mutation in the rod cGMP phosphodiesterase β-subunit gene in autosomal dominant stationary night blindness. Nature Genet. 1994;7:64–68. doi: 10.1038/ng0594-64. [DOI] [PubMed] [Google Scholar]
- 5.Perrault I, Rozet J-M, Calvas P, et al. Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nature Genet. 1996;14:461–464. doi: 10.1038/ng1296-461. [DOI] [PubMed] [Google Scholar]
- 6.Perrault I, Rozet JM, Gerber S, et al. Spectrum of retGC1 mutations in Leber's congenital amaurosis. Eur J Hum Genet. 2000;8:578–582. doi: 10.1038/sj.ejhg.5200503. [DOI] [PubMed] [Google Scholar]
- 7.Payne AM, Downes SM, Bessant DA, et al. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum Mol Genet. 1998;7:273–277. doi: 10.1093/hmg/7.2.273. [DOI] [PubMed] [Google Scholar]
- 8.Downes SM, Holder GE, Fitzke FW, et al. Autosomal dominant cone and cone-rod dystrophy with mutations in the guanylate cyclase activator 1A gene-encoding guanylate cyclase activating protein-1. Arch Ophthalmol. 2001;119:96–105. [PubMed] [Google Scholar]
- 9.Wilkie SE, Li Y, Deery EC, et al. Identification and functional consequences of a new mutation (E155G) in the gene for GCAP1 that causes autosomal dominant cone dystrophy. Am J Hum Genet. 2001;69:471–480. doi: 10.1086/323265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wissinger B, Gamer D, Jagle H, et al. CNGA3 mutations in hereditary cone photoreceptor disorders. Am J Hum Genet. 2001;69:722–737. doi: 10.1086/323613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohl S, Baumann B, Broghammer M, et al. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet. 2000;9:2107–2116. doi: 10.1093/hmg/9.14.2107. [DOI] [PubMed] [Google Scholar]
- 12.Dryja TP, Finn JT, Peng YW, McGee TL, Berson EL, Yau KW. Mutations in the gene encoding the β-subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA. 1995;92:10177–10181. doi: 10.1073/pnas.92.22.10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohl S, Marx T, Giddings I, et al. Total colorblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet. 1998;19:257–259. doi: 10.1038/935. [DOI] [PubMed] [Google Scholar]
- 14.Sundin OH, Yang JM, Li Y, et al. Genetic basis of total colorblindness among the Pingelapese islanders. Nat Genet. 2000;25:289–293. doi: 10.1038/77162. [DOI] [PubMed] [Google Scholar]
- 15.Bareil C, Hamel CP, Delague V, Arnaud B, Demaille J, Claustres M. Segregation of a mutation in CNGB1 encoding the beta-subunit of the rod cGMP-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum Genet. 2001;108:328–334. doi: 10.1007/s004390100496. [DOI] [PubMed] [Google Scholar]
- 16.Rattner A, Sun H, Nathans J. Molecular genetics of human retinal disease. Annu Rev Genet. 1999;33:89–131. doi: 10.1146/annurev.genet.33.1.89. [DOI] [PubMed] [Google Scholar]
- 17.Dryja TP, Li T. Molecular genetics of retinitis pigmentosa. Hum Mol Genet. 1995;4:1739–1743. doi: 10.1093/hmg/4.suppl_1.1739. [DOI] [PubMed] [Google Scholar]
- 18.Imanishi Y, Yang L, Sokal I, Filipek S, Palczewski K, Baehr W. Diversity of guanylate cyclase-activating proteins (GCAPs) in teleost fish: characterization of three novel GCAPs (GCAP4, GCAP5, GCAP7) from zebrafish (Danio rerio) and prediction of eight GCAPs (GCAP1-8) in pufferfish (Fugu rubripes) J Mol Evol. 2004;59:204–217. doi: 10.1007/s00239-004-2614-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imanishi Y, Li N, Sowa ME, et al. Characterization of retinal guanylate cyclase-activating protein 3 (GCAP3) from zebrafish to man. Eur J Neurosci. 2002;15:63–78. doi: 10.1046/j.0953-816x.2001.01835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Surguchov A, Bronson JD, Banerjee P, et al. The human GCAP1 and GCAP2 genes are arranged in a tail-to-tail array on the short arm of chromosome 6 (p21.1) Genomics. 1997;39:312–322. doi: 10.1006/geno.1996.4513. [DOI] [PubMed] [Google Scholar]
- 21.Haeseleer F, Sokal I, Li N, et al. Molecular characterization of a third member of the guanylyl cyclase- activating protein subfamily. J Biol Chem. 1999;274:6526–6535. doi: 10.1074/jbc.274.10.6526. [DOI] [PubMed] [Google Scholar]
- 22.Dizhoor AM, Boikov SG, Olshevskaya E. Constitutive activation of photoreceptor guanylate cyclase by Y99C mutant of GCAP-1. J Biol Chem. 1998;273:17311–17314. doi: 10.1074/jbc.273.28.17311. [DOI] [PubMed] [Google Scholar]
- 23.Sokal I, Li N, Surgucheva I, et al. GCAP1(Y99C) mutant is constitutively active in autosomal dominant cone dystrophy. Mol Cell. 1998;2:129–133. doi: 10.1016/s1097-2765(00)80121-5. [DOI] [PubMed] [Google Scholar]
- 24.Falke JJ, Drake SK, Hazard AL, Peerson OB. Molecular tuning of ion binding to calcium signaling proteins. Q Rev Biophys. 1994;27:219–290. doi: 10.1017/s0033583500003012. [DOI] [PubMed] [Google Scholar]
- 25.Newbold RJ, Deery EC, Walker CE, et al. The destabilization of human GCAP1 by a proline to leucine mutation might cause cone-rod dystrophy. Hum Mol Genet. 2001;10:47–54. doi: 10.1093/hmg/10.1.47. [DOI] [PubMed] [Google Scholar]
- 26.Sokal I, Li N, Verlinde CL, Haeseleer F, Baehr W, Palczewski K. Calcium-binding proteins in the retina: from discovery to etiology of human disease. Biochim Biophys Acta. 2000;1498:233–251. doi: 10.1016/s0167-4889(00)00099-9. [DOI] [PubMed] [Google Scholar]
- 27.Payne AM, Downes SM, Bessant DA, et al. Genetic analysis of the guanylate cyclase activator 1B (GUCA1B) gene in patients with autosomal dominant retinal dystrophies. J Med Genet. 1999;36:691–693. [PMC free article] [PubMed] [Google Scholar]
- 28.Kachi S, Nishizawa Y, Olshevskaya E, et al. Detailed localization of photoreceptor guanylate cyclase activating protein-1 and -2 in mammalian retinas using light and electron microscopy. Exp Eye Res. 1999;68:465–473. doi: 10.1006/exer.1998.0629. [DOI] [PubMed] [Google Scholar]
- 29.Otto-Bruc A, Fariss RN, Haeseleer F, et al. Localization of guanylate cyclase activating protein 2 in mammalian retinas. Proc Natl Acad Sci USA. 1997;94:4727–4732. doi: 10.1073/pnas.94.9.4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berson EL, Gouras P, Gunkel RD. Rod responses in retinitis pigmentosa, dominantly inherited. Arch Ophthalmol. 1968;80:355–388. doi: 10.1001/archopht.1968.00980050060009. [DOI] [PubMed] [Google Scholar]
- 31.Andreasson SO, Sandberg MA, Berson EL. Narrow-band filtering for monitoring low-amplitude cone electroretinograms in retinitis pigmentosa. Am J Ophthalmol. 1988;105:500–503. doi: 10.1016/0002-9394(88)90241-3. [DOI] [PubMed] [Google Scholar]
- 32.Reichel E, Bruce AM, Sandberg MA, Berson EL. An electroretinographic and molecular genetic study of X-linked cone degeneration. Am J Ophthalmol. 1989;108:540–547. doi: 10.1016/0002-9394(89)90431-5. [DOI] [PubMed] [Google Scholar]
- 33.Subbaraya I, Ruiz CC, Helekar BS, et al. Molecular characterization of human and mouse photoreceptor guanylate cyclase activating protein (GCAP) and chromosomal localization of the human gene. J Biol Chem. 1994;269:31080–31089. [PubMed] [Google Scholar]
- 34.Sokal I, Alekseev A, Baehr W, Haeseleer F, Palczewski K. Soluble fusion proteins between single transmembrane photoreceptor guanylyl cyclases and their activators. Biochemistry. 2002;41:251–257. doi: 10.1021/bi015606u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorczyca WA, Polans AS, Surgucheva I, Subbaraya I, Baehr W, Palczewski K. Guanylyl cyclase activating protein: a calcium-sensitive regulator of phototransduction. J Biol Chem. 1995;270:22029–22036. doi: 10.1074/jbc.270.37.22029. [DOI] [PubMed] [Google Scholar]
- 36.Rudnicka-Nawrot M, Surgucheva I, Hulmes JD, et al. Changes in biological activity and folding of guanylate cyclase-activating protein 1 as a function of calcium. Biochemistry. 1998;37:248–257. doi: 10.1021/bi972306x. [DOI] [PubMed] [Google Scholar]
- 37.Papermaster DS. Preparation of retinal rod outer segments. Meth Enzymol. 1982;81:48–52. doi: 10.1016/s0076-6879(82)81010-0. [DOI] [PubMed] [Google Scholar]
- 38.Otto-Bruc A, Buczylko J, Surgucheva I, et al. Functional reconstitution of photoreceptor guanylate cyclase with native and mutant forms of guanylate cyclase activating protein 1. Biochemistry. 1997;36:4295–4302. doi: 10.1021/bi963000d. [DOI] [PubMed] [Google Scholar]
- 39.Schoenmakers TJ, Visser GJ, Flik G, Theuvenet AP. CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. Bio/Tech. 1992;12:870–879. [PubMed] [Google Scholar]
- 40.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 41.Berson EL, Gouras P, Gunkel RD. Progressive cone degeneration, dominantly inherited. Arch Ophthalmol. 1968;80:77–83. doi: 10.1001/archopht.1968.00980050079011. [DOI] [PubMed] [Google Scholar]
- 42.Berson EL, Goldstein EB. Recovery of the human early receptor potential during dark adaptation in hereditary retinal disease. Vision Res. 1970;10:219–226. doi: 10.1016/0042-6989(70)90127-6. [DOI] [PubMed] [Google Scholar]
- 43.Goldstein EB, Berson EL. Rod and cone contributions to the human early receptor potential. Vision Res. 1970;10:207–218. doi: 10.1016/0042-6989(70)90126-4. [DOI] [PubMed] [Google Scholar]
- 44.To K, Adamian M, Jakobiec FA, Berson EL. Histopathologic and immunohistochemical study of dominant cone degeneration. Am J Ophthalmol. 1998;126:140–142. doi: 10.1016/s0002-9394(98)00085-3. [DOI] [PubMed] [Google Scholar]
- 45.Sokal I, Otto-Bruc AE, Surgucheva I, et al. Conformational changes in guanylyl cyclase-activating protein 1 (GCAP1) and its tryptophan mutants as a function of calcium concentration. J Biol Chem. 1999;274:19829–19837. doi: 10.1074/jbc.274.28.19829. [DOI] [PubMed] [Google Scholar]
- 46.Li N, Sokal I, Bronson JD, Palczewski K, Baehr W. Identification and functional regions of guanylate cyclase-activating protein 1 (GCAP1) using GCAP1/GCIP chimeras. Biol Chem. 2001;382:1179–1188. doi: 10.1515/BC.2001.148. [DOI] [PubMed] [Google Scholar]
- 47.Sokal I, Li N, Klug CS, et al. Calcium-sensitive regions of GCAP1 as observed by chemical modifications, fluorescence and EPR spectroscopies. J Biol Chem. 2001;276:43361–43373. doi: 10.1074/jbc.M103614200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ray S, Zozulya S, Niemi GA, et al. Cloning, expression, and crystallization of recoverin, a calcium sensor in vision. Proc Natl Acad Sci USA. 1992;89:5705–5709. doi: 10.1073/pnas.89.13.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ames JB, Tanaka T, Stryer L, Ikura M. Secondary structure of myristoylated recoverin determined by three-dimensional hetero-nuclear NMR: implications for the calcium-myristoyl switch. Biochemistry. 1994;33:10743–10753. doi: 10.1021/bi00201a023. [DOI] [PubMed] [Google Scholar]
- 50.Ames JB, Dizhoor AM, Ikura M, Palczewski K, Stryer L. Three-dimensional structure of guanylyl cyclase activating protein-2, a calcium-sensitive modulator of photoreceptor guanylyl cyclases. J Biol Chem. 1999;274:19329–19337. doi: 10.1074/jbc.274.27.19329. [DOI] [PubMed] [Google Scholar]