

Abstract
New developments in molecular and cellular biology are rapidly increasing knowledge of the mechanisms of androgen action in the prostate. Molecular profiling of recurrent cancer after androgen deprivation therapy has revealed genetic changes related to cell survival and proliferation. It is now clear that loss of the androgen receptor (AR) does not explain androgen-independent growth of a majority of recurrent cancers. Rather, overexpression, mutation, and ligand-independent activation of the AR may confer enhanced sensitivity to androgen or other growth factors. Therefore, the term “androgen-independent” is often a misnomer. This emerging concept is the basis of novel strategies to prevent or slow recurrence.
Key words: Prostate cancer, Androgen, Hormonal therapy
Events leading to the discovery of hormonal ablation therapy for prostate cancer span a period of perhaps several hundred years, culminating in the classic work of Huggins in 1941.1 Androgen deprivation remains the standard of care for men with advanced prostate cancer, with symptomatic or objective responses in 80% of patients. Invariably, progression to androgen-independent disease occurs within a few years. The mechanisms by which tumors recur after androgen deprivation therapy are becoming clear. Several comprehensive reviews discuss current ideas about progression of prostate cancer to androgen independence.2–4 This review focuses on the most recent findings on this topic.
Early Events in the Prostate After Androgen Ablation
An in-depth understanding of the events that occur in the prostate following androgen ablation is undoubtedly essential for identifying the mechanisms by which cancers become resistant to hormonal deprivation. Animal model systems have been useful for studying androgen action, and molecular and cellular events that occur in the normal rat prostate following castration have been extensively documented.5,6 Tissue regression that occurs following androgen deprivation is accompanied by the temporal regulation of several hundred genes, many of them prostate specific and expressed in the epithelium. For the most part, changes in gene expression are related to apoptosis that occurs in the differentiated secretory cells of the normal epithelium in response to androgen depletion. These cells express androgen receptor (AR), and androgen activation of the AR is required for differentiation as well as survival of these cells. Basal epithelial cells for the most part do not express AR, and they are not dependent on androgen for survival. When androgen is restored, the basal cells proliferate and regenerate the differentiated epithelium, presumably through paracrine-acting growth factors mediated by androgenic action on the stroma.
The nature of androgen action on human prostatic cells has by necessity been learned mainly from cultured cells and xenografts. However, clinical application of neoadjuvant androgen ablation therapy has provided a means of studying the events that occur in the human prostate within a relatively short period of time after androgen depletion. By examining specimens obtained by biopsy or radical prostatectomy after 4 to 12 weeks of therapy, investigators have been able to describe a number of events that occur in normal human prostatic tissues as well as in prostatic intraepithelial neoplasia and in tumors. Many of the molecular and cellular changes that occur in the rodent prostate after androgen deprivation also occur in the human prostate. Secretory epithelial cells die by apoptosis, and basal cells remain intact (Figure 1).
Figure 1.

Events occurring in the normal prostate following androgen ablation. At T0, the epithelium and vasculature are intact and androgen is bound to the AR of secretory epithelial cells. At T6h after androgen depletion, the earliest observed event is degeneration of the vasculature. By T48h, secretory epithelial cells undergo apoptosis. At later time points (T>6d), only basal epithelial cells remain.
The effects of neoadjuvant androgen deprivation therapy on cancer in comparison to normal prostatic tissue are interesting to consider. One important finding is that cancer cells may not undergo apoptosis to the same extent that the normal prostatic epithelium does, as there is considerable evidence of residual cancer remaining after androgen deprivation.7–12 This finding is in agreement with the concept that expression of antiapoptotic factors is involved in the development of androgen-independent tumors. If androgen is a survival factor, then expression of factors that protect a cancer cell from androgen-ablation-mediated apoptosis may be a critical feature of the androgen-independent phenotype.13 In this regard, recent observations have provided additional information about the events leading to apoptosis of prostatic epithelial cells upon depletion of androgen. One of the earliest events, occurring prior to the initiation of epithelial apoptosis, is degeneration of the prostatic vasculature. This has been observed in animal models as well as in the human prostate.14,15 This event is mediated at least in part by decreased expression of vascular endothelial growth factor (VEGF), an androgen-regulated gene, in the prostatic epithelium and stroma.16–18 These observations suggest that hypoxia caused by degeneration of the vasculature may be the event that causes death of the epithelium. Those cancer cells that resist apoptosis upon androgen deprivation may therefore do so through expression of factors that protect from hypoxia, which may be different factors from those that protect from apoptosis induced by other stimuli.
In conjunction with characterizing the molecular and cellular changes that occur in prostate cells when androgen is removed, investigators have also tried to delineate the processes that occur upon restoration of androgen. Despite the fact that androgen is considered the major growth factor for prostatic epithelial cells, signaling pathways that mediate androgen-stimulated cell proliferation are poorly understood. Studies that address this issue implicate cell cycle regulators and transcription factors such as CDK4, CDK2, p16, p21, p27, and AP-1.19–22 Other investigations have found roles for protein kinase CK2 and mitogen-activated protein kinase (MAPK) in mitogenic signaling by androgen.23,24 Tactics for sorting out the signaling pathways triggered by androgen include a variety of methods of analyzing the gene expression profiles of control and androgen-treated cell lines such as LNCaP.25
Alterations in the AR Gene in Androgen-Independent Prostate Cancer
Contrary to preconceived notions, loss of the AR gene or its expression is not a common feature of androgen-independent cancer. In fact, a high percentage of androgen-independent cancers appear to maintain expression of the AR gene.26 However, many of these cancers have genetic alterations that change the activity of the receptor (Figure 2). Amplification of the AR is one type of genetic modification, reported to occur in about 20% to 30% of locally recurrent or metastatic androgen-independent prostate cancers.27,28 Amplification of the AR gene was verified to correlate with more expression of RNA transcripts of AR.28 Even hormone-refractory tumors without amplification of the AR gene show more expression of AR mRNA transcripts or protein than androgen-dependent tumors do, suggesting that other mechanisms besides gene amplification may lead to overexpression of the AR.28,29 Investigation of the properties of the AR in model systems of androgen-independent cancer showed that high-level expression of AR was associated with increased stability, constitutive nuclear localization, and increased sensitivity to growth promotion by low levels of dihydrotestosterone.30 Therefore, high levels of AR may allow prostate cancer cells to proliferate in response to the low levels of androgen that remain after many types of androgen-deprivation therapy.
Figure 2.
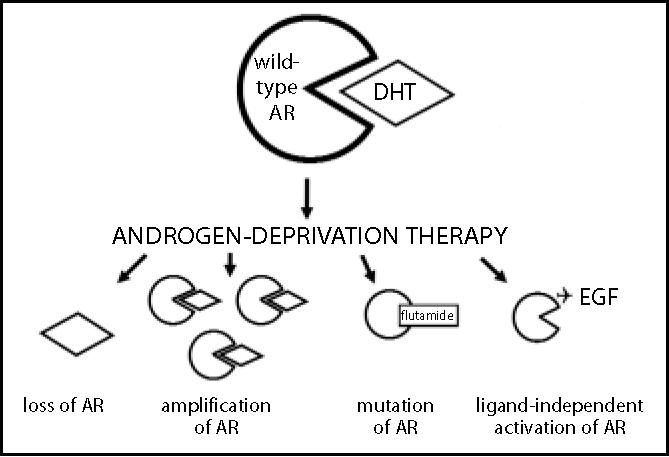
Status of the androgen receptor (AR) in recurrent, hormone-refractory prostate cancer. In a subset of cancers, AR is absent and growth is driven by AR-independent mechanisms. In the majority of cancers, however, AR is present. Amplification of AR makes cells supersensitive to low levels of androgen. Mutation may reduce ligand specificity, permitting activation of the AR by non-androgens. Activity of the wild-type AR may be driven by high levels of peptide growth factors that activate AR by ligand-independent mechanisms.
Activity of AR is dependent on a variety of nuclear receptor coactivators. Analysis of the expression of these cofactors in prostatic tissues has just begun, but a recent study suggests that altered expression of AR coactivators may occur in recurrent cancer. Gregory et al found that overexpression of two coactivators, transcriptional intermediary factor 2 (TIF2) and steroid receptor coactivator 1 (SRC1), accompanied overexpression of the AR in hormone-refractory cancers.29 Furthermore, they demonstrated that overexpression of these coactivators increased AR transactivation of downstream gene targets at physiological concentrations of adrenal androgen. Therefore, overexpression of coactivators is another mechanism by which prostate cancer may continue to grow via androgen-mediated activity in the presence of low levels of androgen.
Mutations in the AR occur in a subset of recurrent cancers, perhaps at a frequency as high as 40% to 50%.31 Buchanan et al recently reviewed the mutations that have been found in the AR and discussed the biological consequences on AR activity for each of these mutations.32 Most of these mutations result in the acquisition of a gain of function for the AR, usually manifested by reduced discrimination for ligand-dependent activation. There is evidence that the type of androgen-ablation therapy used may provide selective pressure for specific mutations. In one study, mutations in the AR were found in 5 of 16 recurrent tumors following therapy with flutamide.33 Molecular studies showed that this particular mutation allowed flutamide to act as an agonist of the AR. Of those patients treated only with monotherapy, only 1 of 17 recurrent tumors had a mutated AR, and this mutation did not confer agonistic activity on flutamide.
Loss of AR expression occurs in some cancers, at a frequency estimated at 20% to 30% by some investigators.34 Methylation silences the AR gene promoter in many of these cases and is linked to loss of expression of AR.34 In these cases, growth of recurrent cancers would indeed be “androgen independent.”
Ligand-Independent Activation of the Androgen Receptor
Steroid hormone receptors, including the AR, can be activated by nonsteroidal factors. Insulin-like growth factor I (IGF-I), epidermal growth factor (EGF), keratinocyte growth factor (KGF), and interleukin-6 (IL-6) are cytokines produced by prostatic stromal or epithelial cells that activate AR.35–37 This effect is specific and is not mediated by certain other growth factors, such as IGF-II or fibroblast growth factor 2 (FGF-2). Activation of the androgen receptor by peptide growth factors is blocked by the androgen receptor antagonist, casodex. These growth factors do not directly bind to androgen receptors but instead activate the receptors via signal transducers such as MAPK and protein kinase A.38–40
Her-2/neu is a nonsteroidal factor that is gaining considerable attention for its possible role in androgen-independent prostate cancer. Overexpression of Her-2/neu, a receptor tyrosine kinase, has been associated with progression to androgen-independent disease.41 Her-2/neu activates the AR by a ligand-independent mechanism,42 like the aforementioned growth factors, and overexpression in a xenograft model of human prostate cancer led to androgen-independent growth.43 These findings are particularly relevant to therapy because antibodies that block Her-2/neu are available and are being tested in clinical trials.
Ligand-independent activation of the AR may not always lead to events that promote cancer growth. The differentiation agent butyrate inhibits the growth of prostate cancer cells yet affects ligand-independent activation of the AR.44 It may be possible to identify agents that differentially promote AR-mediated signaling pathways leading to differentiation instead of proliferation.
Molecular Features of Androgen-Independent Prostate Cancer
Investigators have taken advantage of androgen-responsive prostate cancer cell lines or xenografts to study the development of androgen-independent phenotypes. Growing of cells in vitro or in vivo in androgen-depleted conditions gives rise to androgen-independent variants with an array of different features. Comparative analyses have revealed a number of changes that occurred in the cell lines or xenografts that were selected for androgen-independent growth.45–47 Several of these changes deserve special consideration because of their biological implications. Caveolin, endothelin, and/or bcl-2, for example, are overexpressed in androgen-independent prostate cancer cells or tumors.3,48,49 These proteins are involved in cell survival and resistance to apoptosis. A direct link between changes in expression of caveolin and androgen independence is suggested by restoration of androgen sensitivity in prostate cancer cells by suppression of caveolin expression.50 The association of antiapoptotic genes with recurrent prostate cancer strongly suggests that these genes allow cancer cells to escape apoptosis mediated by androgen deprivation.
Recently, investigators have focused on identifying the earliest changes that occur as prostate cancer cells progress from androgen responsiveness to androgen independence. For example, Myers et al reported that fatty acid synthase was an early marker of progression to androgen independence in the prostate cancer xenograft, CWR22.51 It is interesting that expression of fatty acid synthase originally depends on androgen.52 In fact, a number of studies describe the general phenomenon that the genes differentially expressed in androgen-independent cells are those that were originally regulated by androgen.53 It is also important to note that many sublines selected to grow in the absence of androgen in fact remain androgen responsive, in that they exhibit molecular and cellular changes in response to androgen.54 This is possibly due to the continued expression of AR in these cells. Similarly to the spectrum of changes in the AR in hormone-refractory tumors, prostate cancer cell lines or xenografts show loss of AR, expression of wild-type AR, mutated AR, or amplified AR.55
A transgenic mouse model of prostate cancer driven by the prostate-restricted expression of the SV40 T-antigen revealed relationships between mutation of the AR and emergence of androgen-independent prostate cancer.56 In this model system, following androgen ablation at 12 weeks of age, androgen-independent cancers reproducibly emerge. The majority of these tumors have mutations in the AR, mostly in the AR transactivation domain. All AR variants in this study maintained a strong sensitivity for androgen, and several had increased activity in the absence of ligand. Androgen-independent cancers also emerged in intact animals, and interestingly, these also had mutated AR but the mutations were specifically in the AR ligand-binding domain. These results support the emerging concept that the hormonal environment may select for specific types of mutations in the AR gene that provide a growth advantage in that particular environment.
Androgen-independent tumors derived from patients have also been compared to androgen-dependent tumors in order to identify features consistently associated with androgen independence.57 As might have been expected, a multitude of changes involving factors associated with cell cycle regulation and/or apoptosis have been reported in androgen-independent cancers. Decreased levels of tumor suppressor genes such as RB1 are among the numerous changes reported.58 Certain tumor suppressor genes may in fact directly oppose androgen action. A recent study provided evidence that the tumor suppressor PTEN/MMAC1/TEP-1 represses the transcriptional activity of the AR through inactivation of a protein kinase known as Akt.59 This implies that when PTEN activity is diminished, as it frequently is in advanced prostate cancer due to allelic loss of the PTEN gene, androgen action would be left unopposed. This scenario provides an additional molecular basis for the development of androgen independence and suggests new therapeutic targets, such as Akt.
An additional intriguing observation is that markers of the normal basal epithelium of the prostate, expressed at low or no levels in low-grade or early-stage prostate cancer, appear in androgen-independent prostate cancer.60 Efforts continue to try to sort out which of these changes are causative and which may be secondary or related to increased genetic instability in advanced prostate cancer.
Predictive Markers of Response to Androgen Deprivation Therapy
Molecular markers to predict an individual’s response to androgen ablation therapy are being sought. Given the role of the AR in development of androgen-independent prostate cancer, investigators have evaluated features of the AR or other factors related to androgen action as potential predictive markers of relapse after androgen ablation. Prins et al reported that image analysis of AR immunostaining accurately predicted response to hormonal therapy.61 The predictive feature of the AR was not the intensity of staining per se or the percentage of positive cells but rather was the degree of heterogeneous expression. Others had described a similar phenomenon several years ago.62 On the other hand, Palmberg et al reported that AR gene amplification at primary progression predicted the response to combined androgen blockade as second-line therapy for advanced prostate cancer.63
Other investigators found that polymorphisms in the AR gene not only were associated with prostate cancer risk but also were significant predictors of relapse and survival after androgen ablation. Both the CAG and the GGC repeat lengths in the AR gene were associated with poor response to androgen ablation.64,65
Markers other than the AR have been related to outcome after androgen deprivation therapy. Baretton et al observed that expression of the cell cycle regulatory protein p21WAF1CIP1 had prognostic significance.66 High levels of expression of p21 in tumor tissues before androgen deprivation were associated with poor outcome after therapy. Another factor that regulates cellular growth and differentiation, transforming growth factor-β(TGFβ), was also associated with response to androgen deprivation. Upregulation of TGFβ and its receptor, TβRII, occurs when normal prostatic epithelial cells undergo apoptosis upon the removal of androgen. Prostate cancer that responded to castration by undergoing apoptosis showed an induction of TGFβ and its receptor; cancers that did not respond to castration did not exhibit upregulation of TGFβ and TβRII.11
Stattin et al also evaluated molecular markers associated with apoptosis in prostate cancers before and after castration.67 These investigators were unable to find a correlation between expression of p53, bcl-2, bax, or fas and response to castration, but they did find that the development of a regressive morphology in cancers after castration was strongly related to clinical outcome.
Strategies to Delay Progression to Androgen Independence
As more information is gained about the features of prostate cancer as it progresses from androgen dependent to androgen independent, strategies based on this new knowledge are being developed to slow or prevent such progression. Several strategies are based on the apparent relationship between antiapoptotic factors and androgen independence. For example, expression of bcl-2, an antiapoptotic factor, is significantly more frequent in advanced, androgen-independent prostate cancer than in untreated cancers.68,69 Gleave et al used a prostate cancer xenograft model to show that progression to androgen independence was delayed by adjuvant treatment with antisense bcl-2 oligonucleotides after castration.70
IGF-binding protein 5 (IGFBP-5), like bcl-2, is upregulated by castration. IGFBP-5 potentiates mitogenic activity of the IGFs and accelerates progression to androgen independence in prostate cancer models. Blocking IGFBP-5, like blocking bcl-2, slowed progression to androgen independence in an experimental model.71 Targeting other antiapoptotic factors, such as testosterone-repressed prostate message-2 (TRPM-2), shows similar promising results in preclinical models. 72 Investigators also continue to seek evidence that chemotherapy at the time of androgen ablation can slow or prevent progression to androgen independence.73–76
If androgen-independent growth is in fact being driven by action of the AR, then strategies that block this activity might be effective against recurrent cancer. Drugs being tested include tyrosine kinase inhibitors that block proliferation stimulated by growth factors and/or androgen.77 Down-regulation of the AR might be another effective strategy. This could be accomplished by molecular approaches, such as through application of antisense AR, or by epigenetic means. For example, quercetin and flufenamic acid, compounds that may have chemopreventive activity, were reported to inhibit the expression of the AR.78,79
Conclusions
Based on new developments in our understanding of the molecular phenotype of recurrent prostate cancer, it is perhaps no longer correct to refer to “androgen-independent” disease. In fact, growth of the majority of recurrent cancers quite possibly continues to be driven by androgen. Many recurrent tumors retain AR expression, and there is evidence for mutated AR with altered specificity, amplified AR that confers increased sensitivity, and ligand-independent activation of AR by peptide growth factors. Upregulation of antiapoptotic genes and alterations in cell cycle regulatory molecules contribute to the aggressive phenotype of advanced prostate cancer. Recognition of the molecular basis of recurrent prostate cancer provides a starting point for new strategies to delay or prevent progression.
Main Points.
Androgen deprivation, the standard of care for advanced prostate cancer, achieves initial symptomatic or objective responses in 80% of patients, but disease invariably progresses to androgen independence within a few years.
After androgen deprivation, secretory epithelial cells in the prostate die by apoptosis and basal cells remain intact.
A high percentage of androgen-independent cancers appear to maintain expression of the AR gene, but many have genetic alterations that change receptor activity.
Altered expression of AR coactivators may occur in recurrent cancer, and AR mutations may occur in as many as 40% to 50% of such cancers.
Proteins involved in cell survival and resistance to apoptosis are overexpressed in androgen-independent prostate cancer cells or tumors.
Strategies for delaying progression to androgen independence may involve targeting of antiapoptotic factors, use of chemotherapy at the time of androgen ablation, or blockage or downregulation of AR activity.
References
- 1.Lytton B. Prostate cancer: a brief history and the discovery of hormonal ablation treatment. J Urol. 2001;165:1859–1862. [PubMed] [Google Scholar]
- 2.Koivisto P, Kolmer M, Visakorpi T, Kallioniemi OP. Androgen receptor gene and hormonal therapy failure of prostate cancer. Am J Pathol. 1998;152:1–9. [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson JB. Alternatives to death: understanding androgen-independent prostate cancer. Nat Med. 1998;4:1011–1012. doi: 10.1038/1998. [DOI] [PubMed] [Google Scholar]
- 4.Tang DG, Porter AT. Target to apoptosis: a hopeful weapon for prostate cancer. Prostate. 1997;32:284–293. doi: 10.1002/(sici)1097-0045(19970901)32:4<284::aid-pros9>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 5.Bruyninx M, Ammar H, Reiter E, et al. Genes upregulated during castration-induced rat prostatic apoptosis: cloning and characterization of new cDNAs. BJU Int. 2000;85:1134–1142. doi: 10.1046/j.1464-410x.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- 6.Gubbay J, Doyle JP, Skinner M, Heintz N. Changing patterns of gene expression identify multiple steps during regression of rat prostate in vivo. Endocrinology. 1998;139:2935–2943. doi: 10.1210/endo.139.6.6075. [DOI] [PubMed] [Google Scholar]
- 7.Armas OA, Aprikian AG, Melamed J, et al. Clinical and pathobiological effects of neoadjuvant total androgen ablation therapy on clinically localized prostatic adenocarcinoma. Am J Surg Pathol. 1994;18:979–991. doi: 10.1097/00000478-199410000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Bostwick DG. Immunohistochemical changes in prostate cancer after androgen deprivation therapy. Mol Urol. 2000;4:101–106. discussion 107. [PubMed] [Google Scholar]
- 9.de Matteis A, Guidi A, Di Paolo B, et al. Endothelin-1 in human prostatic carcinoma treated with androgen withdrawal: an immunohistochemical study. Cancer. 2001;91:1933–1939. doi: 10.1002/1097-0142(20010515)91:10<1933::aid-cncr1216>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 10.Westin P, Stattin P, Damber JE, Bergh A. Castration therapy rapidly induces apoptosis in a minority and decreases cell proliferation in a majority of human prostatic tumors. Am J Pathol. 1995;146:1368–1375. [PMC free article] [PubMed] [Google Scholar]
- 11.Wikstrom P, Westin P, Stattin P, et al. Early castration-induced upregulation of transforming growth factor beta1 and its receptors is associated with tumor cell apoptosis and a major decline in serum prostate-specific antigen in prostate cancer patients. Prostate. 1999;38:268–277. doi: 10.1002/(sici)1097-0045(19990301)38:4<268::aid-pros2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 12.Wood DP, Jr, Beaman A, Banerjee M, et al. Effect of neoadjuvant androgen deprivation on circulating prostate cells in the bone marrow of men undergoing radical prostatectomy. Clin Cancer Res. 1998;4:2119–2123. [PubMed] [Google Scholar]
- 13.Lu S, Tsai SY, Tsai MJ. Molecular mechanisms of androgen-independent growth of human prostate cancer LNCaP-AI cells. Endocrinology. 1999;140:5054–5059. doi: 10.1210/endo.140.11.7086. [DOI] [PubMed] [Google Scholar]
- 14.Franck-Lissbrant I, Haggstrom S, Damber JE, Bergh A. Testosterone stimulates angiogenesis and vascular regrowth in the ventral prostate in castrated adult rats. Endocrinology. 1998;139:451–456. doi: 10.1210/endo.139.2.5683. [DOI] [PubMed] [Google Scholar]
- 15.Shabisgh A, Tanji N, D’Agati V, et al. Early effects of castration on the vascular system of the rat ventral prostate gland. Endocrinology. 1999;140:1920–1926. doi: 10.1210/endo.140.4.6644. [DOI] [PubMed] [Google Scholar]
- 16.Burchardt M, Burchardt T, Chen MW, et al. Vascular endothelial growth factor-A expression in the rat ventral prostate gland and the early effects of castration. Prostate. 2000;43:184–194. doi: 10.1002/(sici)1097-0045(20000515)43:3<184::aid-pros4>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 17.Haggstrom S, Wikstrom P, Bergh A, Damber JE. Expression of vascular endothelial growth factor and its receptors in the rat ventral prostate and Dunning R3327 PAP adenocarcinoma before and after castration. Prostate. 1998;36:71–79. doi: 10.1002/(sici)1097-0045(19980701)36:2<71::aid-pros1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 18.Joseph IB, Nelson JB, Denmeade SR, Isaacs JT. Androgens regulate vascular endothelial growth factor content in normal and malignant prostatic tissue. Clin Cancer Res. 1997;3:2507–2511. [PubMed] [Google Scholar]
- 19.Fribourg AF, Knudsen KE, Strobeck MW, et al. Differential requirements for ras and the retinoblastoma tumor suppressor protein in the androgen dependence of prostatic adenocarcinoma cells. Cell Growth Differ. 2000;11:361–372. [PubMed] [Google Scholar]
- 20.Lu S, Jenster G, Epner DE. Androgen induction of cyclin-dependent kinase inhibitor p21 gene: role of androgen receptor and transcription factor Sp1 complex. Mol Endocrinol. 2000;14:753–760. doi: 10.1210/mend.14.5.0461. [DOI] [PubMed] [Google Scholar]
- 21.Ripple MO, Henry WF, Schwarze SR, et al. Effect of antioxidants on androgen-induced AP-1 and NF-kappaB DNA-binding activity in prostate carcinoma cells. J Natl Cancer Inst. 1999;91:1227–1232. doi: 10.1093/jnci/91.14.1227. [DOI] [PubMed] [Google Scholar]
- 22.Waltregny D, Leav I, Signoretti S, et al. Androgen-driven prostate epithelial cell proliferation and differentiation in vivo involve the regulation of p27. Mol Endocrinol. 2001;15:765–782. doi: 10.1210/mend.15.5.0640. [DOI] [PubMed] [Google Scholar]
- 23.Guo C, Luttrell LM, Price DT. Mitogenic signaling in androgen sensitive and insensitive prostate cancer cell lines. J Urol. 2000;163:1027–1032. [PubMed] [Google Scholar]
- 24.Peterziel H, Mink S, Schonert A, et al. Rapid signalling by androgen receptor in prostate cancer cells. Oncogene. 1999;18:6322–6329. doi: 10.1038/sj.onc.1203032. [DOI] [PubMed] [Google Scholar]
- 25.Xu LL, Su YP, Labiche R, et al. Quantitative expression profile of androgen-regulated genes in prostate cancer cells and identification of prostate-specific genes. Int J Cancer. 2001;92:322–328. doi: 10.1002/ijc.1196. [DOI] [PubMed] [Google Scholar]
- 26.Evans BA, Harper ME, Daniells CE, et al. Low incidence of androgen receptor gene mutations in human prostatic tumors using single strand conformation polymorphism analysis. Prostate. 1996;28:162–171. doi: 10.1002/(SICI)1097-0045(199603)28:3<162::AID-PROS3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 27.Bubendorf L, Kononen J, Koivisto P, et al. Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res. 1999;59:803–806. [PubMed] [Google Scholar]
- 28.Linja MJ, Savinainen KJ, Saramaki OR, et al. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–3555. [PubMed] [Google Scholar]
- 29.Gregory CW, He B, Johnson RT, et al. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–4319. [PubMed] [Google Scholar]
- 30.Gregory CW, Johnson RT, Jr, Mohler JL, et al. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–2898. [PubMed] [Google Scholar]
- 31.Bentel JM, Tilley WD. Androgen receptors in prostate cancer. J Endocrinol. 1996;151:1–11. doi: 10.1677/joe.0.1510001. [DOI] [PubMed] [Google Scholar]
- 32.Buchanan G, Greenberg NM, Scher HI, et al. Collocation of androgen receptor gene mutations in prostate cancer. Clin Cancer Res. 2001;7:1273–1281. [PubMed] [Google Scholar]
- 33.Taplin ME, Bubley GJ, Ko YJ, et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511–2515. [PubMed] [Google Scholar]
- 34.Kinoshita H, Shi Y, Sandefur C, et al. Methylation of the androgen receptor minimal promoter silences transcription in human prostate cancer. Cancer Res. 2000;60:3623–3630. [PubMed] [Google Scholar]
- 35.Culig Z, Hobisch A, Cronauer MV, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–5478. [PubMed] [Google Scholar]
- 36.Hobisch A, Eder IE, Putz T, et al. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998;58:4640–4645. [PubMed] [Google Scholar]
- 37.Lin DL, Whitney MC, Yao Z, Keller ET. Interleukin-6 induces androgen responsiveness in prostate cancer cells through up-regulation of androgen receptor expression. Clin Cancer Res. 2001;7:1773–1781. [PubMed] [Google Scholar]
- 38.Abreu-Martin MT, Chari A, Palladino AA, et al. Mitogen-activated protein kinase 1 activates androgen receptor-dependent transcription and apoptosis in prostate cancer. Mol Cell Biol. 1999;19:5143–5154. doi: 10.1128/mcb.19.7.5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen T, Wang LH, Farrar WL. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000;60:2132–2135. [PubMed] [Google Scholar]
- 40.Nazareth LV, Weigel NL. Activation of the human androgen receptor through a protein kinase A signaling pathway. J Biol Chem. 1996;271:19900–19907. doi: 10.1074/jbc.271.33.19900. [DOI] [PubMed] [Google Scholar]
- 41.Signoretti S, Montironi R, Manola J, et al. Her-2-neu expression and progression toward androgen independence in human prostate cancer. J Natl Cancer Inst. 2000;92:1918–1925. doi: 10.1093/jnci/92.23.1918. [DOI] [PubMed] [Google Scholar]
- 42.Wen Y, Hu MC, Makino K, et al. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000;60:6841–6845. [PubMed] [Google Scholar]
- 43.Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5:280–285. doi: 10.1038/6495. [DOI] [PubMed] [Google Scholar]
- 44.Sadar MD, Gleave ME. Ligand-independent activation of the androgen receptor by the differentiation agent butyrate in human prostate cancer cells. Cancer Res. 2000;60:5825–5831. [PubMed] [Google Scholar]
- 45.Patel BJ, Pantuck AJ, Zisman A, et al. CL1-GFP: an androgen independent metastatic tumor model for prostate cancer. J Urol. 2000;164:1420–1425. [PubMed] [Google Scholar]
- 46.Stubbs AP, Abel PD, Golding M, et al. Differentially expressed genes in hormone refractory prostate cancer: association with chromosomal regions involved with genetic aberrations. Am J Pathol. 1999;154:1335–1343. doi: 10.1016/S0002-9440(10)65387-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vaarala MH, Porvari K, Kyllonen A, Vihko P. Differentially expressed genes in two LNCaP prostate cancer cell lines reflecting changes during prostate cancer progression. Lab Invest. 2000;80:1259–1268. doi: 10.1038/labinvest.3780134. [DOI] [PubMed] [Google Scholar]
- 48.Liu AY, Corey E, Bladou F, et al. Prostatic cell lineage markers: emergence of BCL2+ cells of human prostate cancer xenograft LuCaP 23 following castration. Int J Cancer. 1996;65:85–89. doi: 10.1002/(SICI)1097-0215(19960103)65:1<85::AID-IJC15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 49.Tahir SA, Yang G, Ebara S, et al. Secreted caveolin-1 stimulates cell survival/clonal growth and contributes to metastasis in androgen-insensitive prostate cancer. Cancer Res. 2001;61:3882–3885. [PubMed] [Google Scholar]
- 50.Nasu Y, Timme TL, Yang G, et al. Suppression of caveolin expression induces androgen sensitivity in metastatic androgen-insensitive mouse prostate cancer cells. Nat Med. 1998;4:1062–1064. doi: 10.1038/2048. [DOI] [PubMed] [Google Scholar]
- 51.Myers RB, Oelschlager DK, Weiss HL, et al. Fatty acid synthase: an early molecular marker of progression of prostatic adenocarcinoma to androgen independence. J Urol. 2001;165:1027–1032. [PubMed] [Google Scholar]
- 52.Pizer ES, Pflug BR, Bova GS, et al. Increased fatty acid synthase as a therapeutic target in androgen-independent prostate cancer progression. Prostate. 2001;47:102–110. doi: 10.1002/pros.1052. [DOI] [PubMed] [Google Scholar]
- 53.Amler LC, Agus DB, LeDuc C, et al. Dysregulated expression of androgen-responsive and nonresponsive genes in the androgen-independent prostate cancer xenograft model CWR22-R1. Cancer Res. 2000;60:6134–6141. [PubMed] [Google Scholar]
- 54.Chang GT, Blok LJ, Steenbeek M, et al. Differentially expressed genes in androgendependent and -independent prostate carcinomas. Cancer Res. 1997;57:4075–4081. [PubMed] [Google Scholar]
- 55.Stearns ME, Ware JL, Agus DB, et al. Workgroup 2: human xenograft models of prostate cancer. Prostate. 1998;36:56–58. doi: 10.1002/(sici)1097-0045(19980615)36:1<56::aid-pros10>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 56.Han G, Foster BA, Mistry S, et al. Hormone status selects for spontaneous somatic androgen receptor variants that demonstrate specific ligand and cofactor dependent activities in autochthonous prostate cancer. J Biol Chem. 2001;276:11204–11213. doi: 10.1074/jbc.M008207200. [DOI] [PubMed] [Google Scholar]
- 57.Bubendorf L, Kolmer M, Kononen J, et al. Hormone therapy failure in human prostate cancer: analysis by complementary DNA and tissue microarrays. J Natl Cancer Inst. 1999;91:1758–1764. doi: 10.1093/jnci/91.20.1758. [DOI] [PubMed] [Google Scholar]
- 58.Mack PC, Chi SG, Meyers FJ, et al. Increased RB1 abnormalities in human primary prostate cancer following combined androgen blockade. Prostate. 1998;34:145–151. doi: 10.1002/(sici)1097-0045(19980201)34:2<145::aid-pros10>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 59.Li P, Nicosia SV, Bai W. Antagonism between PTEN/MMAC1/TEP-1 and androgen receptor in growth and apoptosis of prostatic cancer cells. J Biol Chem. 2001;276:20444–20450. doi: 10.1074/jbc.M010226200. [DOI] [PubMed] [Google Scholar]
- 60.Gil-Diez de Medina S, Salomon L, Colombel M, et al. Modulation of cytokeratin subtype, EGF receptor, and androgen receptor expression during progression of prostate cancer. Hum Pathol. 1998;29:1005–1012. doi: 10.1016/s0046-8177(98)90208-8. [DOI] [PubMed] [Google Scholar]
- 61.Prins GS, Sklarew RJ, Pertschuk LP. Image analysis of androgen receptor immunostaining in prostate cancer accurately predicts response to hormonal therapy. J Urol. 1998;159:641–649. [PubMed] [Google Scholar]
- 62.Sadi MV, Barrack ER. Androgen receptors and growth fraction in metastatic prostate cancer as predictors of time to tumour progression after hormonal therapy. Cancer Surv. 1991;11:195–215. [PubMed] [Google Scholar]
- 63.Palmberg C, Koivisto P, Kakkola L, et al. Androgen receptor gene amplification at primary progression predicts response to combined androgen blockade as second line therapy for advanced prostate cancer. J Urol. 2000;164:1992–1995. [PubMed] [Google Scholar]
- 64.Bratt O, Borg A, Kristoffersson U, et al. CAG repeat length in the androgen receptor gene is related to age at diagnosis of prostate cancer and response to endocrine therapy, but not to prostate cancer risk. Br J Cancer. 1999;81:672–676. doi: 10.1038/sj.bjc.6690746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Edwards SM, Badzioch MD, Minter R, et al. Androgen receptor polymorphisms: association with prostate cancer risk, relapse and overall survival. Int J Cancer. 1999;84:458–465. doi: 10.1002/(sici)1097-0215(19991022)84:5<458::aid-ijc2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 66.Baretton GB, Klenk U, Diebold J, et al. Proliferation- and apoptosis-associated factors in advanced prostatic carcinomas before and after androgen deprivation therapy: prognostic significance of p21/WAF1/CIP1 expression. Br J Cancer. 1999;80:546–555. doi: 10.1038/sj.bjc.6690390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stattin P, Westin P, Damber JE, Bergh A. Short-term cellular effects induced by castration therapy in relation to clinical outcome in prostate cancer. Br J Cancer. 1998;77:670–675. doi: 10.1038/bjc.1998.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Apakama I, Robinson MC, Walter NM, et al. bcl-2 overexpression combined with p53 protein accumulation correlates with hormone-refractory prostate cancer. Br J Cancer. 1996;74:1258–1262. doi: 10.1038/bjc.1996.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McDonnell TJ, Navone NM, Troncoso P, et al. Expression of bcl-2 oncoprotein and p53 protein accumulation in bone marrow metastases of androgen independent prostate cancer. J Urol. 1997;157:569–574. [PubMed] [Google Scholar]
- 70.Gleave M, Tolcher A, Miyake H, et al. Progression to androgen independence is delayed by adjuvant treatment with antisense Bcl-2 oligodeoxynucleotides after castration in the LNCaP prostate tumor model. Clin Cancer Res. 1999;5:2891–2898. [PubMed] [Google Scholar]
- 71.Miyake H, Pollak M, Gleave ME. Castrationinduced up-regulation of insulin-like growth factor binding protein-5 potentiates insulin-like growth factor-I activity and accelerates progression to androgen independence in prostate cancer models. Cancer Res. 2000;60:3058–3064. [PubMed] [Google Scholar]
- 72.Miyake H, Nelson C, Rennie PS, Gleave ME. Acquisition of chemoresistant phenotype by overexpression of the antiapoptotic gene testosterone-repressed prostate message-2 in prostate cancer xenograft models. Cancer Res. 2000;60:2547–2554. [PubMed] [Google Scholar]
- 73.George DJ, Dionne CA, Jani J, et al. Sustained in vivo regression of Dunning H rat prostate cancers treated with combinations of androgen ablation and Trk tyrosine kinase inhibitors, CEP-751 (KT-6587) or CEP-701 (KT-5555) Cancer Res. 1999;59:2395–2401. [PubMed] [Google Scholar]
- 74.Gleave ME, Sato N, Sadar M, et al. Butyrate analogue, isobutyramide, inhibits tumor growth and time to androgen-independent progression in the human prostate LNCaP tumor model. J Cell Biochem. 1998;69:271–281. [PubMed] [Google Scholar]
- 75.Hartley-Asp B, Vukanovic J, Joseph IB, et al. Anti-angiogenic treatment with linomide as adjuvant to surgical castration in experimental prostate cancer. J Urol. 1997;158:902–907. doi: 10.1097/00005392-199709000-00069. [DOI] [PubMed] [Google Scholar]
- 76.Miyake H, Hara S, Arakawa S, et al. Optimal timing and dosage of chemotherapy as a combined treatment with androgen withdrawal in the human prostate LNCaP tumour model. Br J Cancer. 2001;84:859–863. doi: 10.1054/bjoc.2000.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kondapaka BS, Reddy KB. Tyrosine kinase inhibitor as a novel signal transduction and antiproliferative agent: prostate cancer. Mol Cell Endocrinol. 1996;117:53–58. doi: 10.1016/0303-7207(95)03725-x. [DOI] [PubMed] [Google Scholar]
- 78.Xing N, Chen Y, Mitchell SH, Young CY. Quercetin inhibits the expression and function of the androgen receptor in LNCaP prostate cancer cells. Carcinogenesis. 2001;22:409–414. doi: 10.1093/carcin/22.3.409. [DOI] [PubMed] [Google Scholar]
- 79.Zhu W, Smith A, Young CY. A nonsteroidal anti-inflammatory drug, flufenamic acid, inhibits the expression of the androgen receptor in LNCaP cells. Endocrinology. 1999;140:5451–5454. doi: 10.1210/endo.140.11.7246. [DOI] [PubMed] [Google Scholar]