

Abstract
Carcinoid tumors are low-grade malignant tumors that arise from neuroendocrine cells. Primary renal carcinoid tumors are extremely uncommon. They seem to be more indolent than renal cell carcinomas, although metastases to regional lymph nodes, liver, and bone have been described. The presence of metastases seems to indicate a more malignant course; however, even with metastases a patient might live for 3 or 4 years. Renal carcinoid tumors should be managed by radical or partial nephrectomy, and good outcomes have been obtained for organ-confined disease after radical excision. Conventional methods of imaging are inadequate for detecting smaller carcinoids, so somatostatin receptor scintigraphy should complement computed tomography and magnetic resonance imaging when searching for occult or metastatic disease. Close follow-up after surgery is necessary.
Keywords: Carcinoid tumor, Kidney, Somatostatin receptor scintigraphy
Carcinoid tumors are low-grade malignant tumors that arise from neuroendocrine cells, specifically enterochromaffin or amine precursor uptake and decarboxylation cells. As a result, carcinoid tumors most commonly involve the gastrointestinal tract, although they can occur in a wide range of organs, including the larynx, trachea, bronchi, lung, ovary, testis, biliary duct, gallbladder, and breast. Carcinoid tumor is found in the retroperitoneum only rarely, but not unexpectedly because neuroendocrine cells can be found in the bladder and prostate. Primary renal carcinoid tumors are extremely uncommon lesions of the kidney; their pathogenesis is uncertain because neuroendocrine cells are not found in normal renal parenchyma, pelvis, and ureter. To date, only 43 cases of primary renal carcinoid tumor have been reported in the English-language literature. We report here the 44th case and provide a synopsis of the literature.
Case Report
A 56-year-old Japanese woman was diagnosed with a 9-cm right renal abscess during evaluation for right flank pain and fevers. A computed tomography (CT) scan demonstrated coarse calcification in the periphery of the abscess, as well as air (Figure 1). Additional workup included a urine culture that was positive for Escherichia coli, but all other infectious processes were ruled out, including tuberculosis, coccidiomycosis, malaria, histoplasmosis, and cryptococcus.
Figure 1.
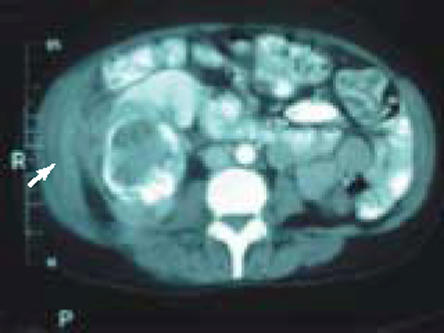
Abdominal computed tomography demonstrates a large, low-density, cystic structure with a thick rim of calcification most consistent with a right renal abscess. There is extension of the inflammatory process to the right lateral abdominal wall (arrow).
The patient underwent an uneventful right radical nephrectomy. The final pathologic examination revealed a well-differentiated neuroendocrine tumor. The tumor histology was typical of carcinoid, with eosinophilic trabeculae intermixed with nests of small, monotonous, cuboidal tumor cells (Figure 2). The tumor cells contained a granular, acidophilic cytoplasm and round to oval nuclei with a finely stippled chromatin pattern. Cells undergoing mitosis were scarcely present. Immuno-histochemistry demonstrated strong cytoplasmic labeling for chromogranin, neuron-specific enolase, and synaptophysin (Figure 3). A large amount of necrotic tissue was present.
Figure 2.

Microscopically, carcinoid tumor cells appear relatively invariable, and they are arranged in a trabecular pattern that is classic of carcinoid tumors.
Figure 3.

Immunohistochemical staining demonstrates that the tumor cells are positive for chromogranin.
In retrospect, the patient had no clinical manifestations of carcinoid syndrome (ie, flushing or diarrhea). The CT scan did not show any evidence of metastatic disease. Approximately 1 month after surgery, somatostatin receptor (SR) scintigraphy with 111-indium-labeled octeotride was performed, and this study confirmed that there was no metastasis or other site of occult disease. The patient is without evidence of disease recurrence at 4 months after the operation.
Discussion
We performed Medline searches of the English-language literature, using the terms “renal carcinoid” and “renal neuroendocrine tumor.” Relevant bibliographies of the literature were manually reviewed for additional material. A summary of the cases reviewed is shown in Table 1.
Table 1.
Patient Demographics, Pathologic Characteristics, and Relevant Clinical Findings in Patients Reported with Primary Renal Carcinoid Tumor
| Characteristic | |
| No. of patients | 43 |
| Mean age at time of diagnosis (y) (range) | 50 (23–79) |
| Gender (male/female) (n) | 20/23 |
| Laterality (left/right) (n) | 13/19 |
| Horseshoe kidney | 11 (25) |
| Mean size (cm) (range) | 7.2 (1.5–21) |
| Presenting symptoms | |
| Abdominal/flank pain | 16 (36.4) |
| Hematuria (gross and microscopic) | 8 (18.2) |
| Constitutional symptoms | 7 (15.9) |
| Asymptomatic | 9 (20.5) |
| Carcinoid syndrome | 6 (13.6) |
| Pathology | |
| Concurrent teratoma | 4 (9.1) |
| Cystic component | 21 (48.8) |
| Necrosis | 11 (25.6) |
| Calcification | 13 (30.2) |
| Metastasis | 10 (22.7) |
| Detected at time of diagnosis | 4 (9.1) |
| Lymph node involvement | 8 (18.2) |
| Liver metastasis | 8 (18.2) |
| Disease recurrence | 4 (9.1) |
| Deaths | 4 (9.1) |
| Mean follow-up (mo) (range) | 27.6 (3–48) |
Data are presented as n (%), unless otherwise noted.
Primary carcinoid tumors of the kidney are very uncommon. The first report was in 1966 by Resnick and colleagues,1 and since then 42 cases have been documented in the English-language literature. Owing to the rarity of this lesion, appropriate management is not well established, and the clinical course of the disease is not well understood. This report summarizes the clinical and pathologic characteristics of primary renal carcinoid tumor, on the basis of our review of the literature.
Renal carcinoid tumors occur predominantly in relatively young adults (mean age 50 years), with no predilection for either sex. The majority (55.8%) of patients presented with abdominal/flank pain and/or hematuria. Only 6 patients (13.6%) presented with carcinoid syndrome. In 9 patients (20.5%), the tumor was incidentally found (they were asymptomatic), and a minority (15.9%) of patients had constitutional symptoms.
Renal carcinoid tumors are typically solid tumors (74%) but are occasionally associated with cystic components (49%). As seen in our case, they often have a significant amount of necrosis (26%) and/or dystrophic calcification (30%). There is, however, no specific finding on CT or magnetic resonance imaging (MRI) that can by itself differentiate renal carcinoid tumors from the other types of renal tumors before surgery. In all cases the treatment was surgical; either a total or partial nephrectomy was performed. The final diagnosis was made histologically by positive reactions to silver stains specific for markers of neuroendocrine tissue, including neuron-specific enolase, synaptophysin, and chromogranin.
Among the 43 reported cases of primary renal carcinoid tumor, 11 (25.6%) originated from a horseshoe kidney,2–4 5 (11.4%) arose from teratoma,5–8 and in 1 case the carcinoid tumor was discovered in a polycystic kidney at the time of autopsy.9 Krishnan and colleagues4 reported an association of horseshoe kidney and the development of renal carcinoid, with a calculated relative risk of 62. They speculate that renal carcinoid tumor arises from pre-existing hyperplasia of neuroendocrine cells found within foci of metaplastic or teratomatous epithelium in the horseshoe kidney. Similarly, regarding the 4 cases of carcinoid found in a renal teratoma: perhaps during organogenesis misplaced progenitor cells form nests within the kidney that later develop into teratomatous intestinal or respiratory epithelium. This is one of the theories on how renal carcinoid originates. Because neuroendocrine cells are not intrinsically present in normal kidneys, the debate over the pathogenesis of primary renal carcinoid tumor continues. Others have hypothesized that renal carcinoid tumors are derived from misplaced neural crest tissue in the hilar aspect of the kidney during embryogenesis.9,10 Another theory suggests that they arise from neuroendocrine cells associated with intestinal metaplasia of the pyelocalyceal urothelium that occurs as a sequela of chronic inflammation.4,11 However, other diseases of the kidney in which chronic inflammation is a prominent feature, such as chronic pyelonephritis, have not been associated with renal carcinoid tumor. To our knowledge, there is no correlation between carcinoid tumor and adult polycystic kidney disease.
The prognosis for primary renal carcinoid tumor is relatively optimistic. In the reviewed cases, 10 patients (23%) developed metastasis, and the most common sites of metastasis were the lymph nodes and liver. Overall, only 4 deaths were reported, with a mean follow-up of 27.6 months (range, 3–48 months). Three of the 4 fatal cases were attributed to renal carcinoid, and the other fatality resulted from complications associated with polycystic kidney disease. One of the patients with disease-specific mortality had evidence of metastatic disease at the time of presentation, whereas the other 2 had lymph node involvement that was discovered at the time of pathologic examination. Three of the 10 patients with metastases died, whereas none of the patients without metastasis died. The patients with metastasis who survived had a mean follow-up of 26.6 months (range, 7–40 months), thus demonstrating that even with metastasis, patients might survive for many months. Kawajiri and colleagues12 compared primary renal carcinoids with carcinoid tumors of other organs and reported a positive correlation between the size of the primary tumor and the incidence of metastasis. The presence of metastasis around the time of diagnosis seems to be an integral prognosticator of survival in cases of renal carcinoid.
All patients underwent either radical nephrectomy or partial nephrectomy. There are no reports of chemotherapy for primary renal carcinoid. Unsatisfactory outcomes of combination chemotherapy have been reported in cases of carcinoid tumor of other sites, and radiation has been found to be only palliative.13,14 Complete surgical excision is the only recommended treatment for localized renal carcinoid tumor, and good results have been reported after excision of primary tumor, with up to 40 months of follow-up.
Whenever a carcinoid tumor is found within the kidney, the need for a thorough search for an unknown primary tumor is indicated. SR scintigraphy is recognized as an integral diagnostic and staging tool in the evaluation of carcinoid tumors, because CT and MRI occasionally lack sensitivity in detecting these tumors. Carcinoid tumors abundantly express SR, and radiolabeled octreotide, a somatostatin analogue that binds SR with high affinity, is useful in detecting even the smallest carcinoid tumors. SR scintigraphy has a reported sensitivity of 85%.13,15 Mufarrij and coworkers16 reported a case of primary carcinoid of the kidney and metastatic disease in the liver undetectable by CT and MRI but identified by SR scintigraphy. McCaffrey and colleagues17 reported a case of a 67-year-old woman who underwent a right nephrectomy and retroperitoneal lymphadenectomy for suspected renal cell carcinoma, which, on further review of the tumor histology, was found to be carcinoid. Workup to detect metastasis included a CT scan of the abdomen, chest radiography, and a bone scan, all of which showed no evidence of disease. SR scintigraphy detected residual disease in the renal bed, which was later identified and followed up with subsequent CT scans. If not for SR scintigraphy, this patient would have been viewed as disease free by the surgeon and by postoperative CT scan. Clearly, SR scintigraphy is a useful adjunct to CT and MRI both before and after surgery in the search for an occult primary, metastatic disease and residual carcinoid tumor.
Conclusion
Despite short-term follow-up in many reports and incomplete documentation, this review has attempted to summarize all the relevant clinicopathologic features of primary carcinoid of the kidney to help describe the clinical behavior of this rare entity. Primary renal carcinoid tumors seem to be more indolent than renal cell carcinomas, although metastases to regional lymph nodes, liver, and bone have been described. The presence of metastases seems to indicate a more malignant course; however, even with metastases a patient might live for a long period. Renal carcinoid tumors should be managed by radical or partial nephrectomy, and good outcomes have been obtained for organ-confined disease after radical excision. Whenever a diagnosis of renal carcinoid is made, it is important to rule out metastatic disease by searching for a primary tumor hidden elsewhere. Conventional methods of imaging are inadequate for detecting smaller carcinoids, and so SR scintigraphy should complement CT and MRI when searching for occult or metastatic disease. Close follow-up after surgery is necessary because recurrences have been observed.
Main Points.
Carcinoid tumors are low-grade malignant tumors that arise from neuroendocrine cells, specifically enterochromaffin or amine precursor uptake and decarboxylation cells.
Primary renal carcinoid tumors are extremely uncommon lesions of the kidney. Their pathogenesis is uncertain because neuroendocrine cells are not found in normal renal parenchyma, pelvis, and ureter.
It has been hypothesized that renal carcinoid tumors are derived from misplaced neural crest tissue in the hilar aspect of the kidney during embryogenesis; another theory suggests that they arise from neuroendocrine cells associated with intestinal metaplasia of the pyelocalyceal urothelium that occurs as a sequela of chronic inflammation.
The prognosis for primary renal carcinoid tumor is relatively optimistic: of the 43 previously reported patients, 10 (23%) developed metastasis, and overall only 4 deaths were reported, with a mean follow-up of 27.6 months.
Complete surgical excision is the only recommended treatment for localized renal carcinoid tumor, and good results have been reported after excision of primary tumor, with up to 40 months of follow-up.
Somatostatin receptor scintigraphy is an integral diagnostic and staging tool in the evaluation of carcinoid tumors, because computed tomography and magnetic resonance imaging occasionally lack sensitivity in detecting carcinoid tumors.
References
- 1.Resnick ME, Unterberger H, McLoughlin PT. Renal carcinoid producing the carcinoid syndrome. Med Times. 1966;94:895–896. [PubMed] [Google Scholar]
- 2.Isobe H, Takashima H, Higashi N, et al. Primary carcinoid tumor in a horseshoe kidney. Int J Urol. 2000;7:184–188. doi: 10.1046/j.1442-2042.2000.00160.x. [DOI] [PubMed] [Google Scholar]
- 3.Begin LR, Guy L, Jacobson SA, Aprikian AG. Renal carcinoid and horseshoe kidney: a frequent association of two rare entities—a case report and review of the literature. J Surg Oncol. 1998;68:113–119. doi: 10.1002/(sici)1096-9098(199806)68:2<113::aid-jso8>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 4.Krishnan B, Truong LD, Saleh G, et al. Horseshoe kidney is associated with an increased relative risk of primary renal carcinoid tumor. J Urol. 1997;157:2059–2066. [PubMed] [Google Scholar]
- 5.Kim JK, Suh K. Primary carcinoid tumor in a mature teratoma of the kidney. J Ultrasound Med. 2004;23:433–437. doi: 10.7863/jum.2004.23.3.433. [DOI] [PubMed] [Google Scholar]
- 6.Kojiro M, Ohishi H, Isobe H. Carcinoid tumor occurring in cystic teratoma of the kidney: a case report. Cancer. 1976;38:1636–1640. doi: 10.1002/1097-0142(197610)38:4<1636::aid-cncr2820380432>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 7.Fetissof F, Benatre A, Dubois MP, et al. Carcinoid tumor occurring in teratoid malformation of the kidney. Cancer. 1984;54:2305–2308. doi: 10.1002/1097-0142(19841115)54:10<2305::aid-cncr2820541042>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 8.Yoo J, Park S, Lee HJ, et al. Primary carcinoid tumor arising in a mature teratoma of the kidney: a case report and review of the literature. Arch Pathol Lab Med. 2002;126:979–981. doi: 10.5858/2002-126-0979-PCTAIA. [DOI] [PubMed] [Google Scholar]
- 9.Shibata R, Okita H, Shimoda M, et al. Primary carcinoid tumor in a polycystic kidney. Pathol Int. 2003;53:317–322. doi: 10.1046/j.1440-1827.2003.01469.x. [DOI] [PubMed] [Google Scholar]
- 10.Zak FG, Jindrak K, Capozzi F. Carcinoidal tumor of the kidney. Ultrastruct Pathol. 1983;4:51–59. doi: 10.3109/01913128309140571. [DOI] [PubMed] [Google Scholar]
- 11.Gordon A. Intestinal metaplasia of the urinary tract epithelium. J Pathol Bacteriol. 1963;85:441–445. doi: 10.1002/path.1700850224. [DOI] [PubMed] [Google Scholar]
- 12.Kawajiri H, Onosa N, Ohira M, et al. Carcinoid tumor of the kidney presenting as a large abdominal mass: report of a case. Surg Today. 2004;34:86–89. doi: 10.1007/s00595-003-2644-x. [DOI] [PubMed] [Google Scholar]
- 13.Kulke MH, Mayer RJ. Carcinoid tumors. N Engl J Med. 1999;340:858–868. doi: 10.1056/NEJM199903183401107. [DOI] [PubMed] [Google Scholar]
- 14.Chakravarthy A, Abrams RA. Radiation therapy in the management of a patient with malignant carcinoid tumors. Cancer. 1995;75:1386–1390. doi: 10.1002/1097-0142(19950315)75:6<1386::aid-cncr2820750622>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 15.Shi W, Johnson CF, Buchanan KD. Localization of neuroendocrine tumours with [111In] DTPA-octreotide scintigraphy (Octreoscan): a comparative study with CT and MR imaging. Q J Med. 1998;91:295–301. doi: 10.1093/qjmed/91.4.295. [DOI] [PubMed] [Google Scholar]
- 16.Mufarrij P, Varkarakis IM, Studeman K, Jarrett TW. Primary renal carcinoid tumor with liver metastases detected with somatostatin receptor imaging. Urology. 2005;65:e19–e21. doi: 10.1016/j.urology.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 17.McCaffrey JA, Reuter VV, Herr HW. Carcinoid tumor of the kidney: the use of somatostatin receptor scintigraphy in diagnosis and management. Urol Oncol. 2000;5:108–111. doi: 10.1016/s1078-1439(99)00050-2. [DOI] [PubMed] [Google Scholar]