

We report a young woman with type Ic congenital disorder of glycosylation (CDG) with new clinical features of idiopathic intracranial hypertension, retinal degeneration, and novel mutations of ALG6. Patients with known or suspected CDG should receive a full ophthalmic examination including dilated fundus examination and electroretinography.
CDG is a rare group of autosomal recessive metabolic disorders. The two major subgroups are type I, caused by dysfunction of glycosylated protein assembly, and type II, caused by abnormal processing of glycosylated protein end products after assembly.1 Patients with type Ic (OMIM #603147) have mental retardation, axial hypotonia, very low factor XI, and seizures; the gene defect in α1,3‐glucosyltransferase (human homologue of ALG6;OMIM *604566) encodes an enzyme that catalyses the transfer of the first glucose residue to the lipid linked oligosaccharide precursor for N‐linked glycosylation. The reported ophthalmic manifestations of type I CDG are summarised in table I. We report a case of type Ic congenital disorder of glycosylation with new clinical features of idiopathic intracranial hypertension, retinal degeneration and novel mutations of ALG6.
Table 1 Reported ophthalmic manifestations in type I congenital disorder of glycosylation (CDG).
| CDG Ia | CDG Ib | CDG Ic | |
|---|---|---|---|
| Strabismus | + | − | + |
| Retinopathy | + | − | − |
| Optic atrophy | + | − | − |
Case report
Our patient, with non‐consanguineous parents, was noted at birth to have incomplete digits on her hands and feet. She had episodes of apnoea early in life and the onset of seizures at age 20 months. Delayed physical and developmental milestones were evident early. Levels of follicle stimulating hormone and luteinising hormone were low and a diagnosis of polycystic ovarian disease was made. She underwent esotropia surgery at age 4 years and at 19 years developed thrombosis of the superficial femoral vein. Coagulation factor studies revealed very low factor XI levels. Isoelectric focusing of serum transferrin was suggestive of a congenital disorder of glycosylation. Sequencing of the ALG6 gene showed a novel three base deletion (897–899 delATT) and an intronic splice site mutation (IVS7+2T>G).2
The patient was obese without signs of abnormal fat distribution. She blinked to light and no strabismus was identified by corneal light reflex. Further ocular examination was not feasible. On examination under anaesthesia at age 20 years, the anterior segment was normal. Dilated ophthalmoscopy showed bilateral optic nerve pallor with elevation of each nerve, diffuse retinal pigment epithelial granularity most notable in the macula, and attenuated retinal vessels (fig 1).
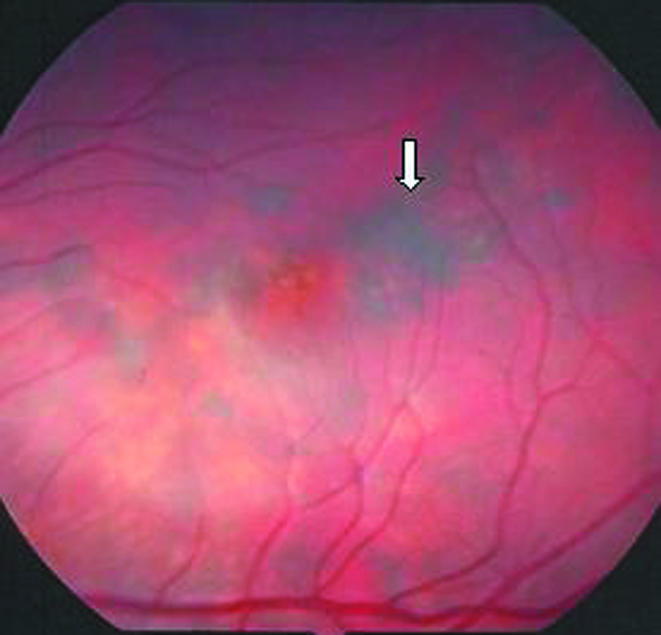
Figure 1 Left fundus showing attenuated vessels with retinal pigment cell granularity of the macula (white arrow).
Electroretinography revealed prolonged implicit times on scotopic and photopic functions. Because of the elevated optic nerve heads, head magnetic resonance imaging was performed and was normal; lumbar puncture revealed an elevated opening pressure of 350 mm H2O (nl 70–180 mm H2O), a glucose of 50 mg/dl (nl 50–75 mg/dl), and a protein of 44 mg/dl (nl 15–45 mg/dl) without white blood cells. A diagnosis of idiopathic intracranial hypertension was made.
Comment
Isoelectric focusing of serum transferrin is the most common screening technique for types I and II CDG.1 Many congenital disorders of glycosylation have been identified based on mutational and enzyme analyses. Type I congenital disorders of glycosylation are caused by reduced functions of the genes for the assembly and processing pathways of N‐glycosylation; a decrease in anodal fractions and an increase of disialotransferrin and asialotransferrin are suggestive of the diagnosis.
Ophthalmic features of the most common congenital disorder of glycosylation, type Ia (OMIM #2I2065) include myopia, attenuated retinal vessels, bone spicules, esotropia, and nyctalopia3; this disease subtype is caused by a deficiency of phosphomanomutase (OMIM #212065) encoded by phosphomannomutase‐2 (PMM2; OMIM *601785) gene. In CDG type Ic, much less common, a homozygous amino acid substitution, A333V substitution, in the gene encoding α1,3‐glucosyltransferase is the most common mutation.4 There is no known biochemical link between CDG and idiopathic intracranial hypertension; the association in our patient may be coincidental. Grunwald and colleagues5 found strabismus to be present in all eight patients with CDG type Ic; neither idiopathic intracranial hypertension nor retinal abnormalities were reported.
The developmental delay evident in our patient was severe. She possessed simple communication skills limiting her ability to complain of visual changes or symptoms of intracranial hypertention. No specific clinical presentation has been identified for the CDG group of diseases. Failure to thrive, unexplained seizures, hypotonia, and developmental delays are all frequently present.
In conclusion, we report an adult patient with CDG type Ic, confirmed by identification of mutations in the ALG6 gene, and describe new systemic and ocular features including idiopathic intracranial hypertension., optic atrophy, and a retinal dystrophy with abnormal electroretinography.
Footnotes
Supported in part by NEI Grant EY 08282 (JBB).
The authors have no interests or disclosures to report.
This is an institutional review board (IRB) exempt single case report with no identifiable patient information.
References
- 1.Jaeken J. Congenital disorders of glycosylation (CDG): it's all in it! J Inherit Metab Dis 20032699–118. [DOI] [PubMed] [Google Scholar]
- 2.Sun L, Eklund E, Van Hove J.et al Clinical and molecular characterization of the first adult congenital disorder of glycosylation (CDG) type Ic patient. Am J Med Genet (accepted) [DOI] [PubMed]
- 3.Jensen H, Kjaergaard S, Klie F.et al Ophthalmic manifestations of congenital disorder of glycosylation type 1a. Ophthal Genet 20032481–88. [DOI] [PubMed] [Google Scholar]
- 4.Imbach T, Burda P, Kuhnert P.et al A mutation in the human ortholog of the Saccharomyces cerevisiae ALG6 gene causes carbohydrate‐deficient glycoprotein syndrome type‐Ic. Proc Natl Acad Sci USA 1999966982–6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grunewald S, Imbach T, Huijben K.et al Clinical and biochemical characteristics of congenital disorder of glycosylation type Ic, the first recognized endoplasmic reticulum defect in N‐glycan synthesis. Ann Neurol 200047776–781. [PubMed] [Google Scholar]