

Abstract
The Hsp110 proteins, exclusively found in the eukaryotic cytosol, have significant sequence homology to the Hsp70 molecular chaperone superfamily. Despite this homology and the cellular abundance of these proteins, the precise functional role has remained undefined. Here, we present the intriguing finding that the yeast homologue, Sse1p, acts as an efficient nucleotide exchange factor (NEF) for both yeast cytosolic Hsp70s, Ssa1p and Ssb1p. The mechanism involves formation of a stable nucleotide-sensitive complex, but does not require ATP hydrolysis by Sse1p. The NEF activity of Sse1p stimulates in vitro Ssa1p-mediated refolding of thermally denatured luciferase, and appears to have an essential role in vivo. Overexpression of the only other described cytosolic NEF, Fes1p, can partially compensate for a lethal sse1,2Δ phenotype, however, the cells are sensitive to stress conditions. Furthermore, in the absence of Sse, the in vivo refolding of thermally denatured model proteins is affected. This is the first report of a nucleotide exchange activity for the Hsp110 class of proteins, and provides a key piece in the puzzle of the cellular chaperone network.
Keywords: chaperone, Hsp70, Hsp110, nucleotide exchange factor, yeast
Introduction
Hsp70 chaperones assist a plethora of folding processes including the folding and assembly of newly synthesized polypeptides, the transport of organellar and secretory proteins across membranes, the refolding of misfolded and aggregated proteins, and the regulated activation of signal transduction proteins (Bukau and Horwich, 1998; Hartl and Hayer-Hartl, 2002). The evolutionary amplification and diversification of HSP70 genes has generated a superfamily comprised of functionally distinct members that employ a large arsenal of co-chaperones to fulfill specific cellular functions.
The canonical Hsp70 proteins transiently associate with hydrophobic peptide segments of protein substrates, thereby affecting their folding. Substrate binding and release is driven by the ATPase activity of Hsp70, which leads to cycles of an ATP-bound state with low affinity for substrate and an ADP-bound state with high affinity (Rudiger et al, 1997; Bukau and Horwich, 1998). The ATP hydrolysis step in the cycle is stimulated by the J-protein co-chaperones; by coupling nucleotide hydrolysis with substrate binding, these proteins target Hsp70s to substrates. The activity of several Hsp70 chaperones is also modulated by nucleotide exchange factors (NEFs), which catalyze nucleotide dissociation and thereby define the lifetime of the Hsp70–substrate complexes. Three general classes of NEFs, differing in their molecular mechanisms of nucleotide release, have been characterized and are typified by GrpE, BAG-1 and HspBP1 (Harrison et al, 1997; Sondermann et al, 2001; Shomura et al, 2005). Additionally, Lhs1p has been described as a NEF for the resident Hsp70 in the yeast endoplasmic reticulum, Kar2p (Steel et al, 2004). Although the structural interactions are undefined, this interaction is thought to represent a novel mechanism of nucleotide release.
The Saccharomyces cerevisiae cytoplasm harbors several members of the Hsp70 superfamily. The classical Hsp70 chaperones include the nonessential Ssb1,2p which transiently associate with ribosomes to facilitate nascent polypeptide folding, and four Ssa homologs (Ssa1–4) (Bush and Meyer, 1996; Kim et al, 1998). The Ssa's are essential for viability and are involved in housekeeping and stress-related protein folding processes. They are activated by at least two distinct J-proteins, Ydj1p and Sis1p (Lu and Cyr, 1998), and are targets for the only confirmed NEF of the yeast cytosol, Fes1p (Kabani et al, 2002).
The yeast cytosol also contains two homologous Hsp110 members, Sse1p and Sse2p. This divergent branch of Hsp70 chaperones are distinguished by an extended C-terminal domain, as well as an acidic region inserted between the terminal strands of the predicted β-sheet structure (Easton et al, 2000; Raviol et al, 2005). Both SSE genes are upregulated upon stress, but only SSE1 is abundantly expressed under normal growth conditions (Mukai et al, 1993). Yeast strains lacking the SSE1 gene demonstrate a severe growth defect, whereas sse2Δ cells have no detectable phenotype. Although it is unclear whether deletion of both SSE genes is lethal, this class of proteins clearly plays a significant role in cell growth (Mukai et al, 1993; Shaner et al, 2004; Shaner et al, 2005; Trott et al, 2005; Yam et al, 2005). Previous in vitro studies revealed that Sse1p can prevent protein aggregation (Brodsky et al, 1999; Goeckeler et al, 2002), whereas others have observed an in vivo role in Hsp90-mediated processes (Liu et al, 1999; Goeckeler et al, 2002; Lee et al, 2004). Furthermore, it has recently been observed that Sse1p forms stable complexes with Ssa1p and Ssb1p (Shaner et al, 2005; Yam et al, 2005), triggering the proposal that Sse1p modulates Hsp70–substrate interactions. However, although the Hsp110 proteins comprise a significant proportion of the cytoplasmic chaperone pool, a clear functional role has not yet been established.
Here, we show that Sse1p acts as a potent nucleotide exchange factor for Ssa1p and Ssb1p. Furthermore, in vivo analyses suggest that specific characteristics of Sse are necessary during the stress response.
Results
Sse1p forms complexes with Ssa1p and Ssb1p
We initially sought to identify functional partners of Sse1p to gain insight regarding the cellular role of this protein. Native Sse1p complexes were purified from a yeast strain with a TAP-tagged SSE1 gene under control of the endogenous promoter (Figure 1A). Two prominent co-isolated proteins were identified by mass spectrometry as Ssa1p and Ssb1p. This agrees with recent reports where His- or FLAG-tagged versions of Sse1p were overproduced in vivo and used to co-precipitate both Hsp70s (Shaner et al, 2005; Yam et al, 2005). To probe the specificity and exclude the possibility of a chaperone–substrate interaction, in vitro complexes of Ssa1p and tritiated Sse1p were incubated with a molar excess of unlabelled Sse1p and/or ATP (Figure 1B). The interaction is specific as addition of unlabelled Sse1p significantly decreased the amount of 3H-Sse1p that co-precipitated with Ssa1p. Interestingly, ATP addition reduced the signal to background levels, indicating that complex formation is sensitive to the nucleotide status of one or both proteins.
Figure 1.
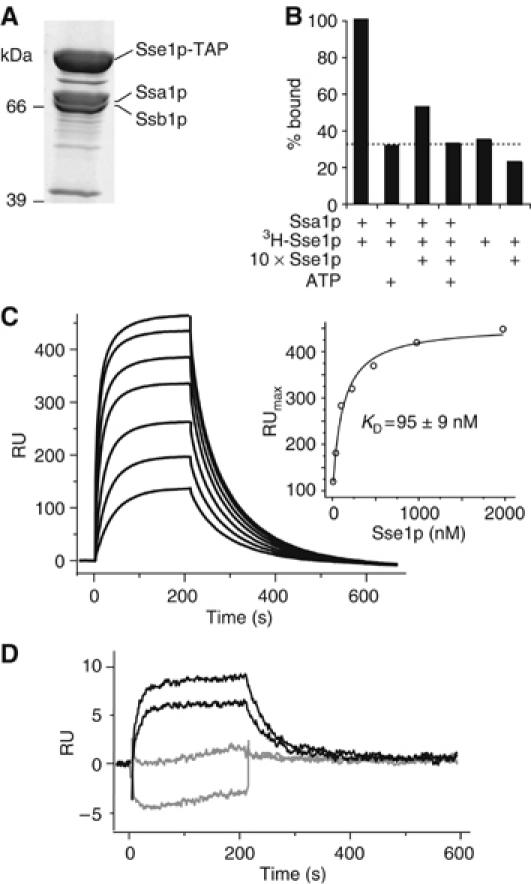
Sse1p forms stable complexes with Ssa1p and Ssb1p. (A) Purification of a TAP-tagged variant of Sse1p reveals an in vivo association with Ssa1p and Ssb1p. (B) Co-immunoprecipitation demonstrates specificity of Ssa1p–Sse1p complexes. Complexes of purified Ssa1p and 3H-Sse1p were incubated with excess unlabelled Sse1p or ATP. Levels of 3H-Sse1p that co-immunoprecpitated with Ssa1p were assessed by scintillation counting of beads. The dotted line indicates the background level of nonspecific binding of 3H-Sse1p. (C) Sensogram of interactions between Ssa1p and Sse1p. In all, 2000 RU of Ssa1p was immobilized and increasing concentrations of Sse1p injected (31.3–2000 nM in two-fold steps). Inset is maximal resonance units plotted against the respective Sse1p concentration. (D) Comparison of interactions between injected Sse1p (31.5 and 62.3 nM) and immobilized Ssa1p (150 RU) in the presence (gray) or absence (black) of 10 mM ATP.
The association between Sse1p and Ssa1p was further characterized using surface plasmon resonance spectroscopy analyses. C-terminally histidine-tagged Ssa1p was immobilized onto a NiNTA chip, and increasing concentrations of Sse1p were injected. The observed interaction was strong with fast association and dissociation rates (Figure 1C). The KD was determined by plotting the maximal resonance units (RU) against the Sse1p concentration, and values of 153±34 and 95±9 nM were obtained with 150 and 2000 RU of immobilized Ssa1p, respectively (Figure 1C inset). The dissociation followed biexponential kinetics with rates of 0.028±0.003 and 0.003±0.002 s−1. The association kinetics could not be satisfactorily fitted with any of the simple interaction model equations, and was therefore calculated from the dissociation equilibrium constant and the dissociation rate constant as 2.3±0.8 × 105 M−1 s−1. In agreement with the co-immunoprecipitation data, inclusion of ATP during the Sse1p injection drastically reduced the interaction with Ssa1p (Figure 1D).
Effects of Sse1p on the ATPase activity of Ssa1p
In support of a functional role for the Sse1p–Hsp70 complexes, SSE1 overexpression partially complements the thermosensitive phenotype of ydj1-151 mutants (Goeckeler et al, 2002). These results suggest that Sse1p and Ydj1p may have similar stimulatory roles in the chaperone network. To further investigate the functional nature of these interactions, we examined whether Ssa1p–Sse1p complex formation influenced steady-state ATPase activities of the involved proteins. Ssa1p was stimulated nine-fold by Ydj1p, whereas the addition of Sse1p resulted in an ATPase activity three-fold greater than combining only Ssa1p and Sse1p (Figure 2A). This synergistic stimulation of the steady-state ATPase activity for Sse1p and Ssa1p is similar to that reported by others (Shaner et al, 2005).
Figure 2.
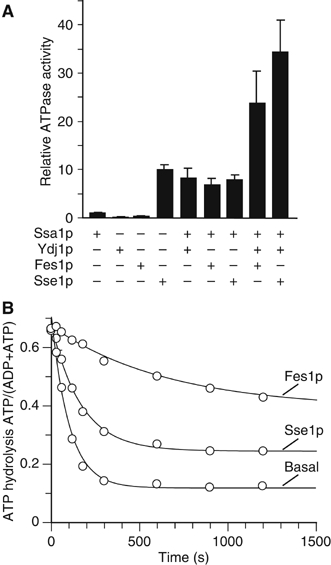
ATPase assays with Ssa1p. (A) Steady-state ATPase assay with Ssa1p, Sse1p, Fes1p, Ydj1p and combinations thereof. The ATPase rates are given relative to the basal activity of Ssa1p (set to one). (B) Single turnover analysis of ATP hydrolysis. [α-32P]ATP–Ssa1p complexes were incubated with buffer or Sse1p (2 μM) or Fes1p (2 μM). The nucleotide status at different time intervals was analyzed by thin-layer chromatography. Shown is a plot of the ATP/(ATP+ADP) values from single turnover assays with Ssa1p alone, or in the presence of Sse1p or Fes1p over time.
To better understand the precise influence of Sse1p on Ssa1p, we examined ATP hydrolysis of preloaded Ssa1p-[α32P]ATP complexes (Figure 2B). Surprisingly, the addition of Sse1p did not significantly affect the single turnover ATP-hydrolysis rate (khyd) of Ssa1p, but diminished the absolute amount of hydrolyzed ATP. This is evident by the different offsets of the fitted curves in the absence and presence of 2 μM Sse1p (Figure 2B). Furthermore, increasing concentrations of Sse1p resulted in a further decrease of hydrolyzed ATP levels (data not shown). Thus, as Sse1p (i) did not affect the khyd of Ssa1p under single turnover conditions, and (ii) did not inhibit, but stimulated catalysis under steady-state conditions, it can be concluded that Sse1p does not inhibit ATP hydrolysis. Rather, Sse1p must act on a different step of the ATPase cycle, such as ATP binding or ADP release. Specifically, the reduced ATP hydrolysis of the single turnover complexes suggests that Sse1p influences ATP release before hydrolysis.
Sse1p is a nucleotide exchange factor for Ssa1p and Ssb1p
The effects of Sse1p on Ssa1p are similar to that of the previously described cytosolic NEF, Fes1p, in that single turnover ATP hydrolysis is inhibited (Kabani et al, 2002; Figure 2B). As well, we find that both proteins stimulate steady-state ATPase rates of Ssa1p/Ydj1p (Figure 2A). For unknown reasons, this differs from the inhibitory effect of Fes1p described by Kabani et al, but is similar to the stimulation of Hsc70 by BAG-1 and HspBP1 (Sondermann et al, 2001; Shomura et al, 2005).
To further investigate a possible NEF activity for Sse1p, we analyzed the dissociation of fluorescent-labelled ADP analogue (MABA-ADP; (N8-(4-N′-methylanthraniloylaminobutyl)-8 aminoadenosine 5′-diphosphate)) from Ssa1p and Ssb1p using stopped-flow instrumentation (Figure 3). Preloaded complexes of MABA-ADP and Ssa1p or Ssb1p were rapidly mixed with an excess of unlabelled ADP in the presence or absence of Sse1p, and MABA-ADP dissociation was tracked by decreases in fluorescence signal. The basal nucleotide dissociation rate of Ssa1p was determined to be 0.53 s−1, which is slightly higher than the published values for mammalian Hsp70 (0.3 s−1) and Hsc70 (0.24 s−1) (Gassler et al, 2001). Addition of a 10-fold molar excess of Sse1p over Ssa1p accelerated the rate by 150-fold (84.6 s−1), whereas Fes1p resulted in only a 12-fold stimulation (7 s−1) (Figure 3A). We found that Sse1p had similar effects on Ssb1p (Figure 3B). The basal MABA-ADP dissociation rate of Ssb1p (0.27 s−1) was stimulated nearly 80-fold by a 10-fold excess of Sse1p (21.2 s−1). In contrast, a 10-fold molar excess of Fes1p had only a weak effect on the nucleotide release of Ssb1, with a less than three-fold stimulation (0.67 s−1). As expected, Ssa1p did not influence the rate of Ssb1p nucleotide release and vice versa (Figure 3A and B).
Figure 3.
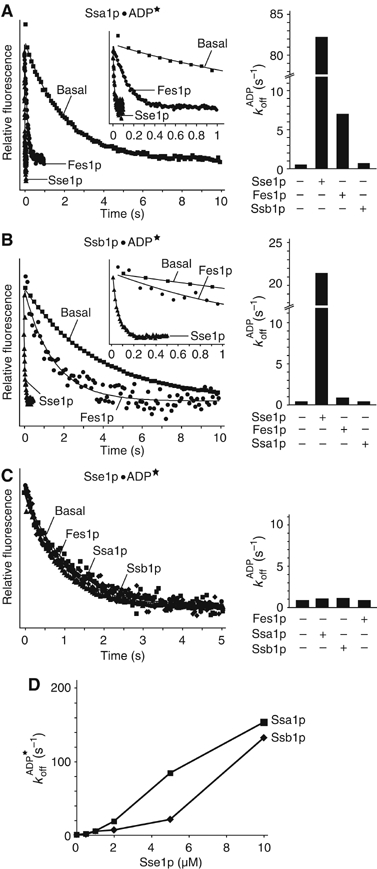
Stopped-flow analysis of MABA (★)-ADP release from Hsp70s. For Ssa1p (A) and Ssb1p (B), release was assessed with a 10-fold excess of Sse1p or Fes1p. Insets are a blow-up of the first second of the MABA-ADP dissociation. (C) Stimulation of release from Sse1p by Fes1p, Ssa1p or Ssb1p. The dissociation curves of MABA-ADP measured with the stopped-flow instrument are shown in the left panel, and the bar diagrams with the calculated rates in the right panel. (D) Titration of Sse1p against Ssa1p/Ssb1p MABA-ADP complexes.
A possible reciprocal functional relationship between Sse1p and the Hsp70s was also investigated. The basal MABA-ADP nucleotide dissociation rate of Sse1p (0.91 s−1) is higher than the rates determined for the Hsp70 homologues, but could not be accelerated by the addition of Ssa1p, Ssb1p or Fes1p at a 10-fold molar excess (Figure 3C). Thus, although the cytosolic Hsp70s have no influence on Sse1p ATPase activity, Sse1p is a highly effective NEF for both Ssa1p and Ssb1p. Notably, the titration curve in Figure 3D could not be fitted by the quadratic equation, and indicates that the maximal Sse1p stimulation of the ADP dissociation from Ssa1p and Ssb1p is significantly higher than the measured rates.
ATP hydrolysis by Sse1p is not required for NEF activity
In general, it is uncertain whether the cycle of ATP binding, -hydrolysis and ADP-release is required for Sse1p functions. ATP-binding deficient variants can support holdase-like activities in vitro, but cannot complement the sse1Δ thermosensitive phenotype (Goeckeler et al, 2002; Shaner et al, 2004). In contrast, an ATP-hydrolysis deficient variant of Sse1p can complement the sse1Δ strain thermosensitivity, as well as rescue a nonviable sse1,2Δ strain (Shaner et al, 2004). In addition, we recently reported that although the Sse1-K69Mp mutant cannot hydrolyze ATP, the binding of either ATP or ADP results in structural changes resembling those of wild-type Sse1p in the ATP-bound rather than the ADP-bound/nucleotide-free state (Raviol et al, 2005). Taken together, it seems that nucleotide binding, but not hydrolysis, by Sse1p may influence its functional state.
To evaluate the role of ATP hydrolysis in Sse1p NEF activity, we assessed the influence of the Sse1-K69Mp protein on Ssa1p nucleotide release (Figure 4A). Remarkably, the rate of MABA-ADP release from Ssa1p was two-fold higher in the presence of the Sse1-K69Mp variant (168 s−1) than in the presence of wild-type Sse1p (86.4 s−1) when both were premixed with ADP. When premixed with ATP, however, wild-type Sse1p also resulted in a two-fold higher nucleotide release rate (168 s−1), whereas the Sse1-K69Mp activity was unchanged (165 s−1). Thus, it seems clear that the ATP-bound state of Sse1p is more efficient at stimulating nucleotide release. Furthermore, a switch between two conformations (ATP-bound versus ADP/nucleotide-free state) is not required for this activity.
Figure 4.
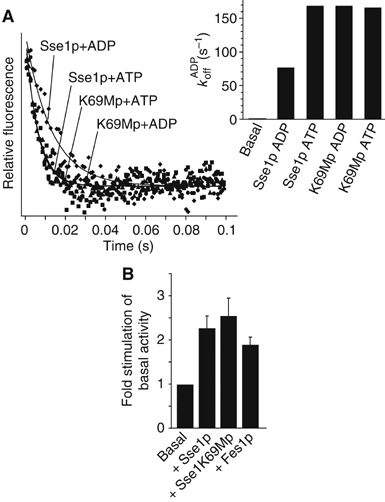
ATP hydrolysis is not required for nucleotide exchange activity of Sse1p. (A) Dissociation curves of MABA-ADP release from Ssa1p in the presence or absence of Sse1-K69Mp. Inset is a bar diagram with the calculated rates of dissociation for Ssa1p–MABA-ADP complexes in the presence of ADP- or ATP-bound Sse1p or Sse1-K69Mp. (B) Sse1p and Sse1-K69Mp are equally efficient at stimulating in vitro refolding of thermally denatured firefly luciferase. The basal level of luciferase refolding in the presence of Ssa1p/Ydj1p was set to 1.
Sse1p stimulates in vitro activities of Ssa1p
To directly assess the influence of Sse1p on Ssa1p-mediated functions, we utilized an in vitro assay for refolding of thermally denatured firefly luciferase. After heat denaturation at 42°C in the presence of chaperones and a recovery interval (30°C, 45 min), luciferase activity was assessed. Titration of Sse1p concentrations indicated that the ratio of 2:1 was optimal for Ssa1p:Sse1p (Figure 1, Supplementary Data). Under these conditions, refolding rates of luciferase were reproducibly stimulated by Sse1p 2–3 fold over Ssa1p/Ydj1p alone (Figure 4B). This effect of Sse1p appears to be mediated through the NEF activity, as addition of Fes1p gave similar results. Interestingly, the stimulation by the Sse1-K69Mp protein was perhaps slightly higher than wild-type Sse1p. This correlates with the stopped flow analyses (see above) indicating that this variant is continually in the ‘on' or ATP-bound state. Thus, the ability of Sse1p to stimulate nucleotide release has a direct stimulatory effect on the Ssa1p-mediated functions.
Combined deletion of SSE1 and SSE2 genes is lethal
We sought to further characterize the in vivo functional role of Sse1p as a nucleotide exchange factor. Whether Sse is essential for viability is unclear, as previous studies have yielded conflicting results. Two reports of viable sse1,2Δ double mutants (Mukai et al, 1993; Yam et al, 2005) are in contrast to the nonviable phenotype observed by the Morano laboratory (Shaner et al, 2005). In light of these conflicting data, we used genetic mating techniques to introduce the sse1,2Δ alleles into the W303 background. Tetrad dissections routinely yielded no more than three viable spores, and no single one inherited markers from both strains (Figure 5A). The slow growth phenotype of sse1Δ cells is remarkably evident in the spore sizes. The absence of four viable spores suggests the sse1,2Δ strain may be synthetically lethal, and further evidence was provided using a plasmid rescue approach. Sporulation and dissection of a diploid strain carrying a URA3-based plasmid constitutively expressing SSE1 (pMPM-CUA-SSE1) yielded spores containing both markers. Only when a different plasmid encoding Sse1p was introduced could the cells survive on 5-FOA media (Figure 5B). This compound is toxic to cells expressing the URA3 gene, and forcibly selects for those that have lost the pMPM-CUA-SSE1 plasmid. Thus, the sse1,2Δ genotype is lethal.
Figure 5.
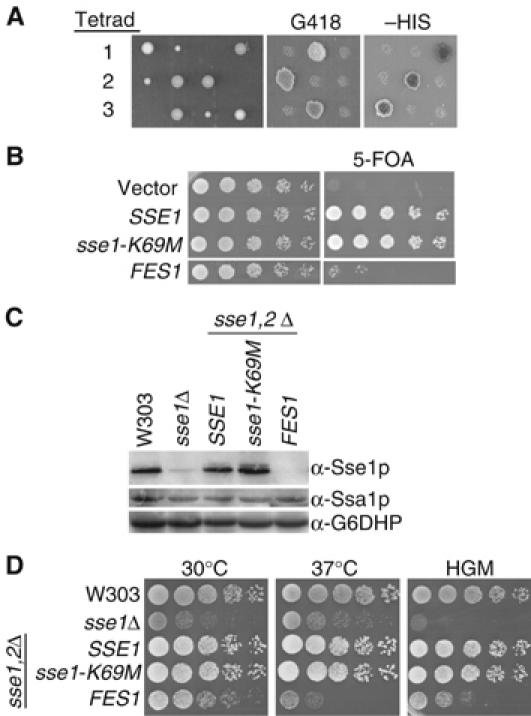
In vivo functions of Sse are essential. (A) Three tetrads generated by mating sse1Δ∷KAN and sse2Δ∷HIS strains were dissected, and viable spores assessed for markers on selective media. (B) Five-fold dilutions of sse1,2Δ cells expressing plasmid-borne SSE1, sse1-K69M or FES1 alleles were spotted onto selective or 5-FOA media (C) Western analysis of Sse1p and Ssa1p levels in W303, sse1Δ and sse1,2Δ strains expressing SSE1, sse1-K69M or FES1 alleles. sse1Δ samples have a small amount of crossreactive Sse2p. Ssa1p levels remain unchanged in all strains. Antibody recognizing glucose 6-phosphate dehydrogenase (Sigma) was a loading control. (D) sse1,2Δ strains expressing plasmid-borne SSE1, sse1-K69M or FES1 alleles were spotted in five-fold dilutions onto YPD media at permissive conditions (30°C), high temperature (37°C) or in the presence of hygromycin (10 μg/ml).
The in vivo effects of the Sse1-K69Mp variant were assessed using the plasmid shuffle technique. A plasmid expressing the sse1-K69M allele was transformed into the sse1,2Δ strain carrying pMPM-CUA-SSE1, and 5-FOA selection forced the loss of the constitutive SSE1 expression. With expression of the sse1-K69M allele as the sole source of Sse protein, cells were viable. The robustness of complementation was examined under conditions of stress. While the sse1,2Δ genotype is lethal, cells deficient only in Sse1p grow slowly and are sensitive to translational inhibitors such as hygromycin (Mukai et al, 1993; Albanese et al, 2006; Figure 5D). Spot test analyses demonstrate that the growth of cells expressing solely the sse1-K69M allele is identical to wild-type cells at both high temperatures and in the presence of hygromycin. Importantly, the sse1,2Δ strain produces both Sse1p or Sse1-K69Mp at similar levels (Figure 5C). The cellular concentration of Sse1p is tightly regulated and cells do not survive when protein levels are aberrant (Shaner et al, 2004); thus, one would expect deregulated production of a mutant protein with a reduced activity. The unchanged levels of the Sse1-K69Mp protein indicates that this mutant does not have a significantly reduced activity, and further supports the notion that ATP hydrolysis is not required for in vivo functions of Sse1p.
The cytosolic NEFs, Sse1p and Fes1p, exhibit partial functional redundancy
Owing to their vital importance in numerous protein folding processes, chaperone networks typically have a degree of functional redundancy, and we wondered whether this also applied to the cytoplasmic NEFs. Remarkably, overexpression of FES1 enabled sse1,2Δ cells to survive upon loss of constitutive SSE1 expression, although not as efficiently as the sse1-K69M allele (Figure 5B). Importantly, the complete absence of Sse1p and Sse2p in these cells was confirmed by Western analysis (Figure 5C). We observed that cells of an sse1,2Δ strain expressing FES1 grow slower at 30°C than cells with wild-type levels of Sse1p. Furthermore, under stress conditions such as heat or hygromycin treatment, the viability of the sse1,2Δ FES1 strain is greatly reduced (Figure 5D). Thus, although these results suggest Sse1p's NEF function is essential for viability, Fes1p can only partially compensate for Sse1p in vivo.
In vivo role of Sse1p in substrate refolding
Sse1p can efficiently promote nucleotide release from the cytoplasmic Hsp70s; however, increased concentrations of another NEF (Fes1p) is insufficient to fully complement sse1,2Δ cells under stressful conditions. Furthermore, based on Sse1p's homology to the Hsp70 chaperone family, and an ability to prevent aggregation of model substrates in vitro (Goeckeler et al, 2002), it seems possible that the cellular role of Sse1p is not restricted to NEF activity.
To further define the required functions of Sse1p, we assessed the in vivo chaperone-dependent refolding of thermally denatured luciferase proteins from bacteria (Vibrio harveyi) and firefly (Photinus pyralis) (Schroder et al, 1993; Frydman et al, 1999; Manukhov et al, 1999) (Figure 6). These proteins can be considered as independent test substrates, as the firefly luciferase (FFL) is a monomeric protein unrelated in sequence or structure to the heterodimeric bacterial protein (LuxAB) (Nathan and Lindquist, 1995). We determined the recovery of protein activity after heat shock of cells. Surprisingly, deletion of the SSE1 gene did not significantly influence refolding of the firefly luciferase substrate, but had a much greater effect on the bacterial luciferase (Figure 6A and C), where only 40–50% of activity was attained as compared to wild-type cells. In contrast, the fes1Δ strain was significantly impaired in refolding of the bacterial substrate, as well as the FFL as previously reported (Ahner et al, 2005). Taken together, it seems that Ssa1p generally requires an NEF to stimulate protein refolding. The different requirements for Sse1p can be explained in two ways; Sse1p may be more involved in the folding of a specific subset of substrates, or alternatively, upregulated Sse2p expression in the absence of Sse1p is sufficient for refolding of firefly but not bacterial luciferase.
Figure 6.
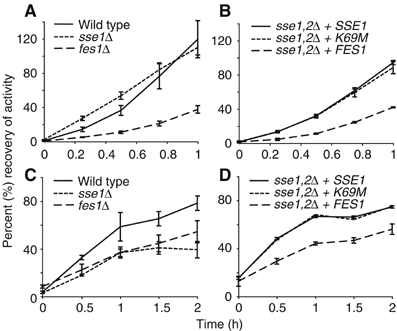
Recovery of thermally denatured firefly luciferase (A, B) and bacterial luciferase (C, D). Cells were grown to mid-logarithmic phase and heat shocked as described in Materials and methods. All strains were assayed for a given substrate in the same experiment, however, are presented separately for clarity: (A, C) wild type, sse1Δ and fes1Δ, (B, D) sse1,2Δ strains expressing SSE1, sse1-K69M or FES1 alleles. Recovery of activity is expressed as a percentage of activity before heat treatments, and is the average of three independent samples. Error bars indicate the standard deviation.
The sse1,2Δ strains rescued by plasmid-borne SSE1, sse1-K69M or FES1 were also examined for their ability to refold each luciferase substrate (Figure 6B and D). As observed for the spot tests analyses, there was little difference between wild-type Sse1p and the Sse1p-K69Mp mutant. Refolding of FFL and LuxAB in sse1,2Δ FES1 cells, however, was slower than in sse1,2Δ cells overexpressing SSE1. At first, this effect on the FFL appears surprising; the refolding in sse1Δ cells did not differ from wild type, whereas lower recovery levels in fes1Δ cells indicate a key role for this protein. Thus, one might expect that FES1 overexpression would be sufficient even in the absence of Sse. However, it appears that Sse2p levels in sse1Δ cells are sufficient for the refolding of FFL but not bacterial luciferase. Taken together, the absence of Sse proteins is detrimental to the refolding of both luciferase substrates. This is suggestive of an additional NEF-independent function for this class of proteins.
Discussion
This is the first report of a nucleotide exchange activity for the Hsp110 class of proteins, and provides insights into the physiological role of the Sse1p homolog, as well as its molecular mode of action. Until now, there has only been one cytoplasmic NEF described to act on Ssa1p (Fes1p;Kabani et al, 2002), and none for the Ssb proteins. The finding that Sse1p is a highly efficient NEF for both of these cytosolic Hsp70s represents a major step towards understanding the functional relationships between different components of the cytosolic Hsp70 network.
The proposition that NEF activity is one of the primary functions of Sse is underscored by genetic analysis of sse1,2Δ mutants. We established that the sse1,2Δ cells are nonviable, consistent with Morano and co-workers (Shaner et al, 2005), and unexpectedly observed that the lethal phenotype is suppressed by overexpression of the unrelated cytosolic NEF, FES1. The compensation for Sse is incomplete, however, as the sse1,2Δ FES1 cells are sensitive to stress and less efficient in the refolding of model proteins. This suggests that either Sse possesses additional NEF-independent activities or, alternatively, that the NEF activity of Fes1p is insufficient to fully compensate for the loss of Sse. Whereas Fes1p was significantly less efficient at stimulating ADP release from Ssa1p, the ability to stimulate in vitro luciferase refolding was only slightly less than Sse1p. Thus, the reason behind the inability of Fes1p to fully compensate for Sse in vivo remains unclear.
The phenotype of the sse1Δ strain clearly demonstrates that Sse1p has some part in the cellular response to stress conditions, and correlates with the incomplete suppression of the sse1,2Δ phenotype by Fes1p. We found that Sse1p forms a stable heterodimeric complex with two major Hsp70 chaperones of the yeast cytosol, Ssa1p and Ssb1p; this is consistent with recent studies (Shaner et al, 2005; Yam et al, 2005), which also observed that virtually the entire pool of cellular Sse1p is engaged in these complexes. These data suggest that the majority, if not all, of Sse1p's cellular functions directly involve the Hsp70s. Taken together with the apparent substrate-specific involvement in refolding processes, we propose that, together with Ssa1p or Ssb1p, the Sse proteins facilitate the folding of a subset of substrates through a mechanism more complicated than the simple stimulation of Hsp70 nucleotide release.
Although the precise mechanism of interaction between Sse1p and the Hsp70s remains unclear, we can form some conclusions regarding the kinetics of complex formation and nucleotide involvement. We observed that Sse1p and Ssa1p interact with a high affinity, and fast kinetics of binding and release. The determined KD of 100–150 nM is remarkably similar to that seen between GrpE and DnaK (KD=64 nM), as well as for interactions between the Hsp70 ATPase domain and HspBP1 (KD=62 nM) and BAG-1 (KD=41 nM) (Chesnokova et al, 2003; Shomura et al, 2005). Furthermore, the calculated association rate constant (2.3 × 105 M−1 s−1) is very similar to the rates reported for GrpE (3.4 × 105 M−1s−1) and BAG-1 (5.3 × 105 M−1 s−1) (Stuart et al, 1998; Chesnokova et al, 2003). The determined dissociation rates (0.028 and 0.003 s−1) are comparable to the rate determined for the DnaK–GrpE interaction (0.033 and 0.0015 s−1), but one order of magnitude smaller than the dissociation rate determined for the Hsc70–Bag1 interaction (0.267 s−1). The similarities of Sse1p–Ssa1p interaction kinetics to that for other NEFs is striking, making it unlikely that complex formation was influenced by the C-terminal tag used to tether Ssa1p. Sse1p did not interact with Ssa1p in the presence of excess ATP, as also observed for other nucleotide exchange factors including Sls1p, Fes1p and HspBP1 (Kabani et al, 2000; Kabani et al, 2002; Shomura et al, 2005). Thus, Sse1p binds Ssa1p molecules in the ADP-bound state with ADP dissociation leading to a high-affinity interaction between the two molecules. The subsequent rebinding of ATP appears to induce dissociation of the Sse1p–Ssa1p complex, as we observed the affinity between the two proteins decreases dramatically in the presence of ATP. Whether substrates bound to Ssa/Ssb or Sse1p affect the interaction between the two molecules is currently unknown, and requires further investigations.
We observed that Sse1p is a more efficient NEF for Ssa1p when in the ATP-bound conformation. This finding is corroborated by the analyses of the ATP-hydrolysis-deficient variant, Sse1-K69Mp, which is constantly in the ATP-bound state. In vitro, this variant protein is capable of acting as an NEF for Ssa1p, and is as functionally active as wild-type protein in vivo. Furthermore, the kinetics of interaction with Ssa1p are unchanged from that of the wild-type protein (data not shown). Interestingly, in this issue, Dragovic et al describe an ATP-binding mutant that is unable to act as an NEF. Thus, it seems that binding, but not hydrolysis of nucleotide by the Hsp110 protein is a prerequisite for its NEF function. It is unclear how the different nucleotide-bound conformations relate to NEF activity, and further work is necessary to clarify the dynamics of the association between Hsp110 and Hsp70 proteins, and delineate the precise mechanism of nucleotide release.
A role for evolutionary divergent Hsp70 members to regulate classical Hsp70 proteins is endorsed by two recent studies. Ssz1p has been reported to facilitate the stimulatory function of the Zuo1p J-protein on Ssb (Huang et al, 2005), whereas Lhs1p, an Hsp170 family member, has a coordinated mechanism of activation with Kar2p in the endoplasmic reticulum (Steel et al, 2004). In fact, Lhs1p is reported to act as a nucleotide exchange factor for Kar2p, whereas Kar2p stimulates ATP hydrolysis of Lhs1p. Hsp110 and Hsp170 proteins form distinct subfamilies from classical Hsp70s based on strong differences in both the length and sequence of the C-terminal regions. Thus, it seems these outliers in the Hsp70 superfamily have independently evolved as a specialized set of co-chaperones for the classical Hsp70 proteins.
We have demonstrated that Sse1p forms a stable complex with the ADP state of Ssa/Ssb and accelerates nucleotide dissociation in vitro. However, the fate of the complex after the exchange of nucleotides bound to Ssa/Ssb remains elusive. Three possible models may be used to describe subsequent events (Figure 7): (i) The ternary complex comprised of Ssa/Ssb, substrate and Sse1p dissolves in response to ATP binding and the substrate is released. (ii) An Sse1p–Ssa/Ssb complex acts in a concerted refolding action, whereby Sse1p may stabilize intermediates that are passed back to Ssa/Ssb for folding. (iii) ATP binding to Ssa/Ssb or Sse1p induces a transfer of the substrate to Sse1p, before being shuttled to other chaperones such as TRiC or Hsp90. In the last two scenarios, Sse1p directly interacts with the Ssa/Ssb-bound substrate, and could account for the substrate-specific effects observed in the in vivo luciferase-refolding experiments. All three models underline the central role of the Sse proteins in the cytosolic chaperone network as essential regulators of Ssa/Ssb function.
Figure 7.
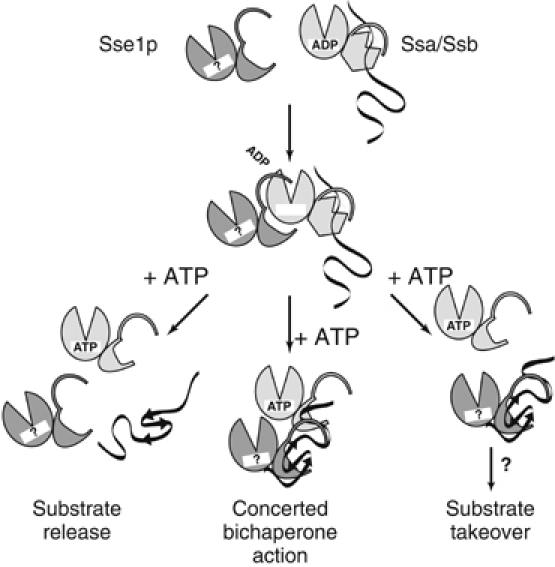
Graphical models for the molecular mechanisms of substrate folding by Sse1p and Ssa/Ssb. Within the framework of the ternary complex formed by Ssa1p, substrate and Sse1p, nucleotide dissociation is triggered, and Ssa1p is free to bind ATP. Three potential pathways leading to the final outcome of a correctly folded substrate: (i) the complex may disassemble into free Sse1p, Ssa1p and folded substrate; (ii) a coordinated bichaperone mechanism where Sse1p and Ssa1p could either work in tandem, or in unison to fold the substrate, (iii) the substrate is handed to Sse1p before a transfer to other cellular chaperones.
Materials and methods
Antibodies and reagents
Rabbit polyclonal antibodies were raised against purified Sse1p and Ssa1p; whole antiserum recognizing glucose-6 phosphate dehydrogenase was obtained from Sigma. Luciferase and luciferin were purchased from Roche, [α32P]ATP was from Amersham Biosciences, and MABA-ADP from TriLink Biotechnologies Inc., San Diego.
Strains and plasmids
A strain bearing the C-terminally TAP-tagged SSE1 integrated into the chromosome under the endogenous promoter was obtained from the EUROSCARF strain collection (Gavin et al, 2002). Isolation of TAP-tagged Sse1p complexes was performed according to Rigaut et al (1999).
The sse1Δ∷KAN strain was generated in the W303 (MATa trp1-1 ura3-1 leu2-3,11 his3-11,15 ade2-1 can1-100) background by direct replacement of the coding region with the KanMX4 cassette, whereas the sse2Δ∷HIS and fes1Δ∷HIS strains were generated by replacement with a His3MX6 cassette. The sse1Δ∷KAN was mated with the sse2Δ∷HIS strain, and the diploid transformed with a centromeric plasmid expressing SSE1 from the ADH promoter (pMPM-CUA-SSE1) before sporulation. Spore phenotypes, consistent with the presence of both KanMX4 and His3MX6 markers, were confirmed to be sse1,2Δ by genomic DNA PCR and Western analysis. Standard protocols were used for transformations, sporulation and propagation.
The SSE1 and FES1 overexpression plasmids contained 500–700 bp of 5′UTR in addition to the coding region to promote gene expression. The SSE1 and sse1-K69M genes were inserted into the episomal pRS425 plasmid, whereas FES1 was on the pRS423 plasmid. The sse1-K69M mutation was generated using two-step PCR overlap mutagenesis.
The firefly luciferase gene lacking the peroxisome-targeting sequence was expressed from a glyceraldehydes-3-phosphate dehydrogenase promoter in the pRS316 plasmid.
Protein purification
Ssa1p and Ssb1p with a TEV-cleavable N-terminal His6-tag were expressed from pProEX Htb vectors (Invitrogen) in BL21 Star™ bacteria (Invitrogen) and purified as described (Shorter and Lindquist, 2004). Ssa1p with a C-terminal His10-tag was expressed from the pET20b vector (Novagen). The protein was isolated as for the N-terminally tagged version, with the exclusion of TEV protease cleavage steps. An N-terminally GST-tagged Fes1p was expressed from the pGEX-4T-2 vector (Amersham Biosciences) and purified as described (Kabani et al, 2002). The purification strategies for the Sse1p and Sse1-K69Mp proteins were as described previously (Raviol et al, 2005).
Co-immunoprecipitation of 3H-Sse1p
Sse1p was tritiated using equimolar amounts of N-succinimidyl-[2,3-3H]propionate (Amersham Biosciences). Equal molar amounts (0.5 μM) of Ssa1p and 3H-Sse1p were incubated in T-buffer (20 mM Tris pH 7.6, 200 mM KCl, 10 mM MgCl2, 5% glycerol, 0.05% Tween-20) at 30°C, for 30 min. Unlabelled Sse1p and/or ATP was added to 5 μM or 5 mM and incubated for a further 10 min. Samples were diluted with cold T-buffer and incubated with α-Ssa1p antibody for 1 h, 4°C. Protein A Sepharose (Amersham Biosciences) was added, and samples were incubated for a further 2 h. Beads were washed with two times 50 volumes of T-buffer, and transferred to scintillation vials for counting. The average of two independent assays is presented.
Biacore analysis
Biacore 3000 (Biacore AB, Uppsala, Sweden) was used for the surface plasmon resonance spectroscopy. All assays were run at 25°C in running buffer (0.01 mM Hepes pH 7.6, 200 mM KCl, 50 mM EDTA and 0.005% Tween 20). In total, 150–2000 RU of Ssa1p with a C-terminal His10-tag were immobilized on an NTA chip (Biacore). Sse1p was dialyzed and diluted into running buffer, and injected concentrations ranged from 31.3 to 2000 nM. The NTA chip was regenerated with successive injections of 3 M GuHCl, 12 mM NaOH and 0.35 M EDTA. After regeneration, the chip was reloaded with NiCl2. BIA evaluation 4.0.1 and GraFit 5.0.1 software were used to fit the data. The KD was determined by fitting a simple one-sided ligand-binding equation to the titration data. The association rate constant was calculated from weighted averages of koff/KD. No significant changes in kinetic parameters were observed between runs with different RUs of immobilized Ssa1p.
ATPase assays
Determination of steady-state ATPase activity was performed as described (Raviol et al, 2005). All proteins were used at a concentration of 1 M. To determine absolute ATP-hydrolysis rates, single-turnover complexes were generated as described (Mayer et al, 1999).
Stopped-flow analysis
Stopped-flow analysis was performed using an Applied Photophysics 18 SX instrument. In all, 0.5 μM (Ssa1p and Ssb1p) or 2.5 μM (Sse1p) of protein was preincubated with MABA-ADP in HKM (25 mM Hepes–KOH pH 7.5, 150 mM KCl, 5 mM MgCl2) buffer for 30 min at 30°C. For basal dissociation rate determination, equal volumes of protein–MABA-ADP complex and 500 μM unlabeled ADP were rapidly mixed in the stopped-flow device and the decrease in fluorescent signal was monitored (time scales ranged from 0.1 to 20 s). Stimulation of the dissociation rate by different concentrations of Sse1p or Fes1p (0.5, 1, 2, 5 and 10 μM) was examined in the presence of 500 μM unlabeled ADP or ATP. Sse1-K69Mp was tested at 5 μM concentration. The kinetic rates were fitted using the GraFit 5.0.1. program.
In vitro luciferase assays
Luciferase (80 nM in 1 M glycylglycine, pH 7.4) was incubated in refolding buffer (25 mM Hepes pH 7.6, 50 mM KCl, 10 mM MgCl2, 10 mM DTT, 2 mM ATP, 0.1 mg/ml BSA) with chaperones at 42°C for 12 min, and then placed at 30°C. The concentrations of Ssa1p and Ydj1p were 1 μM, respectively, whereas Sse1-K69Mp and Fes1p were used at 0.5 μM. After 45 min, luciferase activity was assayed as described (Schroder et al, 1993).
In vivo luciferase assays
Overnight cultures of cells constitutively expressing luciferase proteins were diluted and grown at 30°C to mid-logarithmic phase. After 30 min at 37°C to induce the heat shock response, cells were placed at 46°C for 30 min. Cycloheximide was added to 10 μg/ml to prevent further expression of luciferase protein, and cells were transferred to 30°C to recover. At different intervals during recovery, aliquots of culture were mixed with luciferin (50 μM), and fluorescence was measured in a BioLumat (Berhold). The substrate for bacterial luciferase activity was 5 μl of decanal (Sigma). Recovery is expressed as a percentage of the activity before heat shock treatments. A typical experiment with an average of triplicates is presented for each substrate.
Supplementary Material
Supplementary Figure
Acknowledgments
We thank J Weibezahn for the Ydj1p and Ssa1p antibody, P Tessarz for the firefly luciferase plasmid and Ssa1p purification, R Nikolay for assistance with Biacore analyses and C Andreasson for instrumental discussions. Plasmids for producing Ssa1p and Ssb1p and the bacterial luciferase were kindly provided by S Lindquist (Whitehead Institute, Cambridge). The FES1 expression plasmid was a gift from G Jones (NUI Maynooth, Ireland).
References
- Ahner A, Whyte FM, Brodsky JL (2005) Distinct but overlapping functions of Hsp70, Hsp90, and an Hsp70 nucleotide exchange factor during protein biogenesis in yeast. Arch Biochem Biophys 435: 32–41 [DOI] [PubMed] [Google Scholar]
- Albanese V, Yam AY, Baughman J, Parnot C, Frydman J (2006) Systems analyses reveal two chaperone networks with distinct functions in eukaryotic cells. Cell 124: 75–88 [DOI] [PubMed] [Google Scholar]
- Brodsky JL, Werner ED, Dubas ME, Goeckeler JL, Kruse KB, McCracken AA (1999) The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J Biol Chem 274: 3453–3460 [DOI] [PubMed] [Google Scholar]
- Bukau B, Horwich AL (1998) The Hsp70 and Hsp60 chaperone machines. Cell 92: 351–366 [DOI] [PubMed] [Google Scholar]
- Bush GL, Meyer DI (1996) The refolding activity of the yeast heat shock proteins Ssa1 and Ssa2 defines their role in protein translocation. J Cell Biol 135: 1229–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokova LS, Slepenkov SV, Protasevich II Sehorn MG, Brouillette CG, Witt SN (2003) Deletion of DnaK's lid strengthens binding to the nucleotide exchange factor, GrpE: a kinetic and thermodynamic analysis. Biochemistry 42: 9028–9040 [DOI] [PubMed] [Google Scholar]
- Easton DP, Kaneko Y, Subjeck JR (2000) The hsp110 and Grp1 70 stress proteins: newly recognized relatives of the Hsp70s. Cell Stress Chaperones 5: 276–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frydman J, Erdjument-Bromage H, Tempst P, Hartl FU (1999) Co-translational domain folding as the structural basis for the rapid de novo folding of firefly luciferase. Nat Struct Biol 6: 697–705 [DOI] [PubMed] [Google Scholar]
- Gassler CS, Wiederkehr T, Brehmer D, Bukau B, Mayer MP (2001) Bag-1M accelerates nucleotide release for human Hsc70 and Hsp70 and can act concentration-dependent as positive and negative cofactor. J Biol Chem 276: 32538–32544 [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141–147 [DOI] [PubMed] [Google Scholar]
- Goeckeler JL, Stephens A, Lee P, Caplan AJ, Brodsky JL (2002) Overexpression of yeast Hsp110 homolog Sse1p suppresses ydj1-151 thermosensitivity and restores Hsp90-dependent activity. Mol Biol Cell 13: 2760–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F, Kuriyan J (1997) Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science 276: 431–435 [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295: 1852–1858 [DOI] [PubMed] [Google Scholar]
- Huang P, Gautschi M, Walter W, Rospert S, Craig EA (2005) The Hsp70 Ssz1 modulates the function of the ribosome-associated J-protein Zuo1. Nat Struct Mol Biol 12: 497–504 [DOI] [PubMed] [Google Scholar]
- Kabani M, Beckerich JM, Brodsky JL (2002) Nucleotide exchange factor for the yeast Hsp70 molecular chaperone Ssa1p. Mol Cell Biol 22: 4677–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabani M, Beckerich JM, Gaillardin C (2000) Sls1p stimulates Sec63p-mediated activation of Kar2p in a conformation-dependent manner in the yeast endoplasmic reticulum. Mol Cell Biol 20: 6923–6934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Schilke B, Craig EA, Horwich AL (1998) Folding in vivo of a newly translated yeast cytosolic enzyme is mediated by the SSA class of cytosolic yeast Hsp70 proteins. Proc Natl Acad Sci USA 95: 12860–12865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Shabbir A, Cardozo C, Caplan AJ (2004) Sti1 and Cdc37 can stabilize Hsp90 in chaperone complexes with a protein kinase. Mol Biol Cell 15: 1785–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XD, Morano KA, Thiele DJ (1999) The yeast Hsp110 family member, Sse1, is an Hsp90 cochaperone. J Biol Chem 274: 26654–26660 [DOI] [PubMed] [Google Scholar]
- Lu Z, Cyr DM (1998) Protein folding activity of Hsp70 is modified differentially by the hsp40 co-chaperones Sis1 and Ydj1. J Biol Chem 273: 27824–27830 [DOI] [PubMed] [Google Scholar]
- Manukhov IV, Eroshnikov GE, Vyssokikh MY, Zavilgelsky GB (1999) Folding and refolding of thermolabile and thermostable bacterial luciferases: the role of DnaKJ heat-shock proteins. FEBS Lett 448: 265–268 [DOI] [PubMed] [Google Scholar]
- Mayer MP, Laufen T, Paal K, McCarty JS, Bukau B (1999) Investigation of the interaction between DnaK and DnaJ by surface plasmon resonance spectroscopy. J Mol Biol 289: 1131–1144 [DOI] [PubMed] [Google Scholar]
- Mukai H, Kuno T, Tanaka H, Hirata D, Miyakawa T, Tanaka C (1993) Isolation and characterization of SSE1 and SSE2, new members of the yeast HSP70 multigene family. Gene 132: 57–66 [DOI] [PubMed] [Google Scholar]
- Nathan DF, Lindquist S (1995) Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol Cell Biol 15: 3917–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviol H, Bukau B, Mayer MP (2005) Human and yeast Hsp110 chaperones exhibit functional differences. FEBS Lett 80: 168–174 [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032 [DOI] [PubMed] [Google Scholar]
- Rudiger S, Buchberger A, Bukau B (1997) Interaction of Hsp70 chaperones with substrates. Nat Struct Biol 4: 342–349 [DOI] [PubMed] [Google Scholar]
- Schroder H, Langer T, Hartl FU, Bukau B (1993) DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J 12: 4137–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner L, Trott A, Goeckeler JL, Brodsky JL, Morano KA (2004) The function of the yeast molecular chaperone Sse1 is mechanistically distinct from the closely related hsp70 family. J Biol Chem 279: 21992–22001 [DOI] [PubMed] [Google Scholar]
- Shaner L, Wegele H, Buchner J, Morano KA (2005) The yeast HSP110 SSE1 functionally interacts with the HSP70 chaperones SSA and SSB. J Biol Chem 280: 41262–41269 [DOI] [PubMed] [Google Scholar]
- Shomura Y, Dragovic Z, Chang HC, Tzvetkov N, Young JC, Brodsky JL, Guerriero V, Hartl FU, Bracher A (2005) Regulation of Hsp70 function by HspBP1: structural analysis reveals an alternate mechanism for Hsp70 nucleotide exchange. Mol Cell 17: 367–379 [DOI] [PubMed] [Google Scholar]
- Shorter J, Lindquist S (2004) Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 304: 1793–1797 [DOI] [PubMed] [Google Scholar]
- Sondermann H, Scheufler C, Schneider C, Hohfeld J, Hartl FU, Moarefi I (2001) Structure of a Bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors. Science 291: 1553–1557 [DOI] [PubMed] [Google Scholar]
- Steel GJ, Fullerton DM, Tyson JR, Stirling CJ (2004) Coordinated activation of Hsp70 chaperones. Science 303: 98–101 [DOI] [PubMed] [Google Scholar]
- Stuart JK, Myszka DG, Joss L, Mitchell RS, McDonald SM, Xie Z, Takayama S, Reed JC, Ely KR (1998) Characterization of interactions between the anti-apoptotic protein BAG-1 and Hsc70 molecular chaperones. J Biol Chem 273: 22506–22514 [DOI] [PubMed] [Google Scholar]
- Trott A, Shaner L, Morano KA (2005) The molecular chaperone Sse1 and the growth control protein kinase Sch9 collaborate to regulate protein kinase A activity in Saccharomyces cerevisiae. Genetics 170: 1009–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam AY, Albanese V, Lin HT, Frydman J (2005) HSP110 cooperates with different cytosolic HSP70 systems in a pathway for de novo folding. J Biol Chem 280: 41252–41261 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure