

Abstract
The functions of DNA satellites of centric heterochromatin are difficult to assess with classical molecular biology tools. Using a chemical approach, we demonstrate that synthetic polyamides that specifically target AT-rich satellite repeats of Drosophila melanogaster can be used to study the function of these sequences. The P9 polyamide, which binds the X-chromosome 1.688 g/cm3 satellite III (SAT III), displaces the D1 protein. This displacement in turn results in a selective loss of HP1 and topoisomerase II from SAT III, while these proteins remain bound to the adjacent rDNA repeats and to other regions not targeted by P9. Conversely, targeting of (AAGAG)n satellite V repeats by the P31 polyamide results in the displacement of HP1 from these sequences, indicating that HP1 interactions with chromatin are sensitive to DNA-binding ligands. P9 fed to larvae suppresses the position-effect variegation phenotype of white-mottled adult flies. We propose that this effect is due to displacement of the heterochromatin proteins D1, HP1 and topoisomerase II from SAT III, hence resulting in stochastic chromatin opening and desilencing of the nearby white gene.
Keywords: D1, heterochromatin, HP1, topoisomerase II, 1.688 g/cm3 satellite
Introduction
Polyamides are short polymers composed of aromatic pyrrole and imidazole amino acids that recognize specific DNA sequences with remarkable affinities (Dervan and Edelson, 2003). The use of polyamides provides an alternative approach to the study of DNA satellites (Janssen et al, 2000b), which, unlike regulatory sequences of genes, are difficult to study with classical molecular biology approaches.
It was previously demonstrated that satellite-specific polyamides fed to developing Drosophila melanogaster can induce either gain or loss-of-function phenotypes in adult flies. One of these polyamides, P31, binds with great specificity the (AAGAG)n repeats of the Drosophila satellite V (SAT V) (Janssen et al, 2000b). P31 was found to induce well-defined homeotic transformations in a brown-dominant genetic background that are akin to mutations of the gene encoding GAGA factor (GAF) (Janssen et al, 2000a). The effect of P31 was proposed to be mediated by a redistribution of GAF from its euchromatin-binding sites, where this protein acts as a positive transcription activator to SAT V. The homeotic phenotype is then explained by a reduced availability of GAF for gene expression. In contrast, a gain-of-function phenotype was induced by the P9 polyamide. P9 specifically targets the AT-rich satellites SAT I (AATAT)n and, presumably, SAT III (density 1.688 g/cm3), which is composed of 359-base pairs (bp) repeats. It was observed that feeding P9 to white-mottled (wm4) larvae suppressed the position-effect variegation (PEV) phenotype in adult flies. The wm4 fly strain contains a chromosomal inversion that places the white gene near the centric heterochromatin of the X chromosome, which harbors the rDNA repeats and the enormous 11-Mbp SAT III array (Lohe et al, 1993). PEV is thought to arise through heterochromatin-mediated silencing of the juxtaposed white gene (for a review, see Huisinga et al, 2006). It was proposed that suppression of PEV by P9 (increased expression of the white gene) acts through chromatin opening of SAT III and a subsequent reduction of silencing effects on the nearby white gene. This paper aims to dissect the biological effect of P9 by examining the biochemical composition of SAT III using a combination of in vivo and in vitro experiments.
D1 is an essential protein ubiquitously expressed throughout Drosophila development (Aulner et al, 2002) containing 10 copies of the AT hook motif, a DNA-binding domain that interacts with the minor groove of AT-rich DNA (Reeves and Nissen, 1990). D1 is associated with SAT I and SAT III repeats. Related to D1 is the artificial MATH20 protein that contains 20 AT hook motifs and, as D1, is expected to bind SAT I and SAT III. Overexpression of D1 enhances wm4 PEV (Aulner et al, 2002), while overexpression of MATH20 suppresses it (Girard et al, 1998). To explain these opposing effects, it was suggested that D1 increases, while MATH20 reduces, silencing effects mediated by SAT III (Monod et al, 2002). In contrast, overexpression of a D1 transgene carrying a deletion of its C-terminal domain (D1ΔE) suppresses wm4 PEV, an effect similar to that of MATH20. Because D1ΔE behaves as a dominant negative mutation of D1, we proposed that the C-terminal domain of D1 might serve to recruit proteins required for heterochromatin assembly to SAT I and SAT III.
Heterochromatin protein 1 (HP1) is a well-characterized structural and functional component of heterochromatin. Binding of HP1 to heterochromatin occurs through the specific recognition of histone H3 methylated at the lysine 9 position by the chromo domain of HP1. Mutations in the gene encoding HP1 or in the genes that encode the histone deacetylase and methyl transferase activities required for the creation of an HP1-binding site suppress wm4 PEV (for a review, see Huisinga et al, 2006). The interplay between these proteins provides a rationale for the assembly and propagation of heterochromatin, which might involve other proteins as well.
Topoisomerase II (topo II) might conceivably be implicated in wm4 PEV as it is prominently associated with centric heterochromatin and SAT III repeats, as demonstrated by in vivo cleavage (Käs and Laemmli, 1992) and localization studies (Swedlow et al, 1993). Topo II is a DNA topological handler that relaxes positively and negatively supercoiled DNA and mediates DNA strand passage during catenation/decatenation reactions. Previous studies established that topo II is required for the segregation and assembly of mitotic chromosomes (Adachi et al, 1991). Hence, this enzyme may perhaps play roles in heterochromatin and chromosome structure, in addition to its enzymatic function.
One possibility is that D1 might help to recruit HP1 and topo II to AT-rich satellites and we explore here the mechanism whereby P9 exerts suppression of the wm4 phenotype. Using an immunofluorescence approach, we show that P9 mediates the displacement of D1 from SAT I and SAT III in vivo and in vitro. Moreover, P9 also specifically displaces HP1 and topo II from SAT III, while these proteins remain bound to other genomic sites that are not targeted by P9. Conversely, targeting SAT V repeats with the P31 polyamide results in the selective displacement of HP1 from these sites, supporting the hypothesis that HP1 association with chromatin involves a direct DNA-binding component. We propose that the suppression of the wm4 phenotype by P9 is mediated by displacement of D1, HP1 and topo II from SAT III. To refine our analysis, we studied the effect of topo II inhibitors on wm4 flies and we show that feeding VM26, a topo II poison, to wm4 flies also results in suppression of white-mottled PEV, supporting a structural and/or functional role for this enzyme in heterochromatin-mediated silencing.
Results
Specificity of DNA-binding polyamides for different Drosophila satellites
Previous studies established that the satellite-specific polyamides, P9 and P31, fed to developing flies induce gain or loss-of-function phenotypes, respectively. The binding properties and the biological effects of these polyamides are recapitulated in Supplementary Table I and discussed below.
P31: This compound is known to bind with impressive specificity the (AAGAG)n repeats of satellite V (SAT V) as determined by footprinting and fluorescence microscopy. P31TR, a derivative labeled with Texas red, was demonstrated to bind to the chromocenter of polytene chromosomes and to the 1.7-Mbp (AAGAG)n repeats inserted in the brown-dominant allele of the brown gene (Janssen et al, 2000b). This analysis was extended here to high resolution using mitotic chromosomes obtained from larval neuroblasts (Figure 1A). This panel localizes SAT V repeats to pericentric regions on chromosomes 2, 3 and 4 and to the very tip of the X chromosome. Figure 1A also includes a nucleus, where the signal is well confined to the SAT V repeats congregated in the chromocenter.
Figure 1.
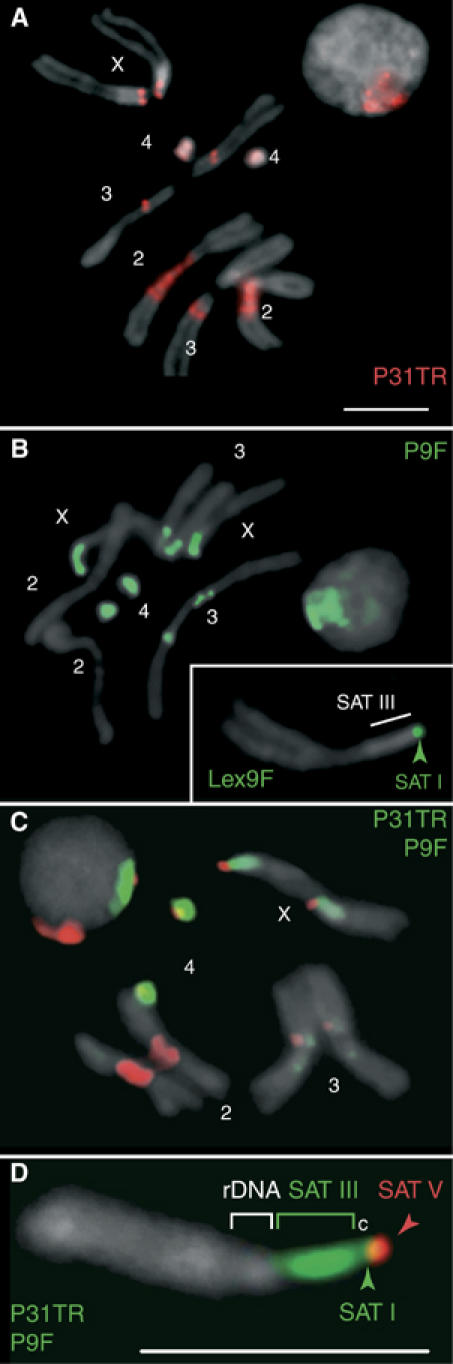
Localization of satellites in diploid cells using fluorescent polyamides. (A–D) Brains dissected from female third-instar larvae were fixed and stained using fluorescent polyamides and counterstained with DAPI (gray). The properties of the drugs are described in Supplementary Table I. P31TR (A) detects satellite V, P9F (B, D) satellites III and I, Lex9F (inset in B) satellite I. SAT I, III and V maps are diagrammed in Figure 4G below. A sample double-stained with P9F and P31TR is shown in (C). The localization of satellite and rDNA repeats is shown for a magnified X chromosome double-stained with P9F and P31TR (D; c: centromere). Scale bars: 5 μm.
P9: This oligopyrrole is specific for AT-rich satellites and, when fed to developing wm4 flies, suppresses the white-mottled PEV phenotype (Janssen et al, 2000a). The biological effect of P9 is most likely exerted through binding to the alpha-like satellite III (SAT III), the major satellite juxtaposed to the inverted white gene of wm4 flies. This notion was examined further here by microscopy of spread mitotic chromosomes stained with the fluorescein-labeled derivative P9F. Figure 1B shows that P9F strongly stains the large (11 Mbp) SAT III abutting the centromere of the X chromosome (see also the enlarged X chromosome in panel D). P9F also detects SAT I (AATAT)n repeats throughout chromosome 4 and related repeats on either side of the centromere of chromosome 3, as well as a short array of SAT I repeats near SAT V (red) on the X chromosomes (Figure 1D). The order of satellites from the tip of the X chromosome to the rDNA locus is SAT V, SAT I and SAT III, as shown in panel D. These assignments are based on staining with Lex9F (inset Figure 1B) and co-staining with P31TR and P9F (Figure 1C). Lex9F (see Supplementary Table I) is a dimeric oligopyrrole that prefers interaction with long (>W9) AT-rich runs. Hence, Lex9F serves to distinguish between the (AATAT)n repeats of SAT I and the shorter AT-runs of SAT III, which it does not bind (inset panel B). These results are in excellent agreement with previous satellite mapping data (Lohe et al, 1993) and are summarized in Figure 4G below.
Figure 4.
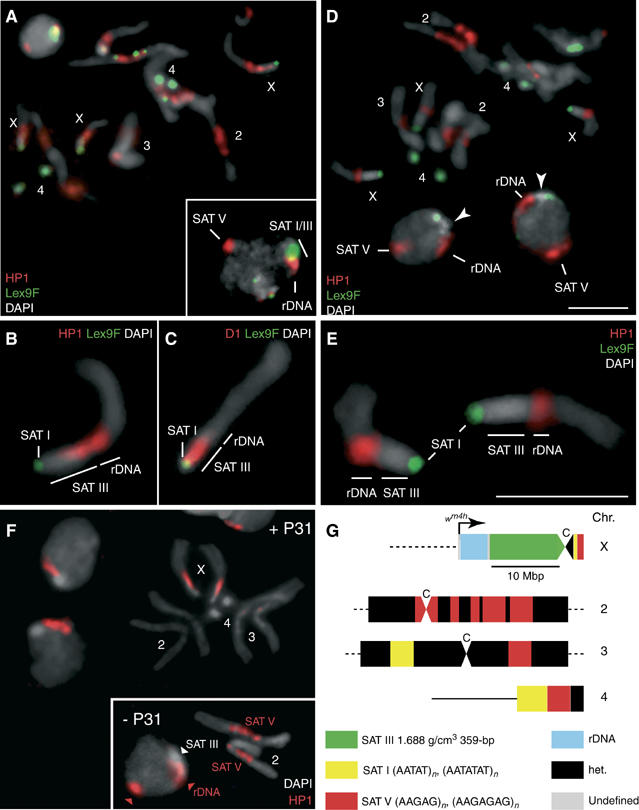
D1 Displacement results in a selective loss of HP1. (A) Localization of HP1 (red) in interphase nuclei and mitotic chromosomes of female diploid larval neuroblasts. Samples were counterstained with Lex9F (green) and DAPI (gray). The inset shows a similarly stained interphase nucleus. A region that stains strongly for HP1 alone corresponds to SAT V repeats on chromosome 2, as assessed by P31TR staining (not shown). rDNA repeats are indicated. (B, C) Magnified X chromosomes after staining for HP1 or D1 (red), respectively, and counterstaining with Lex9F (green) and DAPI (gray). (D–E) Localization of HP1 (red) after treatment with 2.5 μM P9. Magnified X chromosomes are shown in (E). Samples were counterstained with Lex9F (green) and DAPI (gray). In (D), arrowheads indicate the HP1-depleted DAPI-bright region that corresponds to SAT III repeats in interphase nuclei after treatment with P9; SAT V repeats on chromosome 2 are also indicated. (F) The localization of HP1 following treatment with P31. The inset shows untreated chromosomes 2 and a nucleus with the SAT V-associated HP1 signal (arrowhead). A general representation of major satellite blocks is schematized in (G), which also shows the approximate location of the white gene on a wm4 X chromosome, within a type I insertion sequence characteristic of some rDNA repeats. ‘C' denotes the centromere. Scale bars: 5 μm.
The main aim of this report is to elucidate the molecular basis of the gain-of-function phenotype mediated by P9. We proposed that P9 reduces the heterochromatin state of SAT III, presumably as a result of protein displacement. This proposal is explored here, using an experimental approach where polyamides serve two functions: (a) fluorescent derivatives are used to localize the DNA satellites in fixed material (Figure 1) in combination with immunostaining studies and (b) to act as ‘drugs' in vivo and in vitro so as to affect the biochemical composition of heterochromatin.
D1 binds SAT I and SAT III: P9-mediated displacement of D1 in vitro
The D1 protein is associated with SAT I and SAT III repeats and overexpression of D1 enhances PEV of wm4 flies. Figure 2A shows that the immunosignal of D1 (red) colocalizes with the pattern of P9F staining (green, panel B and merged in panel C). Hence, D1 binds SAT I and SAT III repeats in interphase and metaphase chromatin.
Figure 2.
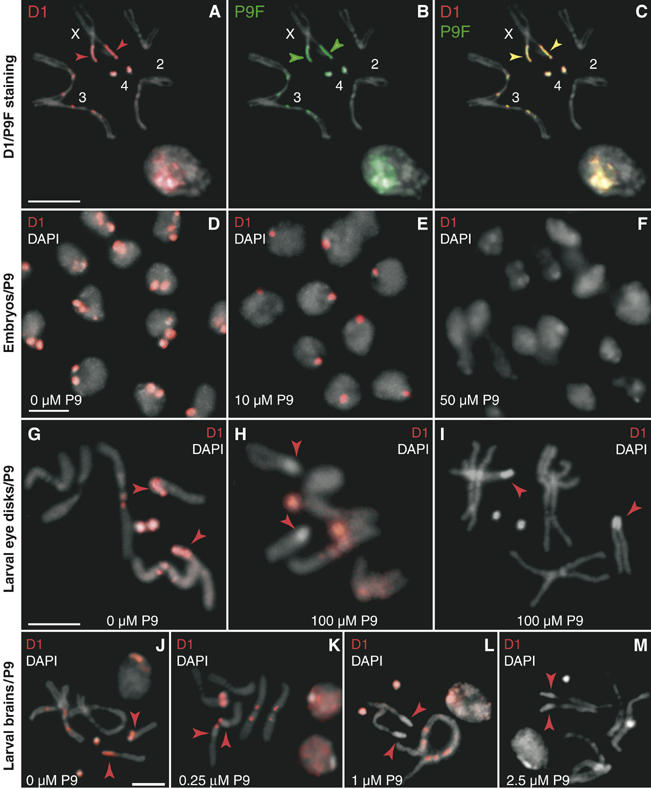
Uptake of oligopyrroles by whole embryos and larvae and displacement of D1. (A–C) Larval neuroblast nuclei and mitotic chromosomes stained with an antibody against D1 (red) and P9F (green) and counterstained with DAPI (gray). (D–F) Cellular blastoderm embryos were treated with 0, 10 or 50 μM P9, immunostained for D1 (red) and mounted in DAPI (gray). Treatment with 10 μM P9 results in a partial loss of the D1 signal, which is complete at 50 μM P9. (G–I) Third-instar larvae were fed colchicine (100 μg/ml) and P9 (0 or 100 μM) to assess displacement of D1 (red signal) from SAT I and SAT III repeats on mitotic chromosomes from eye imaginal disks. Arrowheads indicate the position of SAT III repeats, as determined by P9F staining (not shown). (J–M) The D1 signal in permeabilized larval brains treated with 0, 0.25, 1 or 2.5 μM P9, respectively. Merged DAPI (gray) and D1 (red) signals are shown. Arrowheads show the DAPI-bright SAT III array on the X chromosome. Scale bars: 5 μm.
We explored whether the biological effect of P9 on PEV might arise by displacement of D1. This notion was first studied in vitro with gel retardation/competition experiments. Figure 3A shows that binding of purified D1 to a 359-bp SAT III monomer generates a ladder of retarded complexes reflecting increased binding of D1 (Figure 3A, lanes 1–4). In contrast to D1, addition of P9 (MW=962) resulted in faster migrating complexes (lanes 5–9), due to unbending of the curved SAT III fragment, although retardation was seen at the highest P9 concentration tested (500 nM, lane 10).
Figure 3.
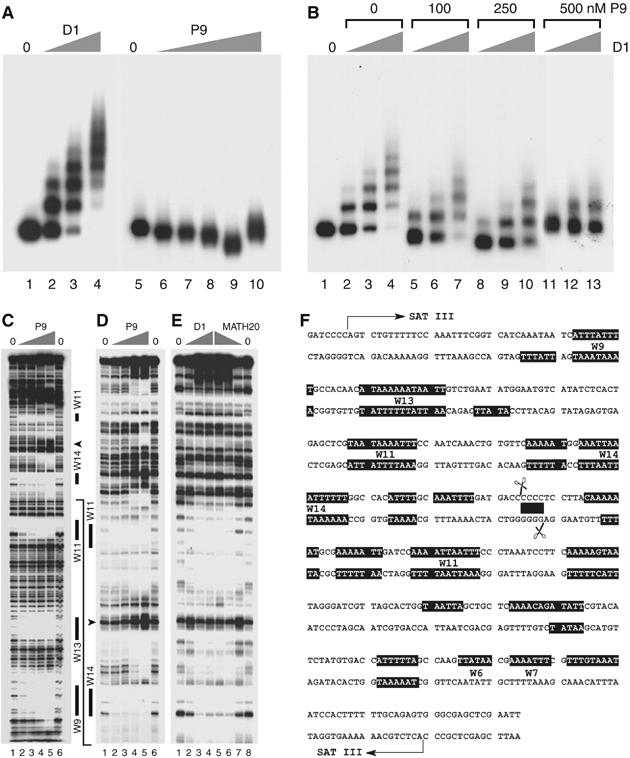
The P9 oligopyrrole competes with D1 for binding to SAT III repeats in vitro. (A) A SAT III monomer was incubated with 2, 5 or 10 ng D1 (2.7, 6.75 and 13.5 nM, lanes 2–4) or P9 (25, 50, 100, 250 or 500 nM, lanes 6–10). No-protein controls are shown in lanes 1 and 5. (B) SAT III was incubated with 1, 5 or 10 ng D1 (1.35, 6.75 or 13.5 nM) in the presence of 0, 100, 250 or 500 nM P9 as shown above the gel. Lane 1 contained no protein. The displacement of D1 by the drug is almost complete at 500 nM P9 (lanes 11–13). (C) SAT III monomer DNA 3′-end labeled on the lower strand was incubated with 0 (lanes 1 and 6) or 50, 100, 250, 500 nM P9 (lanes 2–5) and digested with DNAse I. Solid bars denote regions protected from digestion at the lowest P9 concentrations used and correspond to the largest dA·dT tracts (W) of SAT III (W6/W7–W14, where the number denotes the number of dA·dT bp). Note that only W tracts that are clearly resolved on the gels shown are indicated. The arrowhead indicates a P9-induced hypersensitive site. Binding of the same concentrations of P9 to SAT III (D) was compared to that of D1 and MATH20 (E). Lanes 2–4 of panel (E) correspond to 2, 5 and 10 ng D1 (2.7, 6.75 and 13.5 nM), while lanes 5–7 contained 2.5, 1.5 and 0.5 ng MATH20 (1.5, 0.9 and 0.3 nM). Lanes 1 and 8 are no-protein controls. The solid line starting at the bottom of (D) indicates the SAT III region shown on these gels relative to (C). These results are summarized on the sequence of a SAT III monomer shown in (F). Filled boxes indicate the regions protected by binding of D1, MATH20 and P9. Scissors indicate the sequence cleaved by topoisomerase II in vivo (Borgnetto et al, 1996), the filled box corresponds to the four consecutive dC·dG bp located within the staggered cut. This region spans the P9-induced DNAse I-hypersensitive site and is not bound by D1 or MATH20.
Figure 3B establishes that P9 dissociates D1/SAT III complexes in vitro. Examination of panel B shows that the D1/SAT III ladder diminishes in a concentration-dependent manner upon addition of P9 (compare lanes 5–7, 8–10 and 11–13 with lanes 2–4). This is accompanied by an increase in the mobility of the free DNA and protein-bound fragments indicative of P9 binding. This displacement reaction was independent of the order in which P9 and D1 were added (data not shown). Taken together, these results demonstrate that the P9 oligopyrrole binds SAT III repeats in vitro and displaces the D1 protein bound to these sequences.
DNAse I footprinting experiments demonstrate that the competition between P9 and D1 is the result of nearly identical DNA-binding properties. Figure 3C (lanes 2–5) shows that P9 primarily protects stretches of dA·dT base pairs (W7–W14) of the SAT III repeat from DNAse I digestion. These tracts are also the main interaction sites for D1 (Figure 3D and E). Although both ligands bind similar sequences, some minor differences can be noted, as is manifested by the greater affinity of P9 for longer dA·dT tracts. A similar experiment performed with the artificial MATH20 protein yielded a footprint similar to that of P9 or D1 (lanes 5–7 of panel E). These interaction results are graphically summarized in Figure 3F.
In vivo displacement by P9 of D1 from SAT I and III in embryos
We tested whether P9 displaces D1 from SAT III in vivo by exposing whole embryos at the cellular blastoderm stage to this compound. Panels D–F of Figure 2 show that P9 induces a dose-dependent decrease of the D1 immunosignal in the chromocenter of nuclei. In untreated embryos, D1 (red) was found to localize predominantly to two foci in each nucleus (panel D) that correspond to SAT I and SAT III as verified by P9F binding (not shown). Exposure of embryos to 10 μM P9 (panel E), however, led to a loss of D1 from some but not all foci, typically resulting in the detection of only one D1-positive region in each nucleus. This displacement of D1 was essentially complete at the highest drug concentration tested (50 μM, panel F). These results extend our in vitro competition experiments and demonstrate that P9 displaces D1 from AT-rich satellite repeats in vivo.
D1 displacement from chromosomes of eye disks in vivo
The displacement of D1 was studied in more detail on mitotic chromosomes from eye imaginal disks isolated from P9-fed larvae. As shown above for neuroblast chromosomes (Figure 2A), the immunosignal of D1 is also predominantly confined to SAT III and SAT I in eye disk chromosomes isolated from control larvae (no P9, Figure 2G). In contrast, panels H and I show that D1 is displaced if larvae were fed yeast paste containing 100 μM P9. While displacement by P9 of D1 from SAT III was complete (panel H), we observed a stochastic loss of D1 from SAT I in about 20% of the examined mitotic spreads. Panel I shows such an example, where D1 was completely displaced from both SAT I and III repeats. This experiment shows that P9 displaces D1 from chromosomes isolated from eye disks, where the suppression of wm4 PEV is exerted.
The above experiments were carried out with large amounts of P9. As our polyamide supply is very limited, we used permeabilized larval brains in the following experiments, where lower concentrations can be used. Squashes of larval brains also have the advantage that they yield mitotic chromosome spreads of excellent quality (Figure 2A–C). As shown in panels J–M of Figure 2, the displacement of D1 in permeabilized brains occurs at a much lower P9 concentration. Notably, we observed that a P9 concentration of 250 nM suffices to displace D1 from SAT III (panel K, red arrowheads), while higher concentrations (1–2.5 μM) are required to evict D1 from SAT I (panels L and M).
P9 displaces HP1 from SAT III but not from rDNA repeats
The possible implication of HP1 in the suppression of wm4 PEV by P9 was also studied. Figure 4A shows that HP1 (red) is bound to the pericentric regions of mitotic chromosomes, with a predominant association with SAT III and SAT V repeats of chromosomes X and 2, respectively. In contrast, the HP1 immunosignal is undetectable on the SAT I repeats on chromosomes 4 and Y (not shown) and at the tip of the X chromosome. It is important to note that Lex9F was used to counterstain these panels of Figure 4, as it serves to highlight SAT I (not SAT III) repeats that remain green since their HP1 immunosignal remains undetectable (panel A). This conclusion is also supported by an examination of the staining pattern of interphase nuclei. The inset of panel A shows the intense HP1 staining of SAT V and partial staining of the SAT III chromocenter (yellow), while HP1 is excluded from a region (green) that also harbors SAT I repeats.
Careful inspection of the HP1 immunosignal over the X chromosome at higher magnification (panel B) shows that its signal extends from the rDNA repeats to the adjacent SAT III array, where staining reproducibly appears in the form of a gradient that diminishes toward the centromere. In contrast to the binding of HP1 to the SAT III and rDNA repeats, the immunosignal of D1 shown for comparison is restricted to the centromere-proximal region of SAT III and only partially overlaps with HP1 (panel C).
Panels D and E of Figure 4 show that P9 selectively displaces HP1 from SAT III repeats, but not from the rDNA array. This conclusion is best noted on the enlarged X chromosomes shown in panel E. This micrograph shows the green Lex9F stain of SAT I at the chromosomal tip and the red immunosignal of HP1 over the rDNA repeats, while the SAT III region remains unstained (gray). Hence, following treatment with P9, HP1 is delocalized from the SAT III repeats, but remains associated with the rDNA array. Examination of panel D demonstrates that, in contrast, the protein remains prominently associated with SAT V repeats on chromosome 2. The nuclei shown in panel D also confirm this conclusion. In this case, the loss of HP1 from SAT III is reflected by the appearance of a green–gray–red pattern over the chromocenter, corresponding to the SAT I, SAT III and rDNA repeats, respectively, while the strong HP1 association site on SAT V repeats is unaffected.
We tested next the sensitivity of HP1 binding to chromatin to treatment with the P31 polyamide. As shown in the inset of Figure 4F, HP1 is prominently associated with SAT V repeats on chromosome 2. Incubation with P31 led to a selective displacement of HP1 from the pericentric region of chromosome 2, without affecting the association of the protein with SAT III and rDNA repeats on the X chromosome. We conclude from these results that HP1 can be targeted by polyamides in a sequence-specific fashion that reflects the selectivity of P9 and P31 for SAT III and SAT V, respectively. This suggests that the association of HP1 with satellites repeats may occur, at least in part, via DNA sequence recognition (Zhao et al, 2000; Perrini et al, 2004).
HP1 delocalization results in the invasion of rDNA repeats by D1
The displacement of HP1 from SAT III repeats might be due to direct competition with P9, predicting that D1 and HP1 may compete for binding to SAT III. Displacement of HP1 from heterochromatin should then result in additional D1 binding to sites normally occupied by HP1. We tested this possibility by feeding a histone deacetylase inhibitor to growing third-instar larvae. Treatments with trichostatin A (TSA) have been shown to result in the delocalization of HP1 from heterochromatin (Taddei et al, 2001). Figure 5A shows the HP1 and D1 localization patterns normally observed in larval neuroblast nuclei. HP1 (red) partially associates with SAT III repeats stained with P9F (green) as well as with SAT V repeats and is largely absent from the nucleoplasm. The localization of D1 is strictly correlated with P9F-positive foci, which correspond to SAT I and SAT III repeats. TSA feeding resulted in a loss of HP1 from heterochromatin and a relocalization of the protein to the nucleoplasm (Figure 5B). Significantly, this displacement was accompanied by an extension of the D1 signal: instead of colocalizing with P9F-positive foci, yielding a characteristic yellow signal (Figure 5A), the D1 antibody also stained an immediately adjacent region, yielding both red and yellow signals. The red-only domain occupied by D1 after TSA treatment corresponds to the rDNA repeats normally associated with HP1 (data not shown and see below).
Figure 5.
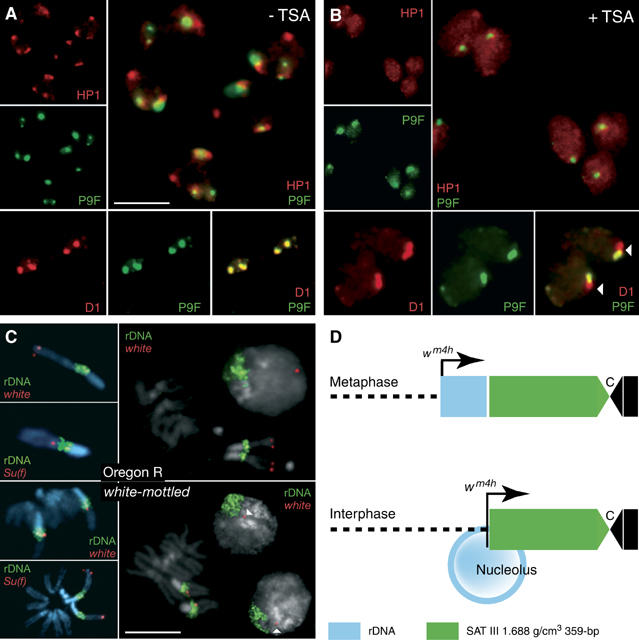
SAT III and rDNA repeats define alternative D1 and HP1 domains. Neuroblasts from control (A) or TSA-fed (B) third-instar larvae were immunostained for HP1 or D1 (red) and counterstained with P9F (green). Individual and merged signals are as indicated in the photographs. TSA feeding induces a delocalization of HP1 to the nucleoplasm and an extension of the D1 signal from SAT III to an adjacent P9F-negative region that corresponds to the rDNA repeats. (C) The results of FISH experiments performed with white and Su(f) probes (red) or a full-length rDNA probe (green). Photographs at the top and bottom show results obtained from wild-type Oregon R or wm4 neuroblasts, respectively. Arrowheads indicate the white signal detected in interphase nuclei. Scale bars: 5 μm. (D) The juxtaposition of the rDNA-linked white gene to SAT III repeats as a result of looping out of rDNA sequences in the nucleolus.
SAT III protein composition affects the white gene across the rDNA repeats
The delocalization of D1 and HP1 from SAT III repeats by P9 correlates with suppression of wm4 PEV. However, as the inverted white gene lies near the centromere-distal end of the rDNA repeats (see Figure 4G), this effect must somehow be transduced across the 3.5-Mbp rDNA locus. How satellite protein composition can affect gene expression over such distances is an important question, which we addressed by performing fluorescence in situ hybridization (FISH) experiments. Hybridization of rDNA and white probes to neuroblast nuclei and chromosomes resulted in well-defined signals. The Suppressor of forked gene (Su(f)), which maps to the heterochromatin region of the X chromosome and is distal to the rDNA repeats, was used as a control. The insets of Figure 4C show hybridization signals for rDNA (green) and white or Su(f) sequences (red) on X chromosomes from Oregon R (wild-type) or white-mottled larvae, clearly showing the inversion of the Su(f) and white genes on wm4 X chromosomes. In this case, the white gene could not be resolved from the rDNA repeats, consistent with its localization within type I insertion sequences located at the distal end of the rDNA locus (Schotta et al, 2003). In interphase nuclei, the white signal in wild-type cells was always localized at the opposite pole of the nucleus relative to rDNA repeats. Significantly, in contrast to the close linkage seen in mitotic chromosomes, the white signal was always well resolved from the rDNA repeats in wm4 interphase nuclei, localizing instead immediately adjacent to DAPI-bright foci that correspond to SAT III repeats. As diagrammed in Figure 5D, these results support a model whereby looping out of rDNA sequences within the nucleolus places the white gene very close to the SAT III repeats where the effects of P9 are exerted.
SAT III-specific displacement of topo II by P9
Topo II is one of the growing numbers of proteins found to be associated with centric heterochromatin. This association is particularly evident in immunofluorescence studies of prophase and mitotic chromosomes, where the topo II signal contains conspicuous centromeric foci in addition to axial, subchromosomal extensions (Maeshima and Laemmli, 2003). While topo II appears clearly enriched in centric heterochromatin of mitotic chromosomes, an important association with heterochromatin is also noted in interphase nuclei. This interaction was previously noted by the prominent cleavage of SAT III repeats by topo II in vivo (Käs and Laemmli, 1992). Is topo II associated with SAT III involved in PEV?
Figure 6A shows the localization of topo II in nuclei and mitotic chromosomes of larval neuroblasts immunostained for topo II (red) and counterstained with DAPI (gray) and P9F (green). Examination of interphase nuclei in panel A shows a granular topo II signal that appears excluded from a prominent DAPI-bright (gray) and P9F-bright (green) region thought to correspond to chromosome 4. On chromosome arms, the topo II signal shows a banded pattern that is reminiscent of observations made with mammalian chromosomes. Topo II is also enriched on SAT III and on the adjacent rDNA repeats (see the enlarged X chromosome in the insets of panel A). A comparison of the P9F and protein staining patterns confirms that SAT III repeats constitute the major chromosomal site (yellow merged color in Figure 6A) where satellite sequences and topo II strictly colocalize. Lack of staining of the fourth chromosome, as in the case of HP1, could be due to a reduced accessibility of the antigen in this compact chromosome.
Figure 6.
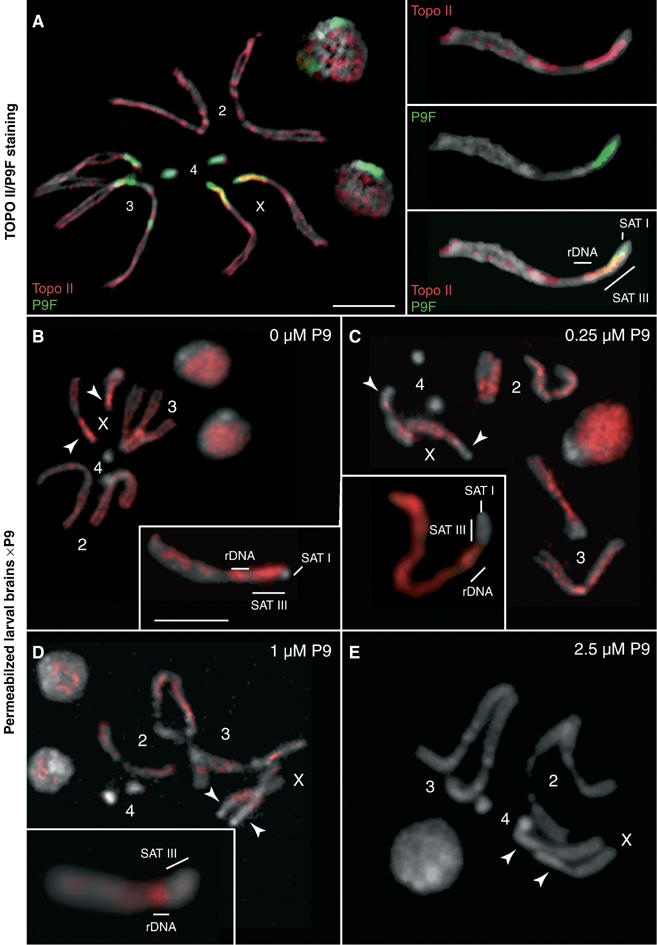
Localization of topoisomerase II and displacement from SAT III by P9. (A) Neuroblasts from female third-instar larvae were immunostained for topo II (red) and counterstained with P9F (green) and DAPI (gray). Individual chromosomes are numbered as shown. The localization of SAT I, SAT III and rDNA repeats is indicated in the inset showing a magnified X chromosome. SAT III repeats are associated with a particularly strong topo II signal, the adjacent region spanning the 3.5-Mbp rDNA repeats is also stained. Photographs to the right show the individual topo II, P9F and merged signals. In panels (B–E), third-instar female larval brains were permeabilized and treated with 0, 0.25, 1 and 2.5 μM P9, respectively. Drug treatment leads to a delocalization and displacement of topo II that is selective for SAT III repeats at low to intermediate concentrations of the drug (250 nM to 1 μM, C and D), while topo II remains associated with rDNA repeats (see magnified X chromosomes in insets). Topo II is completely displaced from nuclei and mitotic chromosomes at the highest drug concentration tested (2.5 μM, E). Arrowheads indicate the position of SAT III repeats. Samples were counterstained with DAPI (gray). Scale bars=5 μm.
Experiments discussed above establish that D1 and HP1 are displaced from SAT III by P9. This observation is extended to topo II next. Exposure of permeabilized brains to P9 in the range of 250 nM to 1 μM resulted in the selective loss of topo II from SAT III repeats (Figure 6C and D and insets with enlarged X chromosomes), while it remained associated with rDNA repeats and the chromosome arms. Note that topo II is completely evicted at the highest concentration of P9 used (2.5 μM, Panel E). These results show that, similarly to HP1, topo II is selectively displaced from SAT III repeats by P9 (250 nM to 1 μM), while it remains bound to the adjacent rDNA repeats. Such a displacement might be due to direct competition between P9 and topo II for binding to AT-rich satellite sequences, or might be a consequence of the loss of D1 that might serve to stabilize the interaction of topo II with SAT III repeats.
Suppression of PEV by a topo II inhibitor
The colocalization of topo II with two heterochromatin proteins, HP1 and D1, on SAT III repeats suggests that topo II might also be a structural/enzymatic component of heterochromatin. Such a notion is supported by our finding that feeding wm4h larvae sublethal doses of VM26, a topo II poison, resulted in a dose-dependent suppression of PEV (Figure 7). Suppression was complete at the highest drug concentrations used, as judged from the recovery of adult flies with wild-type eye color. A similar observation was made by feeding VM26 to flies harboring a variegating mini-white construct (Tp(3;Y)BL2) inserted on the Y chromosome (Lu et al, 1996). In contrast, m-AMSA, another topo II poison structurally unrelated to VM26, had no effect on wm4 PEV (Figure 6): m-AMSA does not induce topo II cleavage in SAT III repeats, although it promotes cleavage at other genomic sites (Borgnetto et al, 1996). We conclude from these results that topo II is implicated in white-mottled heterochromatin-mediated silencing and that this enzyme might more generally play a functional role in the assembly of heterochromatin, as suggested by the effect of VM26 in the Tp(3;Y)BL2 line.
Figure 7.
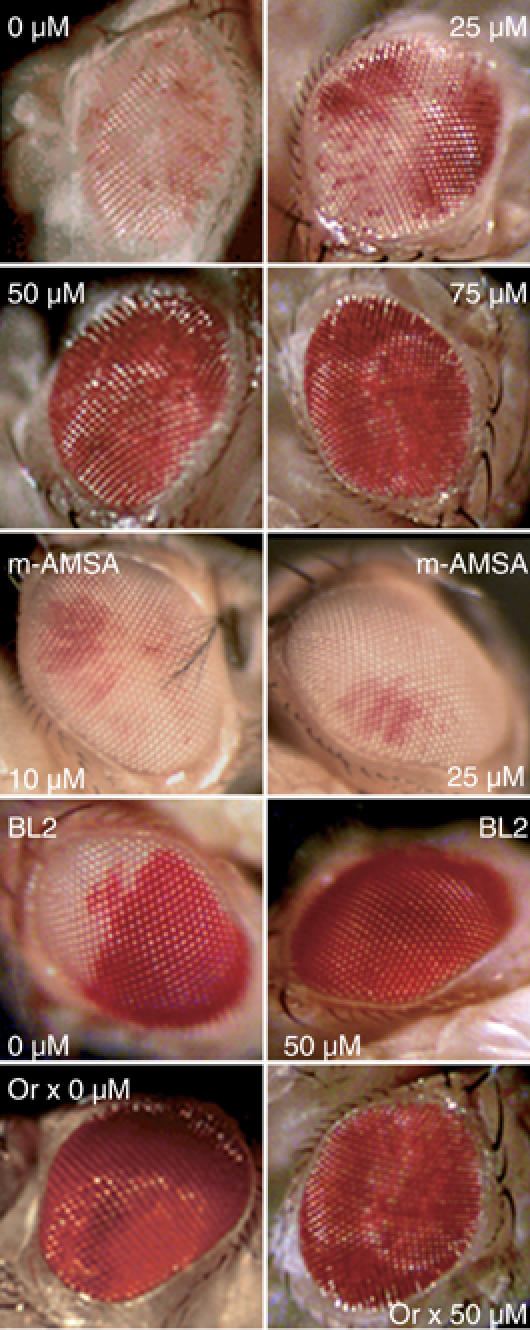
Treatment with VM26, a topoisomerase II poison, suppresses white-mottled PEV. Third-instar larvae from the wm4h, Tp(3;Y)BL2 (‘BL2') and Oregon R (‘Or') lines were fed VM26 or m-AMSA at the indicated concentrations. Representative eyes of 5-day-old adult flies are shown and correspond to those observed in >80% of hatched flies (n=30 for each drug concentration). Eyes from untreated or VM26-treated wild-type Oregon R flies are shown for comparison. Extracted eye pigments from two independent feeding experiments were measured for absorbance at 480 nm and yielded the following values: 0.18±0.03 (wm4h, 0 μM VM26); 0.36±0.05 (wm4h, 25 μM VM26); 0.79±0.05 (wm4h, 50 μM VM26); 0.91±0.03 (wm4h, 75 μM VM26); 0.22±0.06 (wm4h, 10 μM m-AMSA); 0.21±0.03 (wm4h, 25 μM m-AMSA); 0.43±0.05 (BL2, 0 μM VM26); 0.92±0.04 (BL2, 50 μM VM26); 0.92±0.02 (Oregon, 0 μM VM26); 0.89±0.02 (Oregon, 50 μM VM26).
Discussion
P9 is a simple polyamide of linked pyrrole amino acids with a subnanomolar binding preference for long dA·dT-runs (Supplementary Table I). It was previously shown that this compound, if fed to developing flies, suppresses the wm4 phenotype, an effect manifested by a more homogenous, redder appearance of the eyes. This ‘red shift' is due to a generally higher expression of the rearranged white gene (Janssen et al, 2000a). This observation suggested that P9 could work through chromatin opening, yielding a reduced heterochromatic state of SAT III, the main highly repetitive DNA flanking the white gene on the inverted wm4 X chromosome. Results reported here establish that, both in vivo and in vitro, P9 displaces the D1 protein from SAT III. In addition, we show that this compound results in a selective loss of HP1 and topo II from SAT III, while these proteins remain bound to low-affinity targets for P9, such as the rDNA repeats.
The displacement of D1 by P9 is an expected result. Polyamides are deeply buried in the DNA minor groove through specific hydrogen bonding and Van der Waals interactions (Dervan and Edelson, 2003). Similarly, the AT-rich specificity of D1 is mediated through its AT hooks reaching into the DNA minor groove of dA·dT-runs (Slama-Schwok et al, 2000). Oligopyrroles such as P9 mimic the DNA minor groove contacts of AT hooks. Hence, the displacement of D1 by P9 is explained by a molecular competition reaction. The salient observation of this paper is the selective displacement of the heterochromatin proteins HP1 and topo II from P9-targeted DNA such as SAT III. This is an unexpected finding since HP1 is thought to bind chromatin largely through protein–protein contacts mediated by its chromo domain, which recognizes the methylated state of the K9 amino acid of histone H3 (reviewed in Huisinga et al, 2006). It is possible that D1 might recruit the enzymatic activities required to create an HP1-binding site. Other sequence-selective satellite-binding proteins such as PROD (Platero et al, 1998) and SU(VAR)3–7 (Cléard and Spierer, 2001) might play a similar role. Taken together, our data suggest that D1 is directly or indirectly implicated in the association of HP1 and topo II with SAT III repeats and that the effects of P9 are exerted via the selective displacement of these proteins, resulting in a reduction of silencing of the nearby white gene in wm4 flies.
Our conclusions are based on the results of in vitro competition/binding assays and of in vivo P9 bathing and feeding experiments performed on embryos and larvae, respectively. Experimentally, we have followed the chromosome association of D1, HP1 and topo II as a function of P9 treatment by immunofluorescence and with the help of fluorescent polyamide derivatives added during the staining procedure. The latter serve as optical highlighters of the targeted genomic regions (Figure 1). Binding experiments show that P9 competes with D1 for binding to similar sites in SAT III repeats that are also bound by the artificial MATH20 protein (Figure 3). This mimicry extends to the ability of P9, following entry in whole embryos or ingestion by larvae, to displace D1 from its SAT III-binding sites in vivo (Figure 2), one important consequence of which is the loss of HP1 from SAT III (Figure 4) which demonstrates a disassembly most likely caused by the displacement of D1 by P9.
However, our results also indicate the likely significance of HP1/DNA contacts: displacement of HP1 from SAT III or SAT V repeats is strictly determined by the sequences targeted by the P9 or P31 polyamide used, respectively (Figure 4). This is in line with the report that telomeric sequences are bound directly by HP1 (Perrini et al, 2004), an observation which may extend to a more general binding of HP1 to DNA in chromatin (Zhao et al, 2000). The displacement of HP1 from SAT III repeats by P9 might be due to direct competition, a consequence of which is that HP1 and D1 would also compete for binding to similar sites. Alternatively, HP2, a companion of HP1 containing two AT-hook motifs (Shaffer et al, 2002), might be a target for competition by P9 and D1. The opposite gradient-like distributions of HP1 and D1 on SAT III repeats (Figure 4B and C) support such a possibility, suggesting mutually exclusive interactions that might extend to the rDNA repeats. This hypothesis follows from our observation that TSA feeding results in a displacement of HP1 from heterochromatin and the subsequent invasion of the rDNA repeats by D1 (Figure 5). While not a target for P9 (see Figure 1 and the lack of staining of rDNA repeats by P9F), the rDNA repeats are generally AT-rich and may contain binding sites for D1 that are normally occupied by HP1. A destabilization of HP1/heterochromatin interactions by histone hyperacetylation (Taddei et al, 2001) would thus result in an increased accessibility to D1, which is indeed what we observe.
The displacement of topo II from SAT III repeats by P9 (Figure 6) might similarly be due to direct competition with P9 or, alternatively, reflect a secondary delocalization that follows displacement of D1 from that AT-rich sequences. A role for this enzyme in SAT III heterochromatin-mediated silencing, as supported by its localization and the striking suppression of PEV by VM26, a topo II poison, is consistent with the results of recent studies indicating that many modifiers of PEV are involved in several important aspects of chromosome structure and function (Le et al, 2004). We note that a link was previously established between chromatin compaction, topo II and the negative control of gene expression: topo II was shown to be implicated in Polycomb-mediated repression (Lupo et al, 2001). A similar interplay with HP1 might reflect X chromosome-specific silencing phenomena. Genetic screens did not previously identify topo II as a modifier of PEV and flies heterozygous for the Df(2L)TW158 deficiency, which deletes the gene encoding topo II, do not suppress wm4 PEV (Wustmann et al, 1989). However, topo II is a very abundant protein and a 50% reduction in protein levels might not have measurable effects on overall levels of enzymatic activity. Our observations and the failure of Df(2L)TW158 to modify wm4 PEV are therefore not necessarily contradictory.
Feeding VM26 to developing wm4 larvae suppresses PEV, an unexpected result which could be due to a general—but reversible—inhibition of topo II, or to the possible accumulation of topo II/DNA complexes in SAT III repeats that might interfere with dynamic interactions with proteins such as HP1. Synthetic diamidines that promote the relocalization of topo II to AT-rich satellites such as SAT III indeed also suppress PEV when fed to wm4 larvae (Susbielle et al, 2005). We do not rule out alternative mechanisms: trapped complexes might for instance interfere with the bidirectional transcription of SAT III repeats (Aravin et al, 2003), whose role in heterochromatin assembly, structure and function remains to be elucidated. The RNAi machinery has been linked to the variegated expression of mini-white reporters (Pal-Bhadra et al, 2004), but the mutants used were not tested against the wm4 inversion. Preliminary results (data not shown) indicate that piwi and argonaute 1 mutants do not modify wm4 PEV, suggesting that there exist distinct pathways for the initiation of heterochromatin assembly (Danzer and Wallrath, 2004; Huisinga et al, 2006). In addition, SAT III-like sequences are interspersed throughout the D. melanogaster genome (Losada and Villasante, 1996) and the genomic origin of putative SAT III siRNAs has not been established. We are developing a combined DNA/RNA FISH method that can be applied to Drosophila larval tissues to address this important question in the future.
We favor a model whereby the suppression of wm4 PEV by VM26 is a direct result of interfering with topo II activity in SAT III repeats. The suppression of PEV in the BL2 line, in which white is inserted in the largely heterochromatic Y chromosome, further indicates that heterochromatin-mediated silencing in regions other than SAT III is also sensitive to topo II activity levels. Such a requirement for catalytically active topo II in heterochromatin assembly is generally supported by results showing active enzyme localized to centromeric regions and to human alpha-satellites that resemble SAT III sequences (Agostinho et al, 2004).
The strikingly different behavior of HP1 and topo II localized in the rDNA repeats adjacent to the SAT III array is discussed next. The known preference of topo II for association with AT-rich sequences (Maeshima and Laemmli, 2003) might explain its interaction with both SAT III and rDNA repeats. The lack of sensitivity of HP1 to displacement by P9 suggests that different mechanisms can direct HP1 to these closely juxtaposed chromatin domains. The D1-independent association of HP1 with rDNA correlates instead with the methylation of lysine 9 of histone H3 in this region (Ahmad and Henikoff, 2002). This suggests that packaging of the Drosophila rDNA repeats into heterochromatin may serve to regulate their expression. The colocalization of topo II and HP1 in this highly condensed region might reflect a coordinate interaction important for the control of rDNA transcription whose negative regulation involves the recruitment of histone deacetylase and methyl-transferase activities (Santoro et al, 2002). In contrast, resumption of proliferation and activation of ribosomal RNA transcription in Drosophila tissue culture cells is accompanied by the replication-independent replacement of histone H3 methylated on the lysine 9 position by the histone variant H3.3 in rDNA sequences (Ahmad and Henikoff, 2002). We speculate that the stable association of topo II with rDNA repeats might play a role in regulating the topological constraints that are likely to accompany such a replacement process.
Our results demonstrate that interfering with D1, HP1 and topo II interactions with SAT III correlates with P9-induced wm4 gain-of-function. However, the inverted white gene has been mapped very close to—or even within—rDNA repeats (Schotta et al, 2003): suppression of wm4 PEV by P9 acting on SAT III must then somehow be exerted over the distance spanned by the rDNA locus, or approximately 3.5 Mbp. These observations are not consistent with a linear model of heterochromatin spreading and support instead a spatial model of heterochromatin-mediated silencing (Csink and Henikoff (1996); see also Huisinga et al, 2006 and references therein). Our FISH results (Figure 5C), which show that the rDNA-linked inverted white gene is indeed more closely associated with SAT III repeats in interphase chromatin, provide additional support for such a model, in which looping out of the rDNA into the nucleolus would place white in close proximity to SAT III (Figure 5D).
Finally, we note that the stochastic inactivation of the wm4 allele might be a direct reflection of developmentally regulated rDNA expression. The association of D1 and HP1 with SAT III and rDNA repeats is sensitive to histone acetylation levels (Figure 5B) and these proteins may compete for binding to similar sites. Our observations raise the intriguing possibility that a dynamic equilibrium of alternative interactions of D1 and HP1 with SAT III and rDNA repeats might serve to regulate the structure of the centromere-proximal region of the X chromosome and participate in the regulation of rDNA expression.
Materials and methods
Fluorescent drug staining and immunostaining
Brains were dissected from Oregon R third-instar larvae in 0.7% saline and incubated in 0.5% sodium citrate for 15 min. Dissected tissues were carefully transferred to a small drop of 0.5% citrate on a poly-lysine-coated microscope slide. Samples were fixed for 10 min in 2% PFA in PBS containing 0.1% Triton X-100 followed by 10 min in 2% PFA, 45% acetic acid. This fixation protocol yields qualitatively better results than the short acid fixation previously used for D1 localization (Aulner et al, 2002); avoidance of colchicine yields high-quality mitotic chromosomes. Labeling with rabbit polyclonal antibodies against D1 (1:100 dilution) was performed exactly as described (Monod et al, 2002). Samples were stained for 10 min with Lex9F (20 nM), P9F (5 nM) or P31TR (2 nM) in PBS containing 1 mM MgCl2, washed twice in the same buffer, mounted in DAPI and visualized by epifluorescence microscopy. Staining with a polyclonal rabbit antibody against HP1 kindly provided by R Kellum (University of Kentucky) was performed using a 1:5000 dilution and permeabilized larval brains as described in Supplementary data for mock P9 treatments. The mouse C1A9 monoclonal HP1 antibody was also used, yielding identical results. Labeling with rabbit polyclonal antibodies against Drosophila topo II (1:1000 dilution) was performed as described above. Photographs were taken on a Leica DMRB microscope equipped with a CoolSnap HQ CCD camera and processed with Adobe Photoshop. In vivo and ex vivo drug treatments, FISH experiments and DNA binding/footprinting assays were performed as described in Supplementary data.
Supplementary Material
Supplementary Table I
Supplementary Materials and Methods
Acknowledgments
We thank Rebecca Kellum (University of Kentucky) for her gift of antiserum against Drosophila HP1 and Lori Wallrath (University of Iowa) and Sally Elgin (Washington University) for samples of the C1A9 antibody. We thank J Eissenberg, P Geyer, E Kuhn-Parnell, M Grigoriev and M Weber for comments on the manuscript. The Centre de Biologie du Développement provided Drosophila culture facilities and the Institut d'Exploration Fonctionnelle des Génomes microscopy facilities. This work was supported by the Association pour la Recherche sur le Cancer (grant no. 3319), by the ACI Cancéropôle 2004 program ‘Genetic instability as a pejorative outcome in cancer' and by the National Institutes of Health (grant GM29006 to T-sH). R Blattes and G Susbielle are supported by doctoral fellowships from the French Ministry of Research and from the Ligue Nationale Contre le Cancer, respectively.
References
- Adachi Y, Luke M, Laemmli UK (1991) Chromosome assembly in vitro: topoisomerase II is required for condensation. Cell 64: 137–148 [DOI] [PubMed] [Google Scholar]
- Agostinho M, Rino J, Braga J, Ferreira F, Steffensen S, Ferreira J (2004) Human topoisomerase IIalpha: targeting to subchromosomal sites of activity during interphase and mitosis. Mol Biol Cell 15: 2388–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad K, Henikoff S (2002) The histone variant H3*3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell 9: 1191–1200 [DOI] [PubMed] [Google Scholar]
- Aravin AA, Lagos-Quintana M, Yalcin A, Zavolan M, Marks D, Snyder B, Gaasterland T, Meyer J, Tuschl T (2003) The small RNA profile during Drosophila melanogaster development. Dev Cell 5: 337–350 [DOI] [PubMed] [Google Scholar]
- Aulner N, Monod C, Mandicourt G, Jullien D, Cuvier O, Sall A, Janssen S, Laemmli UK, Käs E (2002) The AT-hook protein D1 is essential for Drosophila melanogaster development and is implicated in position-effect variegation. Mol Cell Biol 22: 1218–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgnetto ME, Zunino F, Tinelli S, Käs E, Capranico G (1996) Drug-specific sites of topoisomerase II DNA cleavage in Drosophila chromatin: heterogeneous localization and reversibility. Cancer Res 56: 1855–1862 [PubMed] [Google Scholar]
- Cléard F, Spierer P (2001) Position-effect variegation in Drosophila: the modifier Su(var)3–7 is a modular DNA-binding protein. EMBO Rep 2: 1095–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csink AK, Henikoff S (1996) Genetic modification of heterochromatic association and nuclear organization in Drosophila. Nature 381: 529–531 [DOI] [PubMed] [Google Scholar]
- Danzer JR, Wallrath LL (2004) Mechanisms of HP1-mediated gene silencing in Drosophila. Development 131: 3571–3580 [DOI] [PubMed] [Google Scholar]
- Dervan PB, Edelson BS (2003) Recognition of the DNA minor groove by pyrrole–imidazole polyamides. Curr Opin Struct Biol 13: 284–299 [DOI] [PubMed] [Google Scholar]
- Girard F, Bello B, Laemmli UK, Gehring WJ (1998) In vivo analysis of scaffold-associated regions in Drosophila: a synthetic high-affinity SAR binding protein suppresses position effect variegation. EMBO J 17: 2079–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisinga KL, Brower-Toland B, Elgin SC (2006) The contradictory definitions of heterochromatin: transcription and silencing. Chromosoma 28: 28. [DOI] [PubMed] [Google Scholar]
- Janssen S, Cuvier O, Muller M, Laemmli UK (2000a) Specific gain- and loss-of-function phenotypes induced by satellite-specific DNA-binding drugs fed to Drosophila melanogaster. Mol Cell 6: 1013–1024 [DOI] [PubMed] [Google Scholar]
- Janssen S, Durussel T, Laemmli UK (2000b) Chromatin opening of DNA satellites by targeted sequence-specific drugs. Mol Cell 6: 999–1011 [DOI] [PubMed] [Google Scholar]
- Käs E, Laemmli UK (1992) In vivo topoisomerase II cleavage of the Drosophila histone and satellite III repeats: DNA sequence and structural characteristics. EMBO J 11: 705–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le HD, Donaldson KM, Cook KR, Karpen GH (2004) A high proportion of genes involved in position effect variegation also affect chromosome inheritance. Chromosoma 112: 269–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohe AR, Hilliker AJ, Roberts PA (1993) Mapping simple repeated DNA sequences in heterochromatin of Drosophila melanogaster. Genetics 134: 1149–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A, Villasante A (1996) Autosomal location of a new subtype of 1*688 satellite DNA of Drosophila melanogaster. Chromosome Res 4: 372–383 [DOI] [PubMed] [Google Scholar]
- Lu BY, Bishop CP, Eissenberg JC (1996) Developmental timing and tissue specificity of heterochromatin-mediated silencing. EMBO J 15: 1323–1332 [PMC free article] [PubMed] [Google Scholar]
- Lupo R, Breiling A, Bianchi ME, Orlando V (2001) Drosophila chromosome condensation proteins Topoisomerase II and Barren colocalize with Polycomb and maintain Fab-7 PRE silencing. Mol Cell 7: 127–136 [DOI] [PubMed] [Google Scholar]
- Maeshima K, Laemmli UK (2003) A two-step scaffolding model for mitotic chromosome assembly. Dev Cell 4: 467–480 [DOI] [PubMed] [Google Scholar]
- Monod C, Aulner N, Cuvier O, Käs E (2002) Modification of position-effect variegation by competition for binding to Drosophila satellites. EMBO Rep 3: 747–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal-Bhadra M, Leibovitch BA, Gandhi SG, Rao M, Bhadra U, Birchler JA, Elgin SC (2004) Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science 303: 669–672 [DOI] [PubMed] [Google Scholar]
- Perrini B, Piacentini L, Fanti L, Altieri F, Chichiarelli S, Berloco M, Turano C, Ferraro A, Pimpinelli S (2004) HP1 controls telomere capping, telomere elongation, and telomere silencing by two different mechanisms in Drosophila. Mol Cell 15: 467–476 [DOI] [PubMed] [Google Scholar]
- Platero JS, Csink AK, Quintanilla A, Henikoff S (1998) Changes in chromosomal localization of heterochromatin-binding proteins during the cell cycle in Drosophila. J Cell Biol 140: 1297–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves R, Nissen MS (1990) The A·T-DNA-binding domain of mammalian high mobility group I chromosomal proteins. J Biol Chem 265: 8573–8582 [PubMed] [Google Scholar]
- Santoro R, Li J, Grummt I (2002) The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat Genet 32: 393–396 [DOI] [PubMed] [Google Scholar]
- Schotta G, Ebert A, Dorn R, Reuter G (2003) Position-effect variegation and the genetic dissection of chromatin regulation in Drosophila. Semin Cell Dev Biol 14: 67–75 [DOI] [PubMed] [Google Scholar]
- Shaffer CD, Stephens GE, Thompson BA, Funches L, Bernat JA, Craig CA, Elgin SC (2002) Heterochromatin protein 2 (HP2), a partner of HP1 in Drosophila heterochromatin. Proc Natl Acad Sci USA 99: 14332–14337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slama-Schwok A, Zakrzewska K, Leger G, Leroux Y, Takahashi M, Käs E, Debey P (2000) Structural changes induced by binding of the high-mobility group I protein to a mouse satellite DNA sequence. Biophys J 78: 2543–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susbielle G, Blattes R, Brevet V, Monod C, Käs E (2005) Target practice: aiming at satellite repeats with DNA minor groove binders. Curr Med Chem Anti-canc Agents 5: 409–420 [DOI] [PubMed] [Google Scholar]
- Swedlow JR, Sedat JW, Agard DA (1993) Multiple chromosomal populations of topoisomerase II detected in vivo by time-lapse, three-dimensional wide-field microscopy. Cell 73: 97–108 [DOI] [PubMed] [Google Scholar]
- Taddei A, Maison C, Roche D, Almouzni G (2001) Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat Cell Biol 3: 114–120 [DOI] [PubMed] [Google Scholar]
- Wustmann G, Szidonya J, Taubert H, Reuter G (1989) The genetics of position-effect variegation modifying loci in Drosophila melanogaster. Mol Gen Genet 217: 520–527 [DOI] [PubMed] [Google Scholar]
- Zhao T, Heyduk T, Allis CD, Eissenberg JC (2000) Heterochromatin protein 1 binds to nucleosomes and DNA in vitro. J Biol Chem 275: 28332–28338 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table I
Supplementary Materials and Methods