

Abstract
Tetraploidy can result in cancer-associated aneuploidy. As shown here, freshly generated tetraploid cells arising due to mitotic slippage or failed cytokinesis are prone to undergo Bax-dependent mitochondrial membrane permeabilization and subsequent apoptosis. Knockout of Bax or overexpression of Bcl-2 facilitated the survival of tetraploid cells at least as efficiently as the p53 or p21 knockout. When tetraploid cells were derived from diploid p53 and Bax-proficient precursors, such cells exhibited an enhanced transcription of p53 target genes. Tetraploid cells exhibited an enhanced rate of spontaneous apoptosis that could be suppressed by inhibition of p53 or by knockdown of proapoptotic p53 target genes such as BBC3/Puma, GADD45A and ferredoxin reductase. Unexpectedly, tetraploid cells were more resistant to DNA damaging agents (cisplatin, oxaliplatin and camptothecin) than their diploid counterparts, and this difference disappeared upon inhibition of p53 or knockdown of p53-inducible ribonucleotide reductase. Tetraploid cells were also more resistant against UVC and γ-irradiation. These data indicate the existence of p53-dependent alterations in apoptosis regulation in tetraploid cells.
Keywords: apoptosis, bax, chemoresistance, mitochondria, p53
Introduction
Cancer cells characteristically provide their own growth signals, ignore growth-inhibitory signals, replicate without limit, sustain angiogenesis, invade tissues, proliferate in unnatural locations, and avoid cell death (Hanahan and Weinberg, 2000). Another characteristic of cancer is genomic instability, which is frequently characterized by numeric and structural chromosomal aberrations. Among these hallmarks, the relationship between cell death resistance and genomic instability remains elusive (Zhivotovsky and Kroemer, 2004). Although it is intrinsically difficult to reconstruct the process of aneuploidization by characterizing aneuploid cancer cells (which have survived a Darwinian selection in which most if not all of the intermediates have been lost), there are multiple examples of aneuploid cancer cells generated through asymmetric division of or progressive chromosomal loss from tetraploid precursors (Lin et al, 2001; Castedo et al, 2004; Imkie et al, 2004; Storchova and Pellman, 2004; Watanabe et al, 2004; Fujiwara et al, 2005). This is particularly well documented for cancer development from Barrett's esophagus (Barrett et al, 2003; Maley et al, 2004) and oral leukoplakia (Sudbo et al, 2001; Sudbo et al, 2004).
Tetraploidy can be induced through two distinct processes, namely illicit fusion of two diploid cells (Duelli and Lazebnik, 2003; Ogle et al, 2005) or, more likely, by duplication of the normal chromosomal number in the absence of nuclear and cellular division. This can occur physiologically through endoreplication (DNA replication without mitosis) or endomitosis (karyokinesis without cytokinesis) or, pathologically, through mitotic failure (Storchova and Pellman, 2004). For instance, activation of the spindle assembly checkpoint (SAC) usually arrests mitosis during the metaphase until the problems accounting for SAC activation have been solved and mitotic division can ensue correctly. However, a prolonged arrest due to the impossibility to satisfy the SAC leads to checkpoint ‘adaptation', ‘slippage' or ‘leakage' with the consequent exit of mitosis and fixation of a tetraploid state (Rieder and Maiato, 2004; Weaver and Cleveland, 2005).
Although it has been debated whether a so-called ‘tetraploidy checkpoint' exists (Uetake and Sluder, 2004), it is widely acknowledged that tetraploid cells arrest their cell cycle and that this arrest depends on the tumor suppressor protein p53 (Cross et al, 1995; Yin et al, 1999; Andreassen et al, 2001; Meraldi et al, 2002; Vogel et al, 2004; Sphyris and Harrison, 2005). p53 is a multifunctional transcription factor, which induces multiple target genes. Depending on the cellular context, p53 transactivation can stimulate DNA repair (e.g. by induction of p53R2), cell cycle arrest (e.g. by induction of p21) as well as apoptosis (e.g. by induction of proapoptotic members of the Bcl-2 family such as Bax and Puma/BBC3) (Vogelstein et al, 2000; Vousden and Lu, 2002; Yu et al, 2003). Failure of the ‘tetraploidy checkpoint' due to a missing cell cycle arrest has been accused to be (co)responsible for the genomic instability induced by inactivating p53 mutations (Cross et al, 1995; Yin et al, 1999; Andreassen et al, 2001; Vogel et al, 2004; Sphyris and Harrison, 2005).
Based on these premises, we decided to re-evaluate the relationship between tetraploidization and p53. We discovered that one of the processes that aborts polyploid cells is apoptosis and that suppression of apoptosis by inhibition of mitochondrial outer membrane permeabilization (MOMP)—the major apoptotic checkpoint (Green and Kroemer, 2004; Jiang and Wang, 2004)—is permissive for the survival and propagation of tetraploid cells. When characterizing the epigenetic regulation of tetraploid genomes, we found that tetraploid cells are intrinsically prone to activate p53 and p53 target genes, leading to an increased spontaneous apoptosis and—paradoxically—to an increased resistance against DNA damage-induced cell death.
Results
Apoptosis inhibition is permissive for experimental polyploidization
HCT116 cells exposed to the microtubule poison nocodazole first activate the spindle checkpoint (and hence arrest in the metaphase) and then undergo mitotic slippage to become hyperploid. In response to acute nocodazole treatment (48 h), both cells with a regular DNA content (2–4N) and cells with a hyperploid DNA content (>4N) tended to lose their mitochondrial transmembrane potential (ΔΨm), as detectable with the ΔΨm-sensitive dye DiOC6(3) (Figure 1A), thus demonstrating signs of ongoing cell death (Green and Kroemer, 2004). This cell death was only partially inhibited by the pan-caspase inhibitor Z-VAD-fmk (but not by t Z-VDVAD and Z-FA-fmk, which inhibit caspase-2 and cathepsin B, respectively).
Figure 1.
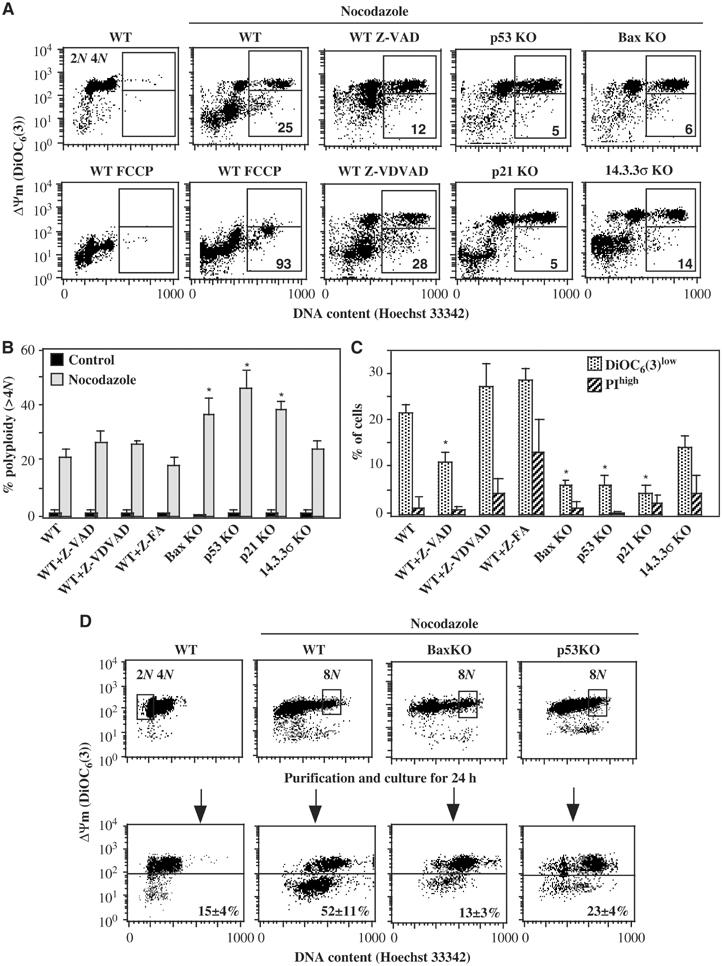
Acute mortality of polyploid cells modulated by p53 and Bax. Wild-type (WT) HCT116 cells or cells manipulated to lose expression of p53, p21, 14.3.3σ or Bax were cultured in the absence or presence of nocodazole, alone or combined with the protonophore FCCP (which dissipates the ΔΨm) or the protease inhibitors Z-VAD-fmk, Z-VDVAD-CHO or Z-FA-fmk for 48 h, followed by three-color staining with Hoechst 33342 (which measures DNA content), DiOC6(3) (which measures ΔΨm) and propidium iodine (PI, a vital dye that incorporates only into dead cells) and cytofluorometric evaluation. (A) Representative pictograms showing the polyploid cells (DNA content >4N) with a normal ΔΨm (upper window), as well as polyploid cells with reduced ΔΨm (lower window). Numbers refer to the percentage of polyploid DiOC6(3)low cells (considering 100% as the sum of all polyploid cells). (B) Percentage of polyploid cells among the total population of cells treated as in (A). (C) Frequency of dying (DiOC6(3)low) and dead (PIhigh) cells among the polyploid population elicited by nocodazole, as determined by FACS analysis. Values are X±s.e.m. of five independent experiments. Asterisks refer to significant effects (paired Student's t-test, P<0.001). (D) Fate of viable polyploid cells elicited by nocodazole. Cells with the indicated genotype were cultured for 30 h in the absence or presence of nocodazole and then stained with Hoechst 33342 and DiOC6(3), followed by FACS purification of the euploid (2N) or polyploidy (>4N) DiOC6(3)high cells (as indicated by the windows in the upper panels). These purified cells were then cultured for 24 h and reanalyzed after restaining with Hoechst 33342 and DiOC6(3), as indicated in the lower panels. Numbers indicate the percentage of cells with a DiOC6(3)low phenotype (X±s.e.m., n=3).
In this setting, the knockout of p53, Bax or p21 (but less than that of 14.3.3σ) strongly reduced cell death and augmented the percentage of viable, >4N cells elicited by a 48-h incubation with nocodazole (Figure 1A–C). Z-VAD-fmk failed to enhance the percentage of polyploid cells (Figure 1B). We FACS-purified viable (ΔΨmhigh) nocodazole-treated cells with an ∼8N DNA content and subjected them to fluorescent in situ hybridization (FISH) with centromere-specific probes for chromosomes 9 and 18. These experiments revealed the presence of four rather than eight FISH-discernible signals per cell for chromosome 9 and 18 (in >90% of the cases). Thus, this population was composed by tetraploid cells in G2/M (before separation of centromeres) rather than by octoploid cells in G1. The FACS-purified population with an ∼8N DNA content was cultured in the absence of nocodazole for 24 h, and the entry of cells into apoptosis was monitored (Figure 1D). These results confirmed that de novo formed tetraploid cells tend to die (as indicated by ΔΨm dissipation) and that the removal of p53 or Bax from the system greatly reduces the death of such cells. Of note, in this setting, nocodazole did not induce a DNA damage response, as indicated by the absence of DNA damage foci staining for phosphorylated histone H2AX (Supplementary Figure 1S). Moreover, the FACS-purified ∼8N population did not increase its DNA content upon re-culture, in line with the FISH data indicating that these cells are in G2/M rather than in the G1 phase of the cell cycle (Figure 1D). Very similar data suggesting that p53 and Bax are required for the death of tetraploid cells were obtained when polyploidization was induced by cytochalasin D, an inhibitor of cytokinesis (Supplementary Figure 2S). Thus, p53 and Bax inhibition are permissive for experimental polyploidization.
Of note, neither p53 nor Bax did influence the expression level of BubR1 and its nocodazole-induced phosphorylation (Supplementary Figure 3S), although BubR1 has been suggested to be a major negative regulator of polyploidization (Shin et al, 2003). Upon nocodazole treatment, Bax-proficient cells with a >4N DNA content translocated cytochrome c from mitochondria and activated caspase-3, while Bax-deficient cells retained cytochrome c in mitochondria and failed to activate caspase-3, as determined by confocal immunofluorescence (Figure 2A). In this system, Z-VAD-fmk only partially inhibited cytochrome c release, although it fully blocked caspase-3 activation (Figure 2A), indicating that MOMP can occur without caspase activation.
Figure 2.
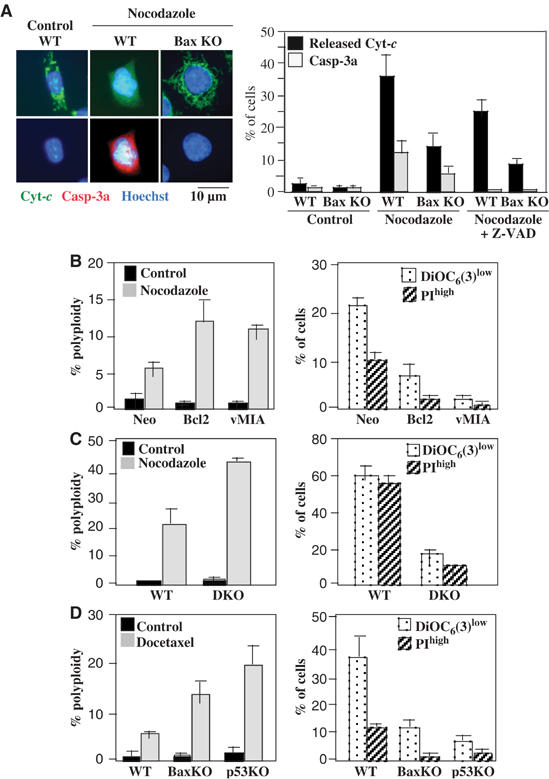
Mitochondrial cell death regulators and the fate of polyploid cells. (A) Evidence for MOMP in nocodazole-treated cells. Untreated control or nocodazole treated HCT116 cells (either wild type or Bax KO) were treated for 48 h with nocodazole alone or in combination with Z-VAD-fmk, followed by confocal immunofluorescence staining with antibodies specific for cytochrome c (Cyt c) and proteolytically active caspase-3 (Casp-3a). The percentage of cells exhibiting diffuse Cyt c staining or positivity for Casp-3a was determined among the entire population in controls and among nocodazole treated cells that exhibited a larger nucleus than controls, and that were considered as polyploid (X±s.e.m., n=3). (B) Effect of Bcl-2 overexpression and vMIA expression on the fate of polyploid cells. HeLa cells transfected with Bcl-2 or vMIA were treated for 48 h with nocodazole. Then, the frequency (X±s.e.m., n=4) of polyploid cells among the total population (left panels) or that of dying (DiOC6(3)low) and dead (PIhigh) cells among the polyploid population elicited by nocodazole (right panels) was determined as in Figure 1A–C. (C) Comparison of wild-type or Bax−/−Bak−/− DKO MEF in their response to nocodazole. Cells were treated and monitored as in (B). (D) Effect of the Bax and p53 knockout on docetaxel-induced polyploidization. HCT116 cells with the indicated genotype were treated for 48 h with 100 nM docetaxel and the frequency (X±s.e.m., n=4) of polyploid cells among the total population (left panels) or that of dying (DiOC6(3)low) and dead (PIhigh) cells among polyploid cells (right panel) was determined.
The survival of cells with a hyperploid DNA content (>4N) was facilitated by Bax inhibition in several experimental systems. Thus, nocodazole-treated HeLa cells that overexpress two distinct Bax antagonists (Bcl-2 or the cytomegalovirus-derived mitochondrial inhibitor of apoptosis, vMIA) (Poncet et al, 2004) did not undergo apoptosis when they accumulated >4N DNA in response to nocodazole (Figure 2B). Mouse embryonic fibroblasts (MEF) in which both Bax and its structural homolog Bak were subjected to a double knockout (DKO) (Wei et al, 2001), also generated more cells with a >4N DNA content in response to nocodazole than wild-type MEF (Figure 2C). When nocodazole was replaced by another spindle poison, docetaxel (Figure 2D), the absence of Bax again facilitated the generation of cells with >4N DNA.
In short-term experiments (48 h), the p53 and the Bax knockout were equivalently permissive for DNA accumulation >4N (Figure 1, Supplementary Figure 2S). However, upon prolonged culture (10 days) of cells transiently exposed to nocodazole (2 days), Bax-negative HCT116 cells had generated more polyploid cells than p53-negative cells, and these Bax-negative polyploid cells were undergoing less spontaneous death than p53-negative polyploid cells (Figure 3A). Note that at this time point (10 days), diploid cells that had been exposed transiently to nocodazole did not undergo a higher rate of apoptosis than untreated control cells, as determined by FACS purification of the cells with a 2N DNA content and re-culture of the cells for 24 h (Figure 3B). The fact that Bax KO protected >4N cells from spontaneous apoptosis more efficiently than the p53 KO (Figure 3A), suggested the existence of a p53-independent Bax activation pathway. Accordingly, a substantial fraction of p53-KO cells with a >4N DNA content demonstrated positivity for activated Bax (detectable with an antibody specific for the N-terminus, which is exposed upon activation and mitochondrial membrane insertion of Bax) as late as 10 days after nocodazole treatment (Figure 3C and D). In contrast, the percentage (but not the absolute number) of tetraploid cells exhibiting the activating phosphorylation of p53 on serine 15 was significantly reduced in Bax KO cells as compared to wild-type controls, 10 days after nocodazole treatment (Figure 3C and D), presumably as a result of the outgrowth of viable tetraploid cells, Altogether, these data indicate that cells with a >4N DNA content are usually aborted by apoptosis executed through a Bax-dependent mitochondrial pathway.
Figure 3.
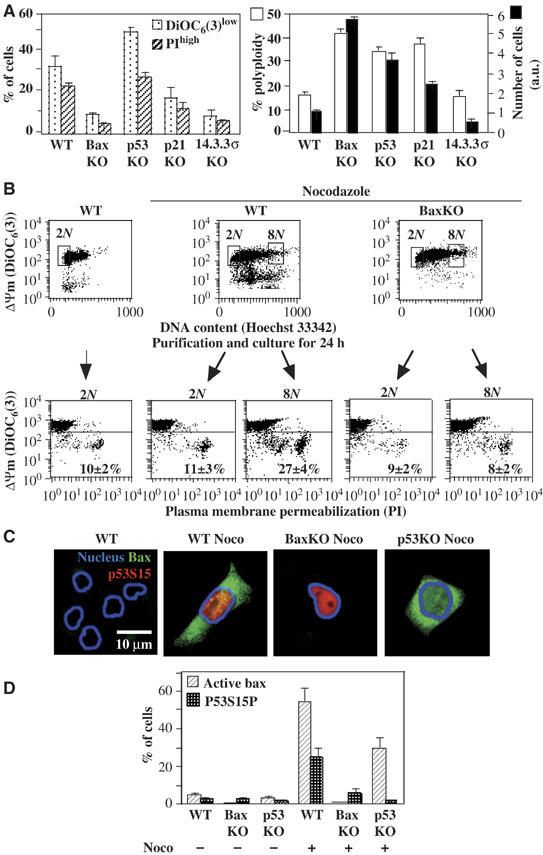
Long-term effects of Bax and p53 on the survival of polyploid cells. (A) Chronic mortality of polyploid cells modulated by p53 and Bax. HCT116 cells with the indicated genotype were treated for 48 h with nocodazole, then washed extensively (5 ×) and cultured during further 8 days in normal culture medium, followed by determination of the frequency (X±s.e.m., n=5) of live, dying and dead polyploid cells as in Figure 1A–C. Note that less than 5% of cells with an ∼2N DNA content were dying (DiOC6(3)low) or dead (PIhigh) cells and that dying or dead cells were almost exclusively found in the polyploid (>4N) population. (B) Fate of viable polyploidy cells induced by nocodazole. HCT116 WT or HCT116 BaxKO cells were transiently exposed to nocodazole for 2 days and then cultured for 8 days without nocodazole, stained with DiOC6(3) versus Hoechst 33342 followed by FACS purification of cells with 2N or 8N DNA content, cultured for 24 h and relabelled with DiOC6(3) and PI to determine the frequency of dying and dead cells. (C, D) p53-dependent and -independent Bax activation in a fraction of polyploid cells. HCT116 cells (WT, Bax KO or p53 KO) were cultured for 48 h in nocodazole and then for 8 days without nocodazole, followed by immunofluorescence staining of activated Bax and S15-phosphorylated p53. Representative micrographs are shown in (C), and data are quantified in (D). The percentage of cells exhibiting a punctate cytoplasmic staining for activated Bax or a nuclear staining for S15-phosphorylated p53 was assessed among the entire population in untreated controls and among nocodazole-exposed, polyploid cells (X±s.e.m., n=3).
Establishment of tetraploid cell lines and characterization of their transcriptome
Although nocodazole or cytochalasin D-treated wild-type HCT116 cells massively succumbed to apoptosis upon accumulation of a >4N DNA content (Figures 1, 2 and 3), we could isolate a few clones of tetraploid HCT116 wild-type cells (Figure 4A). Such cells arose either after treatment with nocodazole (clones N1 and N2) or cytochalasin D (clones C1 and C2) at a frequency of less than 1 in 10 000 and were found to express unmutated, normal Bax levels (not shown). In contrast, we were unable to generate stable clones with a higher-order polyploidy (e.g. octoploidy), irrespective of the HCT116 genotype investigated. We observed that another diploid colon carcinoma cell line, RKO, spontaneously contained ∼5% of tetraploid cells. Limiting dilution-based cloning led to the isolation of several stable tetraploid clones (T1–7) and several diploid clones (D1–D7, which after several passages again contained ∼5% of tetraploid cells). One of these latter clones (D5) was transfected with a histone H2B-GFP fusion construct (which allows to visualize chromosomes and to measure chromatin content) and then FACS-separated to generate H2B-GFP-positive tetraploid (TA, TB, TC, TD) and diploid (DW, DY, DX, DZ) subclones (Figure 4B). Again, there was no consistent difference in the expression level of Bax, Bcl-2, and BubR1 between diploid and tetraploid cells (Figure 4C).
Figure 4.
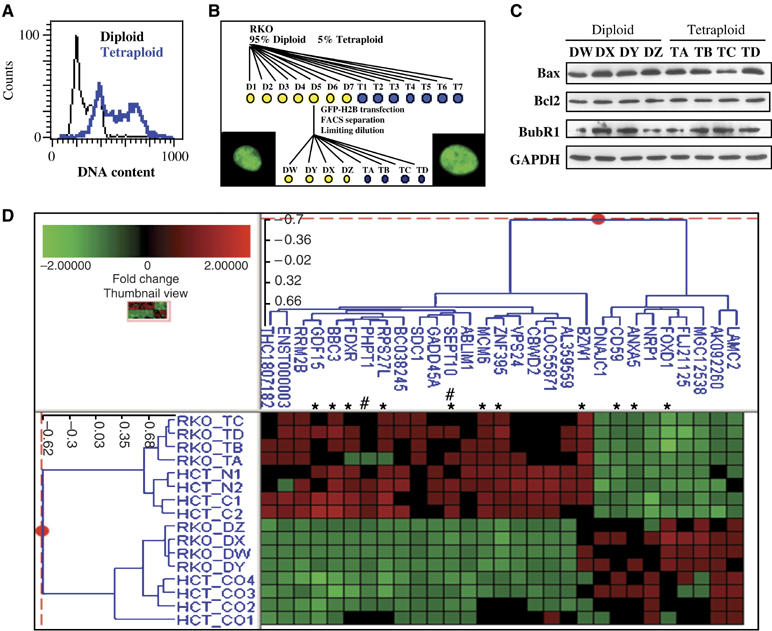
Establishment and characterization of tetraploid cell lines. (A) Comparison of the DNA content of a representative tetraploid HCT116 cell line and of its diploid control. (B) Genealogy of diploid and tetraploid RKO clones. The wild-type control cell line containing ∼5% tetraploid cells was subcloned by limiting dilution into diploid and tetraploid clones; The D5 clone was transfected with a cDNA encoding an H2B-GFP chimera, FACS-separated into subsets of cells enriched in a diploid or tetraploid DNA content, and again subcloned to generate diploid and tetraploid H2B-GFP-expressing clones. Representative fluorescence micrographs of such cells are shown (the width of the squares is 25 μm). (C) Western blot determination of the expression levels of Bax, Bcl-2, p53 and BubR1, with GAPDH as a loading control. (D) Microarray analyses of the transcriptome of diploid versus tetraploid cells. The clones HCT116 C1 and C2 and N1 and N2 are tetraploid, generated upon cytochalasin D or nocodazole treatment, respectively. Clones Co1–4 are diploid. The RKO clones are described in (B). The overlap between the genes coming from an ANOVA test with a threshold P-value of 10−3 on the HCT116 samples (Supplementary Figure 5S) and the same test on the RKO samples (Supplementary Figure 6S) led to the selection of 29 genes. These genes were subjected to a hierarchical cluster analysis using a calculation based on cosine correlation and the agglomerative method of the average link. Each row represents the combination of two dye-swap experimental samples and each column represents a single accession number. *marks genes that carry at least one consensus p53-binding consensus sequence (RRRCWWGYYY), within the 2000 nucleotides upstream of the transcription start. # marks genes the promoter of which contains putative p53 binding sites, as determined by another procedure bases on a sliding profile of four matrixes of the sequence RRRCA/TT/AGYYY, as detailed in the Materials and methods.
Karyograms and comparative genomic hybridization (CGH) performed on representative RKO clones indicate that tetraploid cells contained an exact duplication of the diploid set of chromosomes, without any CGH-detectable duplication or deletion (Supplementary Figure 4S). We then determined whether tetraploidization would induce epigenetic changes in the transcriptome, by performing microarray comparisons of four diploid and four tetraploid HCT116 clones generated by treatment with either nocodazole or cytochalasin D (Supplementary Figure 5S). Similarly, we compared the transcriptome of four diploid and four tetraploid H2B-GFP-positive RKO subclones (Supplementary Figure 6S). Surprisingly, there were little changes in the transcriptome of tetraploid HCT116 or RKO cells. The combined comparison of the 16 diploid and tetraploid clones led to a short list of genes that were consistently up- or downregulated by more than 20% as a function of ploidy. The gene that was most downregulated in tetraploid cells was the winged helix transcription factor FOXD1 (by 40%) and the two genes that were most upregulated were ZNF395, the zinc-finger protein 395 (by 40%), and RRM2B, the gene encoding p53R2, the p53-inducible ribonucleotide reductase-2 (by 30%). These results were confirmed by independent quantitative PCR analyses (not shown). Although the differences were small (20–25%), we found that three previously identified p53-inducible transcripts were significantly (P<0.0001, ANOVA test) incremented in tetraploid cells: BBC3 (which encodes the proapoptotic BH3 only protein Puma) (Nakano and Vousden, 2001; Yu et al, 2001), GADD45A (a cell cycle regulator), and FDXR (which encodes the mitochondrion-localized ferredoxin reductase) (Hwang et al, 2001) (Figure 4D). Intriguingly, 12 out of the 29 genes (41%) contained in the short list of ploidy regulated genes were found to contain a p53 consensus binding site or a putative p53-responsive tetrarepeat motif in the promoter region (Figure 4D). These data point to a major dysregulation of the p53 system in tetraploid cells.
Enhanced spontaneous apoptosis of tetraploid cells
As compared to their diploid precursors, a higher percentage of tetraploid cells exhibited the activating phosphorylation of p53 on serine 15 (p53S15P) or serine 46 (p53S46P), detectable by immunofluorescence staining with antibodies specific for p53 phospho-neoepitopes. This was observed in RKO (Figure 5A–C) and HCT116 cells (not shown). In contrast, we found no difference in the phosphorylation of histone H2AX that would be indicative of DNA damage (not shown). p53 phosphorylation was mainly detected in tetraploid cells that were terminating cytokinesis (Figure 5B), suggesting that p53 activation resulted from deficient mitoses. The increased phosphorylation of p53 on serine 15 was accompanied by a general increase in p53 expression levels, as determined by immunoblot (Figure 5D). An increased percentage of tetraploid cells died spontaneously from apoptosis, as indicated by the ΔΨm loss (Figure 5E), chromatin condensation, and incorporation of the vital dye PI (not shown). The caspase inhibitor Z-VAD-fmk did not affect the spontaneous mortality of tetraploid cells. However, there was an effect of cyclic pifithrin-α, which palliated the overmortality of tetraploid clones down to the diploid level both in RKO (Figure 5E) and in HCT116 cells (not shown). siRNA-mediated knockdown of Puma and of the Puma target Bax also reduced the spontaneous apoptosis of tetraploid cells. Similarly, knockdown of FDXR and GADD45A reduced the spontaneous apoptosis of tetraploid cells (Figure 5F), indicating that these p53 target genes contribute to the death of tetraploid cells.
Figure 5.
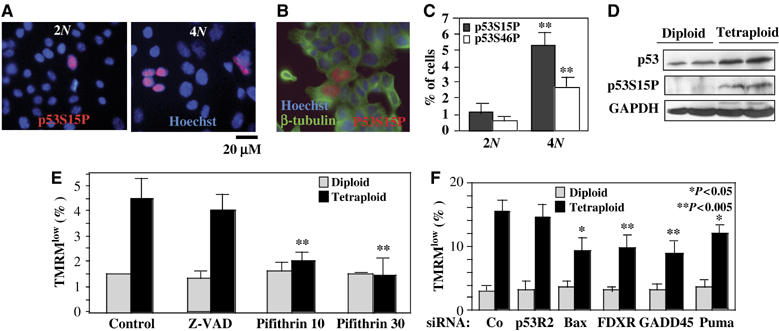
Constitutive p53 activation in tetraploid cells. (A, B) Staining of cells with an antibody recognizing p53 phophorylated on serine 15 (p53S15P). Representative images are shown for diploid and tetraploid RKO populations in (A). In (B), a representative tetraploid cells positive for p53S15P and counterstained with β-tubulin is shown. (C) Frequency of cells with positive nuclear staining for p53S15P or p53 phophorylated on serine 46 (p53S46P) is shown for diploid (n=4) and tetraploid (n=4) RKO cells. Asterisks indicate significant (P<0.01) differences between diploid and tetraploid cells (unpaired Student's t-test). (D) Immunoblot confirmation of the p53 activation. Representative diploid (DW, DY) and tetraploid (TA, TD) cells were subjected to immunoblot detection of p53, p53S15P and GAPDH. (E) Spontaneous cell death in RKO cells and its suppression by cyclic pifithrin-α. Cells were cultured for 2 days in the presence of Z-VAD-fmk or pifithrin-α, and the frequency of dying cells was measured by staining with TMRM. Asterisks indicate significant differences determined by ploidy (P<0.01, Student's t-test, X±s.e.m., n=4). (F) Effect of siRNAs on the spontaneous death of tetraploid RKO cells. Cells were transfected with siRNAs specific for emerin (negative control, Co), ferredoxin reductase (FDXR), GADD45A, Puma, Bax or p53R2 and 72 h later the spontaneous mortality of cells was assessed as in (E). Asterisks in (E) and (F) indicate significant inhibitory effects as compared to control cells (untreated in (E) and Emerin siRNA-transfected in (F)).
Selective resistance of tetraploid cells against DNA-damaging agents
Since p53 controls the cell death induced by DNA damage (Vogelstein et al, 2000; Vousden and Lu, 2002), we evaluated the apoptotic response of diploid and tetraploid cells. While there was no difference in the apoptotic response to the pan-tyrosine kinase inhibitor staurosporine (STS), tetraploid RKO clones without (Figure 6A) or with H2B-GFP (Figure 6B) as well as tetraploid HCT116 clones (Figure 6C) died less in response to cisplatin than their diploid counterparts. When exposed to cisplatin, tetraploid cells manifested a less pronounced ΔΨm dissipation (Figure 6C), Bax activation and proteolytic caspase-3 maturation than their diploid controls (Figure 6D). This relative resistance of tetraploid cells was also observed when cisplatin was replaced by oxaliplatin (which, as cisplatin, is a platinum compound) or the topoisomerase-1 inhibitor camptothecin. In contrast, there was no ploidy-specific difference in the apoptotic response to a number of other toxic compounds including staurosporine (Figure 6A–C), etoposide, doxorubicin and hydroxyurea (Figure 6E). Tetraploid RKO and HCT116 cells were also more resistant against apoptosis induced by UVC or γ-irradiation (Figure 6F). The relative cisplatin resistance was found again in FACS-purified MEF with an 8N DNA content. Such hyperploid cells were obtained by short-term (2 days) culture with 17-allylamino-17-demethoxygeldanamycin, and compared to FACS-purified diploid controls cells that had undergone an identical treatment (Figure 6G). Thus, even acute polyploidization caused cisplatin resistance. When injected into immunodeficient mice, both diploid and tetraploid RKO cells generated tumors (Figure 6H). Tetraploid tumors were relatively more resistant to cisplatin chemotherapy than diploid cancers (Figure 6I). Hence, the increased cisplatin resistance of tetraploid cells could be confirmed in vivo.
Figure 6.
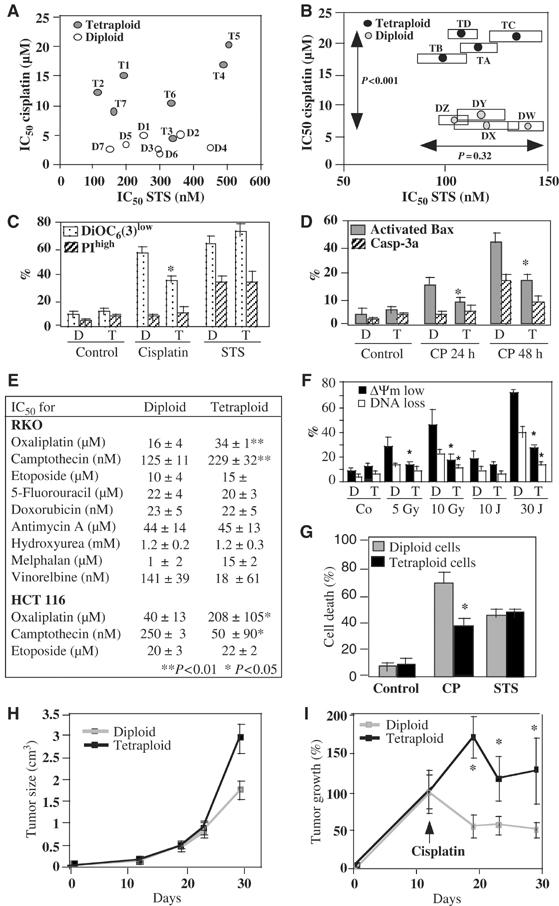
Relative resistance of tetraploid cells against DNA-damaging agents. (A, B) Comparison of the IC50 values of cisplatin and staurosporine (STS) for diploid (D) and tetraploid (T) RKO cells (for the codes of cell lines see scheme in Figure 4B). The IC50 was determined by the MTT assay, 48 h after addition of the drugs. Untransfected cells are shown in (A) and cells expressing GFP-H2B are shown in (B). (C, D). Reduced cisplatin-induced apoptosis in tetraploid cells. Four diploid and four tetraploid HCT116 cell lines were exposed to cisplatin (CP) or STS (48 h in C) and trypsinized, followed by staining with DiOC6(3) (which measures ΔΨm) plus propidium iodine (PI) and FACS analysis (C) or were left on the culture support, fixed, permeabilized and stained for the detection of activated Bax and Casp-3a (like in Figure 3C). Asterisks indicate a significant reduction of apoptotic parameters measured in tetraploid as compared to diploid cells (P<0.01, unpaired Student's t-test, X±s.e.m., n=3). (E) Systematic comparison of distinct cytotoxic compounds on diploid and tetraploid cells. The IC50 values were determined by MTT assays on 4 diploid and tetraploid clones of each cell lines. (F) Apoptotic response of tetraploid cells to UVC and γ-irradiation. Diploid and tetraploid RKO cells were analyzed 3 days post-treatment for loss of the ΔΨm (with TMRM staining) and DNA loss (with Hoechst 33342). Asterisks indicate significant differences dictated by the ploidy status (P<0.01, nonpaired Student's t-test, X±s.e.m., n=4). (G) Acute tetraploidization of MEF and their effect on the cisplatin response. Wild type MEF were treated for 48 h with 17-AAG, which caused an accumulation of ∼60% of cells with an 8N DNA content (as determined by Hoechst 33342 staining. Such cells (as well as cells with 2N DNA content) were FACS purified and treated with CP or STS for further 48 h, followed by determination of cell death with the crystal violet assay. (H, I). Tumor growth of untreated (G) or cisplatin-treated (H) RKO cells in vivo. Diploid (DY) or tetraploid (TA) RKO clones were injected subcutaneously into athymic nu/nu mice (n=10) and tumor growth (X±s.e.m.) was determined with a caliper. The animals (n=10 per group) were treated on day 12 by intraperitoneal injection of cisplatin (in H).
To explore the molecular mechanisms of drug resistance, we investigated the cisplatin response in diploid and tetraploid cells that were cocultured. Diploid RKO clones (H2B-GFP positive) and tetraploid RKO clones (H2B-GFP negative) were admixed at different ratios and then cocultured during several days in the absence or presence of cytotoxic drugs. Cultures initiated at a 50:50 ratio (diploid:tetraploid) shifted to a predominance of diploidy after 5 days (Figure 7A), in accord with the observation that the mean duplication time of diploid cells (23.2 h) was shorter than that of tetraploid cells (26.4 h), and that tetraploid cells exhibit a higher rate of spontaneous apoptosis than diploid controls (see above). This shift towards a predominance of diploid cells, as observed in untreated cultures, was not affected by STS. However, addition of cisplatin favored the relative outgrowth of tetraploid cells, in line with the interpretation that tetraploid cells are fitter than diploid cells in conditions of DNA damage (Figure 7A and B). Similar results were obtained for mixed diploid/tetraploid cultures of HCT116 clones in which either of the two clones was labeled with the stable fluorescence marker CMFDA (not shown) or when cisplatin was replaced by oxaliplatin (Figure 7C). Addition of the p53 inhibitor cyclic pifithrin-α prevented the cisplatin- or oxaliplatin-induced shift in favor of tetraploid cells (Figure 7C), suggesting that p53 target genes determine the differential behavior of diploid versus tetraploid cells. One of the four p53 target genes overexpressed in tetraploid cells, p53R2, has previously been shown to confer cisplatin resistance (Lin et al, 2004; Okumura et al, 2005). We therefore knocked down p53R2 expression by siRNA. Down modulation of p53R2 sensitized tetraploid cells to cisplatin-induced cell death and annihilated the difference in the cisplatin response between diploid and tetraploid cells (Figure 7D). As expected, downmodulation of Bax, Puma, GADD45A and FDXR reduced cisplatin-induced apoptosis, both in diploid and in tetraploid cells (Figure 7E). Altogether, these data indicate that subtle differences in the p53 transcriptome and in particular in p53R2 expression may explain the relative resistance of tetraploid cells against DNA-damaging agents.
Figure 7.
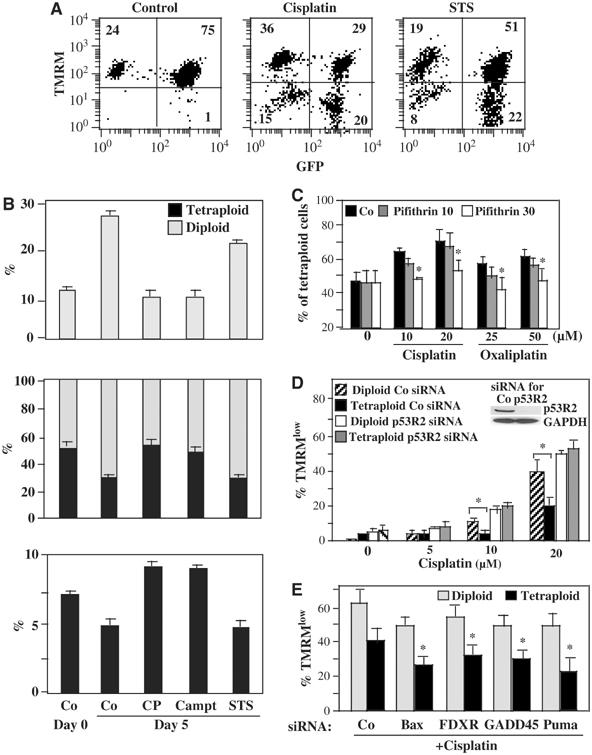
Mechanisms of cisplatin resistance in tetraploid cells. (A, B) Assay for the simultaneous detection of diploid and tetraploid cells dying with cisplatin. The GFP-H2B-expressing diploid RKO clone DY was mixed with the GFP-negative tetraploid RKO clone T1 at a 1:1 ratio (in A), followed by culture for 5 days in the absence of cytotoxic drugs (control) or in the presence of cisplatin or staurosporine (STS), and then stained with TMRM to determine the proportion of dying diploid and tetraploid cells. Note the increase in the relative frequency of diploid cells in untreated control cultures, irrespective of the initial diploid: tetraploid (D:T) ratio (1:1 in (A) or 9:1, 1:1 or 1:9 in (B)), while cisplatin increases the percentage of tetraploid cells in the cultures. Similar results indicating an enhanced resistance of tetraploid cells against cisplatin were obtained when tetraploid H2B-GFP-expressing clones were co-cultured with diploid GFP-negative clones (not shown). (C) Effect of cyclic pifithrin-α (Pif) on the D:T ratio. Cells (inital ratio 1:1) were cultured in the absence of presence of cisplatin, oxaliplatin and/or cyclic pifithrin-α (doses in μg), and the D:T ratio of the cultures was determined after 5 days. (D) Effect of small-interfering RNA designed to downregulate p53R2 on the death of representative diploid (DY, D5) and tetraploid RKO (TA, T1) clones. Cells were transfected with siRNAs specific for p53R2(440) or emerin, and 36 h later the cells were treated with cisplatin for 2 days, and the frequency of dying (TMRMlow) cells was measured. The immunoblot demonstrates the efficacy of p53R2 440 on p53R2 protein expression, as tested on a pool of RKO-derived clones. Similar functional results were obtained for a second p53R2-specific siRNA (UTR, not shown). (E) Effect of other p53 target genes on the cisplatin response. Specific siRNAs were used to downmodulate emerin (Co), FDXR, GADD45A, BAX or Puma, and the frequence of dying (TMRMlow) cells (X±s.e.m., n=3) was determined after addition of 20 μM cisplatin for 2 days. Note that the values of spontaneous apoptosis (obtained in the absence of cisplatin) are contained in Figure 5F. Asterisks in (C–D) indicate significant (P<0.01) apoptosis-inhibitory effects.
Discussion
Based on the data shown in this paper, it appears plausible that disabled apoptosis could be permissive for the generation of tetraploid cells, while tetraploidization has an intrinsic effect on apoptosis regulation. Acute tetraploidization caused activation of p53 (see phosphorylation of p53 in Figure 3D), in accord with the literature (Cross et al, 1995; Yin et al, 1999; Andreassen et al, 2001; Du and Hannon, 2004; Vogel et al, 2004; Sphyris and Harrison, 2005). Acute tetraploidization also resulted in the (partially p53-dependent and partially p53-independent, Figure 3D) activation of Bax, which adopts its proapoptotic conformation (Figures 2A and 3D) and triggers MOMP with cytochrome c release and consequent caspase activation (Figure 2A). MOMP is likely to lead to caspase activation in an amplification loop in which caspase activation, in turn, feeds back on MOMP (Adams, 2003; Danial and Korsmeyer, 2004), explaining the partial inhibitory effects of caspase inhibition (Figures 1A–C and 2A). Since the knockout of Bax or the overexpression of Bax antagonists (such as Bcl-2 or vMIA) blocks cell death and increases the survival of tetraploid cells at least as efficiently as does the knockout of p53 (and that of p21) (Figures 1, 2 and 3), it appears that one mechanisms that suppresses nonphysiological tetraploidization is the apoptotis. In this context, apoptosis would be as a mechanism of quality control (in accord with the rule ‘better dead than wrong') (Thompson, 1995) which, when perturbed, is permissive for the generation of tetraploid cells.
Although disabled apoptosis enhances the probability of stable tetraploidization, we were able to derive tetraploid clones from diploid parental lines that were apoptosis-competent (Figure 4). These tetraploid clones apparently maintained the capacity to undergo apoptosis (and hence remained fully susceptible to cell death induction by staurosporine or antimycin A but also drugs such as etoposide and vinorelbine) (Figure 6), in accord with the observation that they had no major cytogenetic defects (Figure 4S), expressed normal Bax and enhanced p53 levels (Figures 4C and 5D), and exhibited no downregulation of gene products with essential functions in apoptosis (Figure 4D). Tetraploidy-associated changes in the transcriptome were subtle (with the highest variation by less than a factor of 2), which contrasts with reports showing that acute tetraploidization by etoposide treatment has dramatic effects on multiple transcripts (Chen et al, 2003). However, in our system, we investigated the long-term effects of tetraploidization, several weeks after removal of the tetraploidy-inducing agent. We found a significant, yet minor (20–40%) increase in the expression of several p53 target genes (Figure 4D). p53 would be activated among a fraction of tetraploid cells, based on three lines of evidence, namely an activating p53 phosphorylation (on serine 15 and 46) (Figure 5A–D), an increased overall p53 protein level (Figure 5D), and an increased spontaneous mortality, which was blocked by p53 inhibition (Figure 5E) or by inhibition of several proapoptotic p53 target genes (Bax, BBC3/Puma, GADD45A, FDXR) (Figure 5F). The mechanism of the p53 phosphorylation is not clear. However, p53 was phosphorylated in cells that were completing mitosis (Figure 5B), suggesting that p53 activation would be linked to an intrinsic difficulty in managing the division of tetraploid genomes.
Unexpectedly, tetraploid cells did exhibit an increase in the IC50 for DNA-damaging agents such as cisplatin, oxaliplatin and camptothecin, by a factor of approximately two (Figure 6E). This was not a cloning artifact because it could be reproduced in distinct cell lines, as well as in separate sub-subcloning procedures (Figure 6). Moreover, it was probably not related to issues of drug uptake and efflux because tetraploid cells were also more resistant to physical DNA damage by UVC or γ-irradiation (Figure 6F). Inhibition of p53 reversed the relative resistance of tetraploid cells to DNA-damaging agents such as cisplatin (Figure 7C), an effect that could be related to the expression of one particular p53 target gene, p53R2 (Figure 7D). Thus, among the few p53 target genes that are induced in tetraploid cells, the antiapoptotic p53R2 functionally dominates over proapoptotic genes such as BBC3/Puma and FDXR when the cisplatin response is assessed.
Altogether, the data contained in this paper suggest a dual implication of p53-triggered mitochondrial apoptosis in tetraploid cells. First, shortly after tetraploidization, p53 contributes to the activation of the Bax-dependent mitochondrial pathway. At this stage, there are probably also p53-independent mechanisms that can lead to the activation of Bax and to the induction of apoptotic MOMP. Second, when tetraploid cells have become relatively stable and enter a logarithmic phase of growth, p53 is activated at a low level. At this stage, p53 causes the transcriptional activation of several genes, some of which trigger the apoptotic pathway at the mitochondrial level. This applies to FDXR, whose gene product perturbs the mitochondrial redox equilibrium (Hwang et al, 2001), as well as to BBC3/Puma, which is well known to activate Bax through direct physical interactions and hence to stimulate Bax-mediated MOMP (Nakano and Vousden, 2001; Yu et al, 2001). Thus, at this stage, p53 activation accounts for the reduced fitness of tetraploid cells that exhibit an elevated rate of spontaneous apoptosis and proliferate less than diploid parental cells. As a side effect of the enhanced p53 activation and enhanced expression of p53R2, however, tetraploid cells develop a relative resistance against DNA-damaging agents.
In a larger context, our data fit into a scenario in which apoptosis activated through the mitochondrial pathway actively contributes to the elimination of polyploid cells, while tetraploidy causes a shift towards apoptosis resistance. Along the same line, it can be speculated that asymmetric division or chromosome loss from tetraploid cells would trigger apoptosis as a default pathway and that only a few, stress-resistant cells would survive aneuploidization and hence contribute to the oncogenic process.
Materials and methods
Cell lines, culture and treatment
Derivatives of the HCT116 cell line (parental, Bax−/−, p53−/−, p21−/− or 14-3-3σ−/−) were a kind gift by B Vogelstein (Chan et al, 1999; Zhang et al, 2000) and were grown in McCoy's 5A medium supplemented with 10%FCS. HeLa cells transfected with Bcl-2 or vMIA (Poncet et al, 2004) and wild type of Bax−/−Bak−/− DKO MEFs (Wei et al, 2001) were maintained in DMEM and RPMI1640, respectively, with 10% FCS. Cell lines were treated with nocodazole (100 nM, Sigma) or docetaxel (100 nM) for 48 h in the presence or in the absence of Z-VAD-fmk (100 μM) or other protease-inhibitors (Enzyme Systems). Unless specified differently, the standard concentrations were the following: camptothecin: 50 nM; cisplatin: 20 μM; cyclic pifithrin-α: 30 μM; staurosporine:1 μM. To generate tetraploid and diploid clones, the cell line RKO containing ∼5% tetraploid cells was subcloned by limiting dilution into diploid and tetraploid clones. One diploid clone was transfected with a cDNA encoding H2B-GFP (Pharmingen), selected in blasticidine (20 μg/ml, InVitrogene), FACS-separated into subsets of cells enriched in a diploid or tetraploid DNA content to generate diploïd and tetraploid H2B-GFP-expressing clones. Tetraploid HCT116 clones were generated upon treatment with cytochalasin D (0.6 μg/ml, 48 h) or nocodazole (100 nM, 48 h). A colorimetric assay for quantification of cell viability based on the cleavage of the tetrazolium salt WST-1 (Roche Diagnostics, Germany) was used to measure the IC50 for DNA-damaging agents.
siRNAs
The knockdown of Bax, Puma, FDXR, Gadd45A or p53R2 was performed with siRNAs purchased from Proligo (Bax sense 5′-GGUGCCGGAACUGAUCAGATT-3′, anti-sense: 5′UCUGAUCAGUUCCGGCACCTT-3′; FDXR: sense 5′-CUGGAGGCCCUCCUUUUGTT-3′, antisense 5′-ACAAAAGGAGGGCCUCCAGTT-3′; GADD45A: sense 5′-CGACCUGCAGUUUGCAAUATT-3′, antisense 5′-UAUUGCAAACUGCAGGUCGTT-3′); PUMA: sense 5′UCUCAUCAUGGGACUCCUGTT-3′ or 5′-UUGAGGUCGUCCGCCAUCCTT-3′; p53R2 440: sense 5′-GCAGAAGAGGUCGACUUAUTT-3′, antisense 5′-AUAAGUCGACCUCUUCUGCTT-3′; p53R2 UTR : sense 5′-GAACAUGGUAGGGAUUAUUTT-3′, antisense 5′-AAUAAUCCCUACCAUGUUCTT-3′). As a control, an siRNA specific for emerin (Elbashir et al, 2001) as well as scrambled control siRNAs were used. Diploid and tetraploid RKO were cultured in 12-well plates and transfected at 30–40% confluence by adding oligofectamine (Invitrogen) complexed with siRNA (final 150 nM). After 72 h, the efficiency of transfection was determined by immunoblot, yielding in each case >80% of down-modulation of the target gene product.
Microarray analyses and identification of putative p53 binding sites
The microarray data (see Materials and methods in the legend to Supplementary Figure 5S) were submitted to the EBI database and are available under the access code E-TABM-70 IGR_PLOIDY. Putative p53 binding sites were searched within the 2000 nucleotides upstream of the transcription start of each transcript, using two distinct methods. The first method was based on a sliding profile of four matrices of 10 nucleotides in length, spaced between 0 and 13, with each matrix position having a score of 1 for each match but 10 for the central C and G positions (RRRCA/TT/AGYYY). A threshold of 104/112 has been set for this analysis (Bourdon et al, 1997). The second method was based on the identification of at least one p53 consensus binding sequence (RRRCWWGYYY), using the tfsccan program of the EMBOSS suite (Rice et al, 2000).
Staining of live cells and immunofluorescence
Live cells were stained with ΔΨm-sensitive dies (40 nM 3,3′dihexiloxalocarbocyanine iodide, DiOC6(3), or 150 nM tetramethylrhodamine methylester, TMRM, Molecular Probes), vital dies (2μg/ml propidium iodine, PI, Sigma; or 10 μM 4,6-diamino-2 phenylindole, DAPI, Molecular Probes), or Hoechst 33342 (2 μM, Molecular Probes) for 30 min at 37° (Castedo et al, 2002a). Cytofluorometric analyses were performed on a FACS Vantage (Becton Dickinson) equipped with a 70 μm nozzle and CellQuest software. Cells were fixed with paraformaldehyde (4% w:v) and then stained with rabbit antisera specific for p53 phophorylated on serine 15 or serine 46 (Cell Signaling Technology, MA, USA), Casp-3a (Cell Signaling Technology), all revealed with a goat anti-rabbit IgG conjugated to Alexa 568 (red) from Molecular Probes. Cells were also stained for the detection of activated Bax (mAb 6A7, BD Pharmingen), H2AXP (rabbit polyclonal, Trevigen), cytochrome c (mAb, BD Transduction Laboratories), and revealed with an anti-mouse IgG Alexa conjugated to Alexa 488 (green) from Molecular Probes (Castedo et al, 2002b).
Quantitation of protein expression
Protein samples were prepared from HCT116 or RKO cells in lysis buffer. Aliquots of protein extracts (50 μg/lane) were subjected to immunoblots using antibodies specific for p53 (DO-1, Santa Cruz Biotechnology, CA), p53R2 (goat polyclonal N-16, Santa Cruz), Chk1 (rabbit polyclonal antibody FL-476, Santa Cruz), BubR1 (mAb, BD Transduction Laboratories), Bax (N-20, Santa Cruz), Bcl-2 (mAb 100, Santa Cruz), VDAC (anti-porin 31HL, Calbiochem) and GAPDH (Chemicon, CA).
In vivo model
Athymic nu/nu 6-week-old female mice (IGR animal facility) were inoculated s.c. in 200 μl of PBS with 3 × 106 diploid or tetraploid RKO cells into the lower flank. When tumors reached 125 mm3, mice received i.p. either 200 μl of PBS1X or 5 mg/kg of cisplatin three times a week during 3 weeks. Tumor growth was measured with a caliper. The mean of the tumor volume at each point was normalized in each group to the mean volume measured at the first injection. Then, the tumor growth of the diploid and tetraploid treated group was normalized to the growth of the nontreated diploid or tetraploid group, respectively. All animals were maintained in specific pathogen-free conditions and all experiments followed the FELASA guidelines.
Supplementary Material
Supplementary Figure 1S
Supplementary Figure 2S
Supplementary Figure 3S
Supplementary Figure 4S
Supplementary Figure 5S
Supplementary Figure 6S
Supplementary Figure Legend 1S
Supplementary Figure Legend 2S
Supplementary Figure Legend 3S
Supplementary Figure Legend 4S
Supplementary Figure Legend 5S
Supplementary Figure Legend 6S
Acknowledgments
We thank Stanley Korsmeyer (Harvard University, Boston, MA) for Bax/Bak-deficient MEF and Bernd Vogelstein (John Hopkin's University, Baltimore, MD) for genetically modified HCT116 cells. GK is supported by Ligue Nationale contre le cancer, European Community (Impaled, Active p53, RIGHT), and Institut Gustave Roussy. NC received a fellowship from Fondation pour la Recherche Médicale.
References
- Adams JM (2003) Ways of dying: multiple pathways to apoptosis. Genes Dev 17: 2481–2495 [DOI] [PubMed] [Google Scholar]
- Andreassen PR, Lohez OD, Lacroix FB, Margolis RL (2001) Tetraploid state induces p53-dependent arrest of non-transformed mammalian cells in G1. Mol Biol Cell 12: 1315–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett MT, Pritchard D, Palanca-Wessels C, Anderson J, Reid BJ, Rabinovitch PS (2003) Molecular phenotype of spontaneously arising 4N (G2-tetraploid) intermediates of neoplastic progression in Barrett's esophagus. Cancer Res 63: 4211–4217 [PubMed] [Google Scholar]
- Bourdon JC, Deguin-Chambon V, Lelong JC, Dessen P, May P, Debuire B, May E (1997) Further characterisation of the p53 responsive element—identification of new candidate genes for trans-activation by p53. Oncogene 14: 85–94 [DOI] [PubMed] [Google Scholar]
- Castedo M, Ferri K, Roumier T, Metivier D, Zamzami N, Kroemer G (2002a) Quantitation of mitochondrial alterations associated with apoptosis. J Immunol Methods 265: 39–47 [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini J-L, Roumier T, Valent A, Raslova H, Yakushijin K, Horne DA, Feunteun J, Lenoir G, Vainchenker W, Kroemer G (2004) Mitotic catastrophe. A special case of apoptosis preventing aneuploidy. Oncogene 23: 4362–4370 [DOI] [PubMed] [Google Scholar]
- Castedo M, Roumier T, Blanco J, Ferri KF, Barretina J, Andreau K, Perfettini J-L, Armendola A, Nardacci R, LeDuc P, Ingber DE, Este JA, Modjtahedi N, Piacentini M, Kroemer G (2002b) Sequential involvement of Cdk1, mTOR and p53 in apoptosis induced by the human immunodeficiency virus-1 envelope. EMBO J 21: 4070–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B (1999) 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature 401: 616–620 [DOI] [PubMed] [Google Scholar]
- Chen JG, Yang CP, Cammer M, Horwitz SB (2003) Gene expression and mitotic exit induced by microtubule-stabilizing drugs. Cancer Res 63: 7891–7899 [PubMed] [Google Scholar]
- Cross SM, Sanchez CA, Morgan CA, Schimke MK, Ramel S, Idzerda RL, Raskind WH, Reid BJ (1995) A p53-dependent mouse spindle checkpoint. Science 267: 1353–1356 [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer S (2004) Cell death: critical control points. Cell 116: 205–219 [DOI] [PubMed] [Google Scholar]
- Du J, Hannon GJ (2004) Suppression of p160ROCK bypasses cell cycle arrest after Aurora-A/STK15 depletion. Proc Natl Acad Sci USA 101: 8975–8980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duelli D, Lazebnik Y (2003) Cell fusion: a hidden enemy? Cancer Cell 3: 445–448 [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498 [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Natti M, Ivanova EV, Bronson RT, Pellman D (2005) Cytokinesis failure generating tetraploids promotes tumorigenesis in p53 null mice. Nature 437: 1043–1047 [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305: 626–629 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57–70 [DOI] [PubMed] [Google Scholar]
- Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, Kelso GF, Smith RA, Kinzler KW, Vogelstein B (2001) Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med 7: 1111–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imkie M, Davis MK, Persons DL, Cunningham MT (2004) Biphasic acute myeloid leukemia with near-tetraploidy and immunophenotypic transformation. Arch Pathol Lab Med 128: 448–451 [DOI] [PubMed] [Google Scholar]
- Jiang W, Wang X (2004) Cytochrome C-mediated apoptosis. Annu Rev Biochem 73: 87–106 [DOI] [PubMed] [Google Scholar]
- Lin H, de Cavalho P, Kho D, Tai C-Y, Pierre P, Fink GR, Pellman D (2001) Polyploids require Bik1 for kinetochore–microtubule attachment. J Cell Biol 155: 1173–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin ZP, Belcourt MF, Cory JG, Sartorelli AC (2004) Stable suppression of the R2 subunit of ribonucleotide reductase by R2-targeted short interference RNA sensitizes p53(−/−) HCT-116 colon cancer cells to DNA-damaging agents and ribonucleotide reductase inhibitors. J Biol Chem 279: 27030–27038 [DOI] [PubMed] [Google Scholar]
- Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Blount PL, Reid BJ (2004) The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma. Cancer Res 64: 7629–7633 [DOI] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA (2002) Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53(−/−) cells. EMBO J 21: 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694 [DOI] [PubMed] [Google Scholar]
- Ogle BM, Cascalho M, Platt JL (2005) Biological implications of cell fusion. Nat Rev Mol Cell Biol 6: 567–575 [DOI] [PubMed] [Google Scholar]
- Okumura H, Natsugoe S, Matsumoto M, Mataki Y, Takatori H, Ishigami S, Takao S, Aikou T (2005) The predictive value of p53, p53R2, and p21 for the effect of chemoradiation therapy on oesophageal squamous cell carcinoma. Br J Cancer 92: 284–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poncet D, Larochette N, Pauleau AL, Boya P, Jalil AA, Cartron PF, Vallette F, Schnebelen C, Bartle LM, Skaletskaya A, Boutolleau D, Martinou JC, Goldmacher VS, Kroemer G, Zamzami N (2004) An anti-apoptotic viral protein that recruits Bax to mitochondria. J Biol Chem 279: 22605–22614 [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A (2000) EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet 16: 276–277 [DOI] [PubMed] [Google Scholar]
- Rieder CL, Maiato H (2004) Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell 7: 637–651 [DOI] [PubMed] [Google Scholar]
- Shin HJ, Baek KH, Jeon AH, Park MT, Lee SJ, Kang CM, Lee HS, Yoo SH, Chung DH, Sung YS, McKeon F, Lee CW (2003) Dual roles of human BubR1, a mitotic checkpoint kinase, in the monitoring of chromosomal instability. Cancer Cell 4: 483–497 [DOI] [PubMed] [Google Scholar]
- Sphyris N, Harrison DJ (2005) p53 deficiency exacerbates pleiotropic mitotic defects, changes in nuclearity and polyploidy in transdifferentiating pancreatic acinar cells. Oncogene 24: 2184–2194 [DOI] [PubMed] [Google Scholar]
- Storchova Z, Pellman D (2004) From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 5: 45–54 [DOI] [PubMed] [Google Scholar]
- Sudbo J, Kildal W, Risberg B, Koppang HS, Danielsen HE, Reith A (2001) DNA content as a prognostic marker in patients with oral leukoplakia. N Engl J Med 344: 1270–1278 [DOI] [PubMed] [Google Scholar]
- Sudbo J, Lippman SM, Lee JJ, Mao L, Kildal W, Sudbo A, Sagen S, Bryne M, El-Naggar A, Risberg B, Evensen JF, Reith A (2004) The influence of resection and aneuploidy on mortality in oral leukoplakia. N Engl J Med 350: 1405–1413 [DOI] [PubMed] [Google Scholar]
- Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267: 1456–1462 [DOI] [PubMed] [Google Scholar]
- Uetake Y, Sluder G (2004) Cell cycle progression after cleavage failure: mammalian somatic cells do not possess a ‘tetraploidy checkpoint'. J Cell Biol 165: 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Kienitz A, Hofmann I, Muller R, Bastians H (2004) Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene 23: 6845–6853 [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310 [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X (2002) Live or let die: the cell's response to p53. Nat Rev Cancer 2: 594–604 [DOI] [PubMed] [Google Scholar]
- Watanabe A, Inokuchi K, Yamaguchi H, Mizuki T, Tanosaki S, Shimada T, Dan K (2004) Near-triploidy and near-tetraploidy in hematological malignancies and mutation of the p53 gene. Clin Lab Haematol 26: 25–30 [DOI] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW (2005) Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell 8: 7–12 [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong W-X, Cheng EH-Y, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin XY, Grove L, Datta NS, Long MW, Prochownik EV (1999) C-myc overexpression and p53 loss cooperate to promote genomic instability. Oncogene 18: 1177–1184 [DOI] [PubMed] [Google Scholar]
- Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L (2003) PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci USA 100: 1931–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler JW, Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7: 673–682 [DOI] [PubMed] [Google Scholar]
- Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B (2000) Role of BAX in the apoptotic response to anticancer agents. Science 290: 989–992 [DOI] [PubMed] [Google Scholar]
- Zhivotovsky B, Kroemer G (2004) Apoptosis and genomic instability. Nat Rev Mol Cell Biol 117: 4461–4468 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1S
Supplementary Figure 2S
Supplementary Figure 3S
Supplementary Figure 4S
Supplementary Figure 5S
Supplementary Figure 6S
Supplementary Figure Legend 1S
Supplementary Figure Legend 2S
Supplementary Figure Legend 3S
Supplementary Figure Legend 4S
Supplementary Figure Legend 5S
Supplementary Figure Legend 6S