

Abstract
In yeast, separation of duplicated spindle pole bodies (SPBs) (centrosomes in higher eukaryotes) is an indispensable step in the assembly of mitotic spindle and is triggered by severing of the bridge that connects the sister SPBs. This process requires Cdk1 (Cdc28) activation by Tyrosine 19 dephosphorylation. We show that cells that fail to activate Cdk1 are devoid of spindles due to persistently active APCCdh1, which targets microtubule-associated proteins Cin8, Kip1 and Ase1 for degradation. Tyrosine 19 dephosphorylation of Cdk1 is necessary to specifically prevent proteolysis of these proteins. Interestingly, SPB separation is dependent on the microtubule-bundling activity of Cin8 but not on its motor function. Since ectopic expression of proteolysis-resistant Cin8, Kip1 or Ase1 is sufficient for SPB separation even in the absence of Cdc28-Clb activity, we suggest that stabilization of these mechanical force-generating proteins is the predominant role of Cdc28-Clb in centrosome separation.
Keywords: Cdh1, centrosomes, mitosis, spindle, SPB
Introduction
Assembly of a bipolar spindle is essential for accurate segregation of replicated chromosomes to daughter cells during cell division. Both temporal and spatial regulation is critical for construction of a functional bipolar spindle since failure to do so can lead to genomic instability and aneuploidy, often associated with cancers. In budding yeast Saccharomyces cerevisiae, spindle pole bodies (SPBs, the centrosome equivalent) nucleate microtubules and are central to the establishment of spindle bipolarity (Byers and Goetsch, 1975).
The SPB, a multitiered cylindrical organelle embedded in the nuclear envelope throughout the cell cycle, comprises three main layers: outer, central and inner plaques (Jaspersen and Winey, 2004). While the outer plaque faces the cytoplasm and nucleates cytoplasmic microtubules, the inner plaque is oriented towards the nucleoplasm and extends nuclear microtubules. The central plaque spans the nuclear membrane and contains proteins such as Spc42 and Spc29. It also bears an electron-dense structure known as the half-bridge that plays an important role in spindle development. Cdc31 (centrin homologue), Kar1, Mps3 and Sfi1 are specifically localized to this structure (Jaspersen and Winey, 2004). While these proteins are necessary for half-bridge integrity, it is unclear how this structure itself is assembled. The bridge is also the site of new SPB assembly and eventually tethers the duplicated SPBs.
Spindle assembly is a cell cycle-regulated, multistep process. Yeast cells inherit one SPB, bearing a half-bridge, from their mothers. During G1 (prior to START), the tip of the cytoplasmic side of half-bridge acquires the SPB precursor known as ‘satellite' (Byers and Goetsch, 1975). As cells traverse START, the satellite expands into a duplication plaque, similar in structure to the cytoplasmic side of a mature SPB. This requires self-assembly of Spc42 in which Spc42 dimers trimerize to construct the plaque (Bullitt et al, 1997). About the same time, the half-bridge elongates under the duplication plaque and forms a complete bridge by fusion of its cytoplasmic and nuclear fronts. The partial retraction of the half-bridge allows insertion of duplication plaque into the nuclear membrane and construction of nucleoplasmic-side of SPB (Adams and Kilmartin, 1999). Eventually, old and the newly duplicated SPBs lie side-by-side, linked by a complete bridge (O'Toole et al, 1999). Some time in late S phase, the bridge is severed and the SPBs move away from each other, finally facing each other, separated by interdigitating parallel microtubules (Winey et al, 1995). This configuration is what is generally referred to as a short spindle.
Severing of the inter-SPB bridge is one of the critical steps in mitotic spindle assembly; however, this process has remained largely obscure. Cells that fail to sever the bridge are unable to assemble a spindle and arrest in mitosis with side-by-side SPBs. In budding yeast, the following conditions give rise to this phenotype: (i) cells lacking kinesin motor proteins Cin8 and Kip1 (Hoyt et al, 1992; Roof et al, 1992); (ii) cells with combined deficiency of B-type cyclins, Clb3, Clb4 and Clb5 (Yeong et al, 2001); (iii) cells with combined deficiency of Clb1, Clb2, Clb3 and Clb4 (Fitch et al, 1992); (iv) cells unable to activate Cdc28 by tyrosine 19 dephosphorylation (Y19) catalyzed by Mih1 (homolog of Cdc25 phosphatase) and (v) cells expressing cdc28Y19E or overexpressing Wee1 homologue Swe1 (Lim et al, 1996). These observations suggest that SPB separation requires microtubule-associated kinesin motors Cin8 and Kip1, and Cdc28/Clb kinase. Involvement of motor proteins and kinases in centrosome separation has also been reported in other systems. Cdc2-mediated phosphorylation of kinesin motor human Eg5 allows it to localize to centrosomes, where it participates in their separation (Blangy et al, 1995; Sawin and Mitchison, 1995). In both Drosophila and Xenopus, loss of aurora-A kinase activity leads to failure in centrosome separation (Glover et al, 1995; Giet et al, 1999). Similarly, inhibition of polo kinase in Drosophila and human cells results in an inability to separate centrosomes (Sunkel and Glover, 1988; Lane and Nigg, 1996).
Although the role of Cin8 and Kip1 in SPB separation in yeast can be envisioned as force generation, the exact mechanics by which separation is achieved remain elusive. The function of Cdc28/Clb kinases in SPB separation is equally intriguing. It is also unclear whether Clb kinase complexes contribute to SPB separation via a process distinct from the one catalyzed by motor proteins. One possibility is the involvement of Clb kinases in severing of the bridge by a yet unknown mechanism, followed by crosslinking and sliding of interdigitated microtubules by motor proteins, pushing the newly disconnected SPBs further apart (Hoyt et al, 1992; Jaspersen and Winey, 2004). Alternatively, Clb kinases may help to activate motor proteins that in turn generate sufficient mechanical ‘strength' to force duplicated SPBs apart.
Here we examined the regulatory role of Cdc28/Clb kinase in SPB separation. We find that microtubule-associated proteins Cin8, Kip1 and Ase1 are highly unstable in cells that fail to dephosphorylate Cdc28 on Tyr19 (cdc28Y19E mutant), resulting in failure to separate SPBs. This instability is due to persistently active Cdh1 in these cells. Our results imply that Tyr19 dephosphorylation of Cdc28 is required to inactivate APCCdh1 specifically for stabilization of microtubule-associated proteins. Moreover, Cdh1 inactivated by active Cdc28 (Tyr19 dephosphorylated) fails to enter nucleus, ensuring accumulation of these proteins. Strikingly, while the bundling activity of Cin8 is necessary for SPB separation, its motor activity is not. Collectively, our observations suggest that tyrosine dephosphorylation of Cdc28 acts as a ‘regulatory switch' that controls the assembly of mitotic spindle.
Results
cdc28Y19E and cdc28-as1 cells are unable to separate SPBs
clb1Δ clb2Δ clb3Δ clb4Δ and clb3Δ clb4Δ clb5Δ mutants fail to assemble a spindle and arrest in G2/M with side-by-side SPBs (Fitch et al, 1992; Yeong et al, 2001), suggesting that Cdc28-Clb kinase activity is essential for spindle biogenesis. However, since these strains are inconvenient for experimental purposes, we used cdc28Y19E (US3143) and cdc28-as1 (US3558).
The cdc28Y19E strain carries a temperature-sensitive cdc28-4 allele at the native locus and four copies of native promoter-driven cdc28Y19E alleles integrated at URA3 locus (to overcome the sluggishness with which cells carrying one copy of cdc29Y19E traverse START and S phase). At 37°C, cell cycle progression in these cells is driven by cdc28Y19E allele since cdc28-4 allele is inactive. Although slow in their progression, these cells eventually arrest with a large bud, 2C DNA and duplicated SPBs connected by a bridge (Figure 1A, top panel). Inability of these cells to assemble a spindle is not due to lack of Cdc28-Clb mitotic kinase activity since cdc28YE19 cells exhibit substantial histone H1 kinase activity (Figure 1A, lower panel). Moreover, GAL-driven cdc28Y19E exhibit H1 kinase activity equivalent to that of wild type but yet fail to assemble a spindle (Lim et al, 1996). Hence, failure to separate SPBs is specifically due to the inability to dephosphorylate Tyrosine 19.
Figure 1.
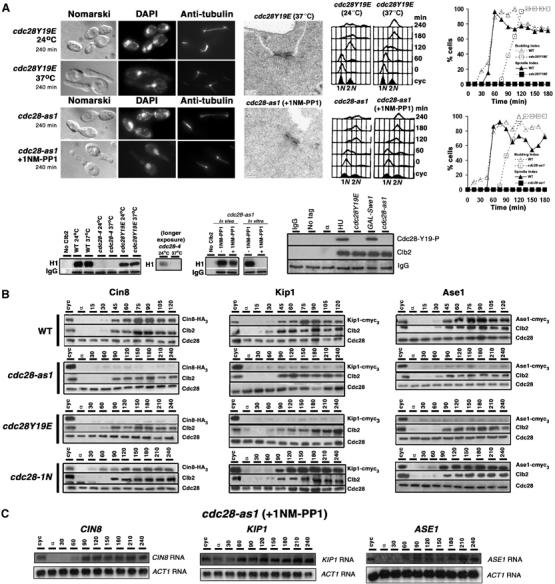
Low endogenous levels of microtubule-associated proteins in cdc28–as1 and cdc28Y19E cells. (A) cdc28Y19E (top panel) and cdc28-as1 (middle panel) cells were arrested with α factor and released at 37°C and 24°C in presence of 1NM-PP1, respectively. Electron micrographs show state of SPBs at 240 min. Plots show spindle and budding index. (Lower panel) Western blots show histone H1 kinase activity and tyrosine phosphorylation status. (B) Wild-type (WT), cdc28-as1, cdc28-Y19E and cdc28-1N cells carrying endogenously tagged Cin8-HA3, Kip1-cmyc3 or Ase1-cmyc3 were synchronized in G1 with α factor treatment and then released into at 24°C in medium containing 1NM-PP1 (for cdc28-as1) or 37°C (for cdc28Y19E and cdc28-1N). Samples for Western blotting were taken at 15-min intervals for WT and 30-min intervals for cdc28 mutants for comparison (doubling time in cdc28 mutants is approximately twice that of WT). (C) G1-synchronised cdc28-as1 cells were released in the presence of 1NM-PP1. Samples were collected at 30-min intervals for RNA isolation and subsequent Northern blotting.
cdc28-as1 allele carries an F88G substitution that alters the ATP-binding pocket and confers sensitivity to a bulky ATP analog, 1NM-PP1 (Bishop et al, 2000). cdc28-as1 cells progress normally through the cell cycle; however, in the presence of 500 nM of 1NM-PP1, they arrest in G2/M with 2C DNA and fail to assemble a mitotic spindle (Figure 1A, middle panel; Bishop et al, 2000). Electron microscopic analysis of these cells revealed side-by-side SPBs (Figure 1A, lower micrograph). This phenotype is similar to that of cdc28Y19E cells, even though Tyr19 is dephosphorylated in terminally arrested cdc28-as1 cells (Figure 1A, bottom panel), suggesting that F88G substitution in cdc28-as1 leads to conformation changes similar to that caused by Tyr19-to-glutamate substitution in ATP-binding site.
Previous studies have implicated Cin8 and Kip1 in SPB separation (Hoyt et al, 1992; Roof et al, 1992). Cin8 also exhibits synthetic lethal interaction with another microtubule-associated protein Ase1 (Schuyler et al, 2003). We have observed spindle formation defects in cin8Δ ase1Δ, suggesting a possible role for Ase1 in spindle assembly (data not shown). It is possible that failure to separate SPBs in cdc28Y19E and cdc28-as1 cells is due to low abundance of these proteins. To test this, G1-synchronized cdc28Y19E cells expressing Cin8-HA3, Kip1-cmyc3 or Ase1-cmyc3 from their respective loci were released into fresh medium at 37°C and protein levels monitored by Western blotting. cdc28-as1 cells carrying identical constructs were released at 24°C into medium containing 1NM-PP1. A wild-type strain and cdc28-1N mutant (arrests with short spindles; Surana et al, 1991) were used as controls and released into medium at 24°C and 37°C, respectively. As shown in Figure 1B, extremely low levels of Cin8, Kip1 and Ase1 were observed in cdc28Y19E and cdc28-as1, while these proteins were easily detectable in wild-type and cdc28-1N cells. Clb2, however, remained detectable in all strains (Figure 1B). To determine if low abundance of these proteins is due to reduced transcription in the mutant strains, G1-synchronized cdc28-as1 cells were allowed to resume cell cycle progression in the presence or absence of 1NM-PP1. PolyA RNA was isolated from cell samples withdrawn at different times and CIN8, KIP1 and ASE1 transcripts were analyzed by Northern blotting using riboprobes. The abundance of the three transcripts in the presence of 1NM-PP1 is comparable to that in the cycling culture (Figure 1C), suggesting that low levels of Cin8, Kip1 and Ase1 proteins in 1NM-PP1-grown cdc28-as1 cells is not due to severely compromised transcription.
Defect in SPB separation is due to proteasomal degradation of microtubule-associated proteins
To assess whether low abundance of these proteins is due to enhanced proteolysis, cdc28Y19E and cdc28-as1 cells carrying epitope-tagged CIN8, KIP1 or ASE1 under control of GAL1 promoter (US3999, US3419, US4175; US4000, US3722, US4099) were synchronized in G1 and subsequently released into nonpermissive conditions in galactose medium. After 60 min, cells were transferred to glucose medium with 1 mg/ml cycloheximide and the protein pulse was monitored. Although the protein pulses remained stable in wild-type and cdc28-1N cells, the abundance of all three proteins declined rapidly in both cdc28Y19E and cdc28-as1 mutants (Figure 2A). However, the Clb2 pulse remained stable in all four strains (Supplementary Figure S1).
Figure 2.

Proteasomal degradation of microtubule-associated proteins in cdc28 mutants (A) WT, cdc28Y19E, cdc28-as1 and cdc28-1N carrying either GAL-CIN8-cmyc3, GAL-KIP1-cmyc3 or GAL-ASE1-cmyc3 on a CEN plasmid were arrested in YEP+Raff containing α factor and then released into YEP+Raff+Gal at respective arrest conditions (as in Figure 1) for 90 min to induce protein expression. Cells were then transferred to YEP+Glu containing 1 mg/ml cycloheximide for shut-off of protein synthesis and the fate of the protein pulse was monitored by Western blotting. Numbers below Western blots indicate Protein/Cdc28 ratio measured by densitometry. (B) G1-synchronized cdc28-as1 cells carrying endogenous Cin8-HA3 and SPC42-GFP integrated at TRP1 locus were released at 24°C into medium containing 1NM-PP1. Proteasome inhibitor MG132 was added 100 min after release from α factor and samples were collected for Western blot and microscopic analysis. (C) (Top left panel) Scheme illustrating principle of BRET between Cin8-Rluc and GFP-ubiquitin. (Left graph) Plot for BRET measured in cdc28-as1 cells in 1NM-PP1-expressing Cin8-Rluc alone, Cin8-Rluc and GFP-monoUbi (from GAL1 promoter) or Cin8-Rluc and GFP-UbiAA (from GAL1 promoter). Sample at 150 min after the release from alpha-factor is shown. (Right graph) Plot of BRET ratios of cells co-expressing Cin8-Rluc and GFPmonoUbi (from GAL1 promoter) or Cin8-Rluc and GFP-UbiAA (from GAL1 promoter). BRET ratios were calculated as defined in Materials and methods. Data represent the mean of three independent experiments. Samples were also analyzed by Western blotting for Cin8 levels in cdc28-as1 cells expressing Cin8-Rluc and cdc28-as1 cells co-expressing Cin8-Rluc and GFP-UbiAA (from GAL1 promoter) (Top left panel).
We asked if failure to form spindles could be rescued by increasing the abundance of microtubule-associated proteins. G1-synchronized cdc28-as1 cells carrying CIN8-HA3 at its native locus and SPC42-GFP at the TRP1 locus were released in medium containing 1NM-PP1 at 24°C. After cells had entered S phase (upon Sic1 degradation, estimated to be 100 min), proteasome inhibitor MG132 was added to prevent Cin8 degradation. In the presence of MG132, cells arrested in G2/M with 2C DNA content, detectable levels of Cin8 and well-separated Spc42-GFP spots (Figure 2B). The fact that MG132 did not appreciably change the level of Clb2-dependent histone H1 kinase activity shows that spindles formed, not because of altered levels of kinase activity, but because of accumulation of Cin8 (Figure 2B). Thus, failure to assemble a spindle in cdc28-as1 (and by inference, cdc28Y19E) cells is due to proteasome-dependent degradation of microtubule-associated proteins.
To further extend these observations, ubiquitylation of Cin8 in live cells was measured using the Bioluminescence Resonance Energy Transfer (BRET2) assay (Figure 2C, top panel) (Perroy et al, 2004). cdc28-as1 carrying endogenous CIN8 tagged with Renilla luciferase (CIN8-Rluc) and GAL promoter-driven GFP2-tagged monoubiquitin (GFP2-monoUb) on a CEN plasmid (US4629) was used. cdc28-as1 carrying CIN8-Rluc and GAL-GFP2-UbiAA (a mutant ubiquitin with G75A and G76A substitutions; unable to covalently attach to the substrate) (US4630) served as a negative control. While cells co-expressing GFP2-monoUbi show an additional peak at 510 nm (i.e. BRET), those co-expressing GFP2-UbiAA and those carrying only Cin8-Rluc do not (Figure 2C, left graph). A continuous rise in BRET ratio suggests a progressive increase in Cin8 ubiquitylation (Figure 2C, right graph). Western blotting showed low abundance of Cin8 in cdc28-as1 cells but higher levels in cells expressing GFP-UbiAA (Figure 2). Collectively, these results suggest that cdc28-as1 and cdc28Y19E cells are unable to separate SPBs due to low levels of microtubule-associated proteins caused by ubiquitin-mediated proteolysis.
Prompted by these observations, we tested if expression of nondegradable versions of Cin8, Kip1 and Ase1 can alleviate spindle formation defect in the cdc28-as1 mutant. While endogenous Cin8, Kip1 and Ase1 are highly unstable in G1, the native promoter-driven nondestructible versions are stable (Figure 3A, rightmost panel; see Materials and methods for mutant alleles' construction). Spc42-GFP expressing cdc28-as1 cells either expressing wild-type versions of Cin8, Kip1 and Ase1 or nondegradable versions Cin8nd, Kip1nd or Ase1nd (from their respective native promoters) were synchronized in G1 by α factor treatment and then released at 24°C in presence of 1NM-PP1. Expression of Cin8nd, Kip1nd or Ase1nd allowed 46–64% of cells to separate SPBs (Figure 3A). As expected, cdc28-as1 cells expressing wild-type version of Cin8, Kip1 or Ase1 failed to do so (Figure 3A).
Figure 3.
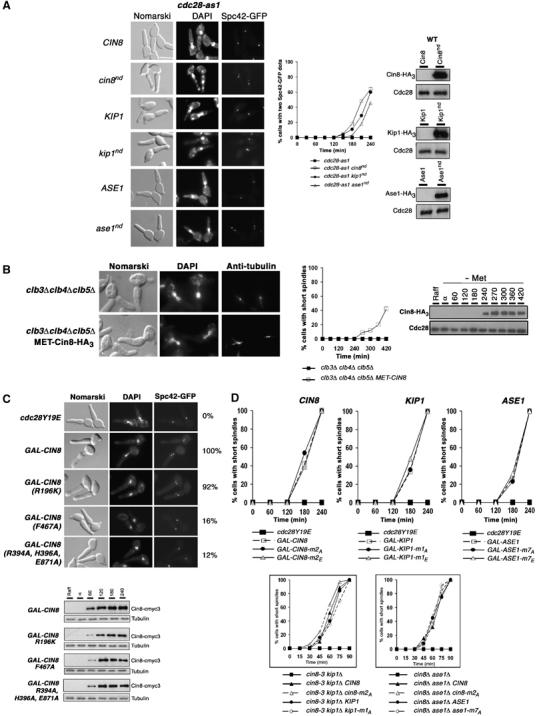
Cin8-bundling activity, not its motor activity or phosphorylation, is required for SPB separation. (A) G1-synchronized cdc28-as1 expressing nondegradable versions of Cin8, Kip1 and Ase1, from their respective native promoter-driven constructs on a CEN vector, were released in the presence of 1NM-PP1. Cells at 240 min after release are shown. The Western blot (rightmost panel) shows stability of the nondegradable versions in G1. (B) clb3Δ clb4Δ clb5Δ GAL-CLB5 cells carrying MET3-CIN8-HA3 were synchronized in G1 in Raff+Gal+Methionine medium and released into methionine-deficient glucose medium. (C) G1-synchronised cdc28Y19E carrying SPC42-GFP at the TRP1 locus and expressing from CEN vector GAL-CIN8-cmyc3, GAL-CIN8 (R196K)-cmyc3 (affecting motor activity), GAL-CIN8 (F467A)-cmyc3 (affecting microtubule binding) or GAL-CIN8 (R394A, H396A, E871A)-cmyc3 (affecting bundling) were released into YEP+Raff+Gal at 37°C. Numbers indicate percentage of cells with two SPC42-GFP dots at 240 min. Western blots show the extent of Cin8 expression. (D) (Top panel) Kinetics of spindle formation in cdc28Y19E expressing GAL-CIN8-cmyc3, GAL-KIP1-cmyc3 or GAL-ASE1-cmyc3 on a CEN plasmid or versions carrying mutations at Cdc28 consensus sites mimicking phosphorylated (E) or unphosphorylated forms (A). (Lower panel) Spindle kinetics in cin8-3 kip1Δ and cin8Δ ase1Δ cells expressing phospho-deficient versions at the Cdc28 consensus sites (A).
The clb3Δ clb4Δ clb5Δ triple mutant, being effectively clb3Δ clb4Δ clb5Δ clb1−clb2−, is largely devoid of Cdc28-Clb kinase activity and fails to assemble a spindle (Yeong et al, 2001). Interestingly, MET3 promoter-driven expression of Cin8 allows spindle assembly in clb3Δ clb4Δ clb5Δ cells (US4615), implying that ectopic expression of Cin8 is sufficient to induce SPB-separation even in the absence of Cdc28-Clb kinase activity (Figure 3B).
Microtubule-bundling activity, not motor activity, is required for SPB separation
While Cin8 is a motor protein and has microtubule-bundling activity, Ase1 is a nonmotor protein that exhibits bundling activity in vitro (Schuyler et al, 2003); yet overexpression of both proteins can induce SPB separation in cdc28Y19E cells. To ask if bundling activity was sufficient to induce SPB separation, mutations characterized in a previous study (Gheber et al, 1999) were utilized which individually affect three different aspects of Cin8 functions: R394A, H396A and E871A substitutions abolish microtubule-bundling activity, K467A eliminates microtubule binding, whereas R196K eliminate motor activity. A CEN vector carrying each construct driven by GAL1-promoter was introduced in cdc28-Y19E cells expressing the GFP-tagged Spc42 (SPB marker) to test their ability to induce SPB separation (US3943, US3944 and US3945). While Cin8 deficient in motor activity was able to separate SPBs (two Spc42-GFP spots), versions defective in microtubule-binding or microtubule-bundling activity were unable to do so (Figure 3C). The distance between separated SPC42-GFP dots (pole-to-pole distance) for cells deficient in Cin8 motor activity was similar to that of normal pre-anaphase short spindles (240 min), implying that both separation and migration of SPBs were efficiently accomplished (data not shown). The difference in the ability of these three versions to promote spindle formation is not due to the level of their expression because all three versions were expressed to comparable extent in these experiments (Figure 3C, lower panel). These observations suggest that motor activity of Cin8 is dispensable for SPB separation but microtubule-binding and microtubule-bundling activities are not.
Microtubule-associated proteins in budding yeast are potential substrates of Cdc28-Clb kinase (Ubersax et al, 2003). There are two consensus Cdc28 phosphorylation sites in Cin8 (S455 and S972), one in Kip1 (S388) and seven in Ase1 (T55, S64, S198, T675, S707, S803, S819). To investigate if phosphorylation of these proteins by Cdc28 regulates SPB separation, the serine/threonine residue in each site was mutated to either alanine or glutamate, under the control of GAL1 promoter and introduced into cdc28Y19E and cdc28-as1 mutants. The modified alleles of all three proteins could induce spindle formation with similar efficiency irrespective of the amino-acid substitution (Figure 3D), suggesting that although SPB separation requires Cdc28, as well as Cin8, Kip1 and Ase1 activities, it is not accomplished through Cdc28-mediated phosphorylation of these proteins. The fact that these modified alleles (with alanine substitutions), when expressed from their respective native promoters, can also induce SPB separation in cin8-3 kip1Δ and cin8Δ ase1Δ double mutants further strengthen this notion (Figure 3D, lower graphs).
APCCdh1 prevents SPB separation
The anaphase-promoting complex (APC, a multi-subunit ubiquitin ligase activated by Cdc20 or Cdh1) mediates the proteolysis of proteins involved in spindle function (Zachariae and Nasmyth, 1999; Castro et al, 2005). Cin8 and Ase1 are targeted for proteolysis by APCCdh1 at the end of mitosis through G1 (Juang et al, 1997; Hildebrandt and Hoyt, 2001), and Kip1 by APCCdc20 (Gordon and Roof, 2001). We find that inactivation of Cdc23 (APC component) in cdc28Y19E and cdc28-as1 mutants leads to stabilization of these proteins and concomitant spindle formation (data not shown). To determine which APC activator(s) is involved, cdc28Y19E and cdc28-as1 cells with or without cdh1Δ were grown at 37°C or at 24°C in the presence of 1NM-PP1, respectively. While both cdc28Y19E and cdc28-as1 with functional CDH1 arrest in G2/M with no spindle, CDH1 deletion allows spindle formation in both strains; however, inactivation of Cdc20 does not lead to spindle formation (Figure 4A). Interestingly, cdc28-as1 cdh1Δ cells not only assembled a short spindle, but unlike cdc28Y19E cdh1Δ cells, proceeded to complete anaphase and arrested with long spindles (Figure 4A), implying that APCCdh1 is specifically responsible for the ‘no spindle' phenotype in cdc28Y19E and cdc28-as1 mutants.
Figure 4.
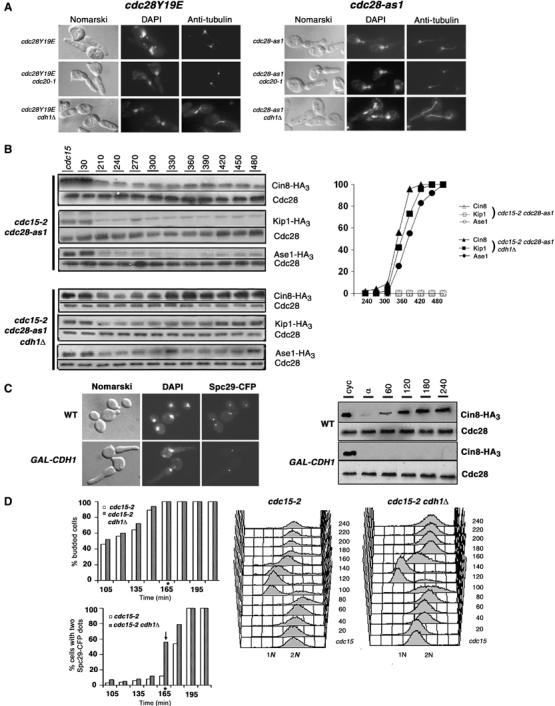
APCCdh1-mediated degradation of microtubule-associated proteins prevents spindle assembly. (A) (Left panel) cdc28Y19E cdh1Δ cells or cdc28Y19E cdc20-1 were arrested with α-factor and released at 37°C. (Right panel) cdc28-as1 cells with similar mutations were released in the presence of 1NM-PP1. Cells are shown 240 min after release. (B) cdc28-as1 and cdc28-as1 cdh1Δ cells carrying endogenously tagged Cin8-HA3, Kip1-HA3 or Ase1-HA3 were arrested in telophase at 37°C by a cdc15-2 mutation, and released in the presence of 1NM-PP1 at 24°C. (C) G1-synchronised WT cells tagged endogenously with Cin8-HA3 with or without GAL-Cdh1-cmyc3 on a CEN plasmid were released from YEP+Raff into YEP+Raff+Gal medium. (D) cdc15-2 and cdc15-2 cdh1Δ cells expressing Spc29-CFP were synchronized in telophase at 37°C, released into fresh medium at 24°C and were monitored for kinetics of spindle assembly. ‘*'Indicate the time at which a significant difference in the percentage of cells-with-spindles was observed between the two strains.
To test if Cdh1 absence allows microtubule-associated proteins to accumulate, we used cdc15-2 mutation, which causes telophase arrest at nonpermissive temperature, as cdh1Δ cells do not arrest well upon α-factor treatment (Schwab et al, 1997). cdc28-as1 cdc15-2 (US4434) and cdc28-as1 cdc15-2 cdh1Δ(US4432) cells expressing Cin8-HA3 from its native locus were first shifted to 37°C to allow arrest and then released into fresh medium containing 1NM-PP1 at 24°C. While cdc28-as1 cells showed low levels of Cin8, Kip1 and Ase1 as they proceeded to arrest in G2/M of the subsequent cycle, cdc28-as1 cdh1Δ cells accumulated discernible levels of all three proteins and were able to assemble a spindle (Figure 4B). These results suggest that Cdh1 is primarily responsible for low levels of Cin8, Ase1 and Kip1 in cdc28Y19E and cdc28-as1 cells. This is consistent with our observation that Cdh1 overexpression in wild-type cells causes severe deficit in Cin8 levels resulting in a failure to separate SPBs (Figure 4C). It is noteworthy that although Kip1 is reported to be a substrate of APCCdc20, these observations suggest that it may also be targeted by APCCdh1 for degradation.
Does Cdh1 influence timing of spindle formation? To test this, we compared kinetics of spindle assembly in cdc15-2 and cdc15-2 cdh1Δ cells expressing CFP-tagged SPB component Spc29, after release from telophase arrest. cdc15-2 strain released from telophase earlier than cdc15-2 cdh1Δ, but both strains showed no significant difference in their timing of budding and completion of S phase. However, SPBs separated 15 min earlier in cdc15-2 cdh1Δ cells (56% at 165 min) compared to cdc15-2 cells (12%) (Figure 4D), indicating that spindle forms somewhat earlier in the absence of Cdh1.
Cdh1 phosphorylation and Cin8 ubiquitylation in cdc28 mutants defective in spindle assembly
It is known that APCCdh1 is inactivated by Cdc28/Clb3, 4, 5 kinase complexes assembled during S phase, allowing cells to progressively accumulate mitotic cyclin Clb2 (Zachariae et al, 1998; Huang et al, 2001; Yeong et al, 2001). Cdh1 phosphorylation persists until it is activated by the Cdc14 phosphatase in late mitosis (Visintin et al, 1998; Jaspersen et al, 1999). Given our observations, it is possible that cdc28Y19E and cdc28-as1 cells are unable to efficiently phosphorylate Cdh1 and thereby fully inactivate it. Hence, we compared phosphorylation status of Cdh1 in clb3Δ clb4Δ clb5Δ, cdc28Y19E and cdc28-as1 strains (incapable of forming spindles), as well as in cdc28-1N and cdc28Y19F (able to form spindles) each harboring cmyc9-Cdh1 at its native locus. As controls, wild-type cells arrested in G1 (by α treatment) and mitosis (by nocodazole) were used since Cdh1 is dephosphorylated in G1 and highly phosphorylated in mitosis (Zachariae et al, 1998). Cells were released from G1 arrest into fresh medium at their respective restrictive growth conditions and phosphorylation status of cmyc9-Cdh1 was determined.
High abundance of slow-migrating forms of Cdh1 can be seen in cdc28-1N and cdc28Y19F cells (Figure 5A). However, these forms of Cdh1 were absent in clb3Δ clb4Δ clb5Δ mutants and present in very low amounts in cdc28Y19E and cdc28-as1 mutants (perhaps ‘partial' phosphorylation). As expected, no slow-migrating Cdh1 forms could be detected in G1-arrested wild-type cells, whereas multiple low-mobility forms (‘full' phosphorylation) were observed in nocodazole-arrested cells. Upon phosphatase treatment, all high molecular weight forms were converted into faster-migrating single bands indicating that these are phosphorylated forms (Figure 5A). The amount of Cdh1 in G1-arrested cells, clb3Δ clb4Δ clb5Δ and cdc28 mutants is consistently lower compared to other strains; at present, the reasons remain unclear. These results show a strong correlation between phosphorylation status of Cdh1 and spindle assembly phenotype in cdc28 mutants.
Figure 5.
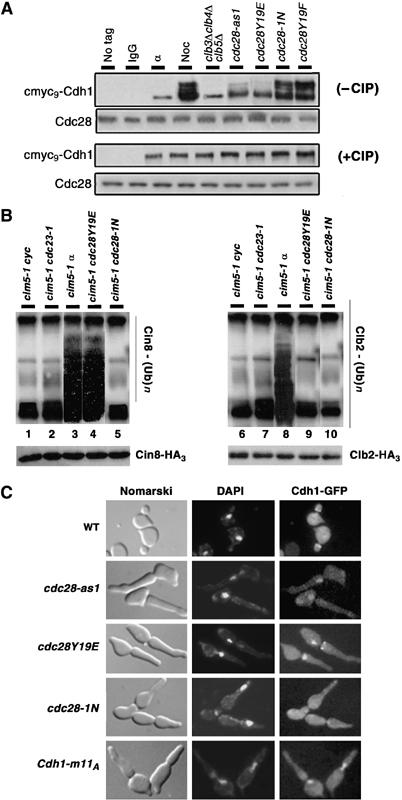
Phosphorylation status of Cdh1 determines its subcellular localization and Cin8 ubiquitylation. (A) Extracts were prepared from cmyc9-Cdh1 carrying WT cells grown in α factor (α) and nocodazole (Noc), and from clb3Δ clb4Δ clb5Δ GAL-CLB5, cdc28-as1, cdc28Y19E, cdc28-1N and cdc28Y19F arrested at their terminal points. Cdh1 was immunoprecipitated using anti-myc agarose beads, treated with and without alkaline phosphatase (CIP) and separated on a 10% SDS gel. Extracts from untagged WT strains (WT) as well as from nocodazole-arrested WT strain carrying cmyc9-Cdh1 immunoprecipitated with IgG beads (IgG) were used as negative controls. (B) cim5-1 cdc23-1 (lanes 2 and 7), cim5-1 cdc28Y19E (lanes 4 and 9), cim5-1 cdc28-1N (lanes 5 and 10) cells were synchronized in G1 with α factor and then released into fresh YPD medium at 37°C to reach their respective terminal phenotype. Since cim5-1 cdc28Y19E and cim5-1 cdc28-1N are synthetic lethal, they were kept alive by GAL-CDC28. Cycling (lanes 1 and 6) or α factor arrested (lanes 3 and 8) cim5-1 cells were used as controls. All strain carried endogenously tagged Cin8-HA3 or Clb2-HA3. Extracts were prepared, Clb2 and Cin8 were immunoprecipitated with anti-HA agarose beads, and ubiquitin conjagates were detected with anti-ubiquitin antibodies. Total amounts of Clb2-HA3 or Cin8-HA3 were detected using anti-HA antibodies. (C) WT, cdc28-as1, cdc28Y19E, cdc28-1N and WT overexpressing Cdh1-m11A (under the weak GALL promoter), all carrying Cdh1-GFP, were released from G1 arrest at their respective nonpermissive growth condition.
To determine if Cdh1 phosphorylation status in various strains correlates with the extent of Cin8 and Clb2 ubiquitylation, We used cdc28Y19E, cdc28-1N, cdc23-1 and wild-type cells carrying CIN8-HA3 at its native locus. To prevent proteolysis of ubiquitylated Cin8 and Clb2, each strain carried temperature-sensitive allele of Cim5 (cim5-1), a component of the 19S regulatory particle of 26S proteasome. Cells were synchronized in G1 by α factor treatment at 24°C and then were allowed to resume cell cycle progression at 37°C. Samples were collected after 210 min and analyzed for Cin8 and Clb2 ubiquitylation. As shown in Figure 5B, the extent of Cin8 ubiquitylation is clearly higher in G1-arrested wild-type and cdc28Y19E strains (lanes 3 and 4), but very low in cdc23-1 and cdc28-1N cells (lanes 2 and 5). The extent of Clb2 ubiquitylation is high in G1-arrested wild-type cells (lane 8) and very low cdc28Y19E, cdc23-1 and cdc28-1N strains (lanes 7, 9 and 10). Hence, there is a consistent correlation between Cdh1 phosphorylation, Cin8 ubiquitylation and the cells' ability to assemble a spindle.
Cdc28/Clb activity controls Cdh1 subcellular localization and spindle assembly
An added dimension to Cdh1 inactivation by Cdk1-dependent phosphorylation is the regulation of its cellular location. During G1, Cdh1 is in the nucleus; it is exported from the nucleus in Cdc28-Clb kinase-dependent manner later in the cell cycle (Jaquenoud et al, 2002). Since Cin8, Kip1 and Ase1 are localized in the nucleus and are targeted for destruction by Cdh1 in cdc28Y19E and cdc28-as1, Cdh1 is expected to be nuclear. To test this, cdc28Y19E and cdc28-as1 cells carrying the weaker GALL-promoter-driven CDH1-GFP on a CEN plasmid were synchronized in G1 by α factor treatment and then released at 37°C or at 24°C in galactose medium containing 500 nM 1NM-PP1. In parallel, cdc28-1N cells (with spindles) carrying GALL-CDH1-GFP and a wild-type strain expressing constitutively active Cdh1-m11A-GFP (in which S/T in all 11 Cdk1 sites have been altered to alanine) from GALL promoter were used as controls. Cdh1-GFP clearly localized to the nucleus in both cdc28Y19E and cdc28-as1, whereas in cdc28-1N it is not seen in the nucleus (Figure 5C). As expected, Cdh1-m11A-GFP remained in the nucleus in wild-type cells. These results imply that Cdc28Y19E and Cdc28-as1 form of the Clb kinases are not only unable to inactivate Cdh1 but also fail to cause its export from the nucleus.
Cdc28-phosphorylation sites in Cdh1 and stability of Cin8 and Clb2
Cdh1 contains 11 Cdc28-phosphorylatable sites (Figure 6). We conducted site-directed mutagenesis to identify phosphorylation sites important for stabilization of Cin8. Serines and threonines in these sites were mutated in different combinations to either alanine or glutamate and resultant mutants were expressed from GALL promoter in wild-type cells. Although it was not an exhaustive search through all possible combinations, we find that Cdh1 carrying alanine substitution in seven phosphorylation sites (S16, S42, T157, S169, T173, T176, S239) precludes spindle assembly when expressed in wild-type cells (US4627) (Figure 6). The levels of Clb2 and Cin8 (both APCCdh1 substrates) were very low in these cells. Conversely, expression of Cdh1 carrying all seven sites substituted with glutamate (US4628) did not interfere with spindle assembly and had detectable amounts of Cin8 and Clb2.
Figure 6.
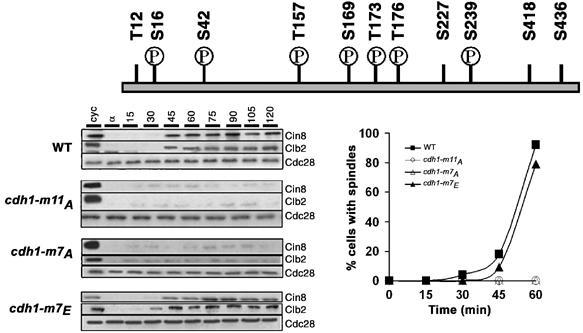
Phosphorylation sites in Cdh1 essential for spindle formation. Cdc28 phosphorylation sites in Cdh1 are schematically shown. ‘P' denotes residues whose phosphorylation is essential for spindle formation. WT and WT expressing Cdh1-m11A, Cdh1-m7A, Cdh1-m7E under the weak GALL promoter (all carrying endogenously tagged Cin8-HA3) were synchronized in G1 in YEP+Raff and released into YEP+Raff+Gal. Cin8 and Clb2 levels were determined by Western blotting. Graph shows proportion of cells with short spindles.
Tyrosine dephosphorylation of Cdc28 temporally precedes spindle assembly during a normal cell cycle
The preceding sections suggest that inhibitory Tyr19 phosphorylation of Cdc28 results in its inability to inactivate Cdh1, which in turn persistently degrade microtubule-associated proteins to preclude spindle assembly. To determine whether such a correspondence exists during normal cell cycle progression, cells expressing Cin8-HA3 and cmyc9-Cdh1 from their respective loci were released from G1 arrest at 24°C. Tyrosine-phosphorylated Cdc28, Cdh1, Cin8 and Clb2 were detected by Western blotting. As shown in Figure 7A, Cin8 abundance reaches its peak when tyrosine phosphorylation of Cdc28 disappears; this coincides with corresponding increase in Cdh1 phosphorylation at 130 min after release. This timing also correlates well with the assembly of short mitotic spindle. Thus, a strong correlation exists between dephosphorylation of Cdc28 at Tyr19, inactivation of Cdh1 by Cdc28-dependent phosphorylation, stabilization of microtubule-associated proteins and SPB separation (spindle assembly).
Figure 7.
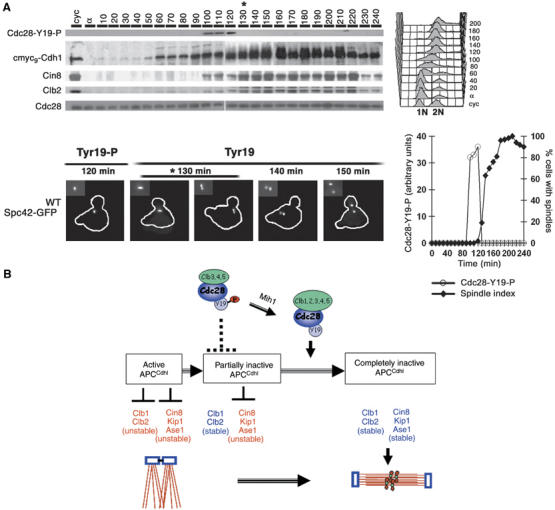
Tyrosine dephosphorylation correlates with the timing of SPB separation during normal cell cycle. (A) G1-synchronized WT cells carrying SPC42-GFP were released into fresh medium at 20°C. Samples were withdrawn at 10 min interval and analyzed for tyrosine dephosphorylation of Cdc28, Cdh1 phosphorylation and Cin8, Clb2 levels. ‘*'Denotes timing of Tyr19-Cdc28 dephosphorylation. The photomicrograph below shows cells at intermediate time points and the graph represents extent of tyrosine 19 phosphorylation. (B) A scheme illustrating the role of activated Cdc28 in SPB separation (see Discussion).
Discussion
In most organisms, the timing of mitotic spindle assembly correlates well with entry into mitosis and requires Cdc28/cdc2 mitotic kinase activity (Blangy et al, 1995; Sawin and Mitchison, 1995). What role does mitotic kinase, and specifically its Y19-dephosphorylated form, serve in SPB separation? In this study, we have shown that cdc28Y19E (mimicking Cdc28 constitutively phosphorylated on Y19) and cdc28-as1 cells are unable to accumulate the microtubule-binding proteins Cin8, Kip1 and Ase1 (Figure 2). The instability of these proteins is dependent on Cdh1 since deletion of CDH1 allows accumulation of these proteins and assembly of a short spindle in these mutants (Figure 4). That Cin8 is ubiquitylated to a significant extent in cdc28Y19E but to a much lesser extent in cdc28-1N and cdc23-1 strains (with spindle) (Figure 5B) and spindles form somewhat-prematurely in cdh1Δ cells (Figure 4D) are consistent with this notion. Thus, our results strongly suggest that dephosphorylation of Cdc28 at Y19 is important for restraining APCCdh1 to allow stabilization of microtubule-associated proteins and hence spindle biogenesis.
This notion is consistent with the correlation between Cdc28 dephosphorylation at Tyr 19, phosphorylation of Cdh1 and stabilization of microtubule-associated proteins during progression through normal cell cycle (Figure 7). If this is true then how do yeast cells, arrested in early S phase by hydroxyurea (HU) treatment, assemble a short spindle when Cdc28 is phosphorylated on Tyr 19? It has been suggested previously that only a proportion of Cdc28 may be phosphorylated on Tyr 19 in HU-arrested cells (Krishnan and Surana, 2005); hence, the unphosphorylated fraction of Cdc28 is perhaps responsible for increased stability of microtubule-associated proteins and, in turn, spindle formation. This idea is consistent with the observation that HU-treated cdc28Y19E cells (mimicking fully phosphorylated Cdc28 at Tyr 19) are unable to assemble a spindle (Lim et al, 1996).
Cdh1 has been implicated in targeting both Clb2 and Cin8 for proteolysis (Schwab et al, 1997; Hildebrandt and Hoyt, 2001). Yet, cdc28Y19E cells possess substantial amounts of Clb2 (Lim et al, 1996) but not of Cin8, Ase1 and Kip1 (Figure 1). One possibility is that in these cells Cdh1 is inactivated only partially by Cdc28Y19E form of Clb kinase such that it cannot target Clb2 for degradation, but continues to cause destruction of microtubule-associated proteins such as Cin8. This is reflected in a relatively low abundance of phosphorylated forms of Cdh1 in cdc28Y19E and high abundance in cdc28-1N and cdc28Y19F cells (with spindles) (Figure 5). As Cdh1 contains 11 potential Cdk1 phosphorylation sites, these results raise the possibility that while phosphorylation of some sites inhibit Cdh1 from targeting Clb2, phosphorylation of some other sites may prevent Cdh1 specifically from targeting microtubule-associated proteins. Our attempts to mutate the phosphorylation sites in various combinations have not revealed such target-specific phosphorylation sites (Figure 6). Hence, it is possible that this specificity is quantitative in nature such that ‘partial phosphorylation' of Cdh1 prevents it from targeting Clb2 while ‘complete phosphorylation' inhibits it from targeting both Clb2 and microtubule-associated proteins. While this issue awaits resolution, our results lead to two main conclusions: (1) dephosphorylation of Cdc28 at Tyr19 is essential for stabilization of Cin8, Kip1 and Ase1; (2) that Cin8 overexpression leads to spindle formation in clb3Δ clb4Δ clb5Δ cells even though they are almost completely devoid of Cdc28-Clb kinase activity implies that stabilization of microtubule-associated proteins via Cdh1 inactivation may be the predominant role of Cdc28-Clb kinase in SPB separation.
It is clear that Cdk1's predominant role in SPB separation is to stabilize microtubule-binding proteins such as Cin8, Kip1 and Ase1. Once stabilized, how do these proteins accomplish the task of breaking the bridge that links the duplicated SPBs? In mammalian cells, microtubule-dependent motors are required for centrosome separation (Blangy et al, 1995; Lane and Nigg, 1996). However, it is not known how these motors induce centrosome separation. We find that SPB separation can be induced by a mutant version of Cin8 that lacks motor activity but not by the one deficient in microtubule-binding or -bundling activity (Figure 3C). SPB separation is accompanied by the conversion of interdigitating microtubules emanating from ‘side-by-side' SPB into a parallel array, forming a short spindle. It is possible that bundling activity of proteins such as Cin8 and Ase1 generates mechanical force of sufficient strength that catalyzes this conversion and, in the process, breaks the inter-SPB bridge. However, involvement of processes other than mechanical ones may also be important. While our results suggest that bundling activity is sufficient for SPB separation, compensation by other microtubule-associated motors cannot be unequivocally discounted.
Our findings are consistent with a regulatory scheme for SPB separation (Figure 7B) in which Cdc28-Clb kinase mediates stabilization of microtubule-associated proteins (Cin8, Kip1 and Ase1) by inactivating Cdh1 via phosphorylation. While the mitotically inactive form of Cdc28 (phosphorylated on Tyr19) can inactivate Cdh1 resulting in Clb2 stability, only the mitotically active form of Cdc28 (with dephosphorylated Tyr19) is able to prevent Cdh1-mediated degradation of microtubule-associated proteins. Hence, one of the essential roles of Tyr15/19 dephosphorylation (the evolutionarily conserved way of activating mitotic kinase Cdc2/Cdc28) is to protect microtubule-binding proteins from Cdh1-mediated proteolysis during S phase, allowing SPB separation and spindle formation. Interestingly, in human cells, constitutive activation of APCCdh1 also results in failure to separate centrosomes (Sørensen et al, 2000). We suspect that requirement of Cdc2/Cdc28 activation by Tyr15/Tyr19 dephosphorylation to prevent Cdh1-mediated proteolysis of microtubule-associated proteins and centrosome separation is not restricted only to budding yeast but may be more widespread among other eukaryotes.
Materials and methods
Yeast media and reagents
Strains used in this study are congenic to the WT strain W303. Cells were grown in yeast extract peptone (YEP) supplemented with 2% glucose or 4% raffinose+2% galactose. Methionine was added (at a final concentration of 24 μg/ml) in experiments requiring repression of the MET3 promoter. cdc28-as1 cells were treated with 500 nM of inhibitor 1NM-PP1 (Cellular Genomics Inc) to obtain G2/M arrest (Bishop et al, 2000). Cells treated with proteasome inhibitor MG132 carried an erg6 disruption to facilitate permeability of the inhibitor.
Strains and plasmids
Standard molecular genetic techniques such as gene transplacement, gene disruption, tetrad dissection, PCR-based site-directed mutagenesis and tagging of endogenous genes were used to construct plasmids and strains with various genotypes (Supplementary Table 1). Southern blot analysis or PCR-based genotyping was used to confirm gene disruptions and transplacements.
To construct nondegradable versions of Cin8, Kip1 and Ase1, the following substitution mutations were introduced: amino acids 932–938 (KENATKD to AAAATKA) and amino acids 994–1000 (RKMLKIE to RNLSKIE) in Cin8, amino acids R3A, L6A, R438A, L441A, R647A, L650A, R755A, F758A, K1083A, R1084A, R1085A in Kip1 and amino acids 760–768 (RQLFIPLN to AQLAPIPLA) in Ase1. The modified versions under their respective native promoter were cloned on CEN vectors.
Cell synchronization, Western and Northern blotting, immuno-precipitation and kinase assay
For synchronization at G1, exponential phase cells were grown in medium at 24°C containing either 1 μg/μl α factor for bar1Δ cells or 5 μg/μl for BAR1 cells. Cells were filtered, washed and resuspended in fresh media prewarmed at the appropriate temperature.
Preparation of whole-cell extracts for Western blot analysis was performed as described (Yeong et al, 2001). Immunodetection of Cdc28, Clb2 and α-tubulin was carried out using anti-Cdc28 polyclonal antibodies (1:1000 dilution), anti-Clb2 polyclonal antibodies (1:1000 dilution) and anti α-tubulin monoclonal antibodies (1:2500 dilution), respectively. Anti-mouse HA (1:1000) and anti-rabbit-MYC (1:1000) antibodies were used to detect HA or myc epitope-tagged fusion proteins (Santa Cruz Biotechnology), respectively. The enhanced chemiluminescence kit from Santa Cruz Biotechnology was used for all Western blot analyses according to the manufacturer's instructions.
CIN8, KIP1 and ASE1 polyA mRNA was isolated using the Oligotex mRNA kit (Qiagen) and detected with the Riboprobe in vitro Transcription Kit (Promega) according to the manufacturer's instructions.
Precleared cell lysates in RIPA buffer with inhibitors were treated for immunoprecipitation according to Surana et al (1991). Sepharose beads conjugated to monoclonal goat anti-HA antibody or rabbit anti-myc antibody (Santa Cruz Biotechnology) were used for co-immunoprecipitation. Detection of Cdc28 tyrosine phosphorylation was carried out according to Lim et al (1996). Histone H1 kinase assay was performed as described by Lim et al (1996).
Detection of ubiquitin conjugates in vivo
G1-synchronized cim5-1 cells were released into restrictive growth conditions. Cells were harvested and lysed in RIPA buffer (as above) containing 10 mM N-ethylmaleimide to inhibit deubiquitylation. Cin8-HA3 or Clb2-HA3 was immunoprecipitated using HA-antibody conjugated Sepharose beads from 2 mg of precleared protein extracts, separated on a 7.5% gel and analyzed with anti-ubiquitin antibody (1:100; Sigma) to detect ubiquitin conjugates. The membrane was autoclaved at 121°C for 10 min after Western transfer to expose buried polyubiquitin epitopes. Whole-cell lysate (25 μg) was used to detect HA-tagged proteins.
BRET2 assay
G1-synchronized cells were released into raffinose medium containing 1NM-PP1. After 80 min, galactose was added to induce expression of ubiquitin under the GAL1 promoter. 105 cells were harvested at 10-min intervals starting from 100 min, washed and resuspended in 1 ml BRET Buffer (Perkin Elmer) and transferred to a cuvette. DeepBlueC coelenterazine was added (Final concentration 20 μM) and the cuvette immediately placed in a Fluorolog spectrofluorometer (SPEX). Emission spectra were collected between wavelengths 360 and 580 nm and recorded at 395 nm (Rluc optimum) and 510 nm (GFP2 optimum). The BRET2 ratio was calculated as follows: (sample 510 nm/sample395 nm)−(Cin8-Rluc alone 510 nm/Cin8-Rluc alone395 nm). All assays were performed in triplicate.
Flow cytometry, immunofluorescence and electron microscopy (EM)
Analysis of DNA content by flow cytometry, and staining and visualization of spindles (α-tubulin) by immunofluorescence was carried out according to Yeong et al (2001). Visualization of fluorescent-tagged cells was carried out according to Lim et al (2003). Exposure time for SPC42-GFP and SPC29-CFP was 400 ms, and that for Cdh1-GFP was 800 ms.
Cells were prepared for EM as described by Lim et al (1996). Serial thin sections were viewed on a Philips CM10 electron microscope, and images were captured with Gatan digital camera and viewed with Digital Micrograph software.
Supplementary Material
Supplementary Figure S1
Supplementary Table 1
Acknowledgments
We thank Drs David Morgan, David Pellman, Matthias Peter, Wolfgang Zachariae and Mark Hall for providing various strains and constructs, and Michel Bouvier for BRET reagents. We thank Vaidehi Krishnan and Hong Hwa Lim for their comments on the manuscript. This work was supported by the Biomedical Research Council of Singapore.
References
- Adams IR, Kilmartin JV (1999) Localization of core spindle pole body (SPB) components during SPB duplication in Saccharomyces cerevisiae. J Cell Biol 145: 809–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shinizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM (2000) A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407: 395–401 [DOI] [PubMed] [Google Scholar]
- Blangy A, Lane HA, d'Herin P, Harper M, Kress M, Nigg EA (1995) Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 83: 1159–1169 [DOI] [PubMed] [Google Scholar]
- Bullitt E, Rout MP, Kilmartin JV, Akey CW (1997) The yeast spindle pole body is assembled around a central crystal of Spc42p. Cell 89: 1077–1086 [DOI] [PubMed] [Google Scholar]
- Byers B, Goetsch L (1975) Behavior of spindles and spindle plaques in the cell cycle and conjugation of Saccharomyces cerevisiae. J Bacteriol 124: 511–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro A, Bernis C, Vigneron S, Labbe JC, Lorca Y (2005) The anaphase-promoting complex: a key factor in the regulation of cell cycle. Oncogene 24: 314–325 [DOI] [PubMed] [Google Scholar]
- Fitch I, Dahmann C, Surana U, Amon A, Nasmyth K, Goetsch L, Byers B, Futcher B (1992) Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol Biol Cell 3: 805–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheber L, Kuo SC, Hoyt MA (1999) Motile properties of the kinesin-related Cin8p spindle motor extracted from Saccharomyces cerevisiae cells. J Biol Chem 274: 9564–9572 [DOI] [PubMed] [Google Scholar]
- Giet R, Uzbekov R, Cubizolles F, Le Guellec K, Prigent C (1999) The Xenopus laevis aurora-related protein kinase pEg2 associates with and phosphorylates the kinesin-related protein XlEg5. J Biol Chem 274: 15005–15013 [DOI] [PubMed] [Google Scholar]
- Glover DM, Leibowitz MH, McLean DA, Parry H (1995) Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell 81: 95–105 [DOI] [PubMed] [Google Scholar]
- Gordon DM, Roof DM (2001) Degradation of the kinesin Kip1p at anaphase onset is mediated by the anaphase-promoting complex and Cdc20p. Proc Natl Acad Sci USA 98: 12515–12520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt ER, Hoyt MA (2001) Cell cycle-dependent degradation of the Saccharomyces cerevisiae spindle motor Cin8p requires APC(Cdh1) and a bipartite destruction sequence. Mol Biol Cell 12: 3402–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, He L, Loo KK, Saunders WS (1992) Two Saccharomyces cerevisiae kinesin-related gene products required for mitotic spindle assembly. J Cell Biol 118: 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JN, Park I, Ellingson E, Littlepage LE, Pellman D (2001) Activity of the APCCdh1 form of the anaphase-promoting complex persists until S-phase and prevents the premature expression of Cdc20p. J Cell Biol 154: 85–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaquenoud M, van Drogen F, Peter M (2002) Cell cycle-dependent nuclear export of Cdh1p may contribute to the inactivation of APC/C(Cdh1). EMBO J 21: 6515–6526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Morgan DO (1999) Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol 9: 227–236 [DOI] [PubMed] [Google Scholar]
- Jaspersen SL, Winey M (2004) The budding yeast spindle pole body: structure, duplication, and function. Annu Rev Cell Dev Biol 20: 1–28 [DOI] [PubMed] [Google Scholar]
- Juang YL, Huang J, Peters JM, McLaughlin ME, Tai CY, Pellman D (1997) APC-mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science 275: 1311–1314 [DOI] [PubMed] [Google Scholar]
- Krishnan V, Surana U (2005) Taming the spindle for containing the chromosomes. Cell Cycle 4: 376–379 [DOI] [PubMed] [Google Scholar]
- Lane HA, Nigg EA (1996) Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol 135: 1701–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HH, Goh PY, Surana U (1996) Spindle pole body separation in Saccharomyces cerevisiae requires dephosphorylation of the tyrosine 19 residue of Cdc28. Mol Cell Biol 16: 6385–6397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HH, Yeong FM, Surana U (2003) Inactivation of mitotic kinase triggers translocation of MEN components to mother–daughter neck in budding yeast. Mol Biol Cell 14: 4734–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole ET, Winey M, McIntosh JR (1999) High voltage electron tomography of spindle pole bodies and early mitotic spindles in the yeast. Mol Biol Cell 10: 2017–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perroy J, Ponier S, Charest PG, Aubry M, Bouvier M (2004) Real-time monitoring of ubiquitination in living cells by BRET. Nat Methods 1: 203–208 [DOI] [PubMed] [Google Scholar]
- Roof DM, Meluh PB, Rose MD (1992) Kinesin-related proteins required for assembly of the mitotic spindle. J Cell Biol 118: 95–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin KE, Mitchison TJ (1995) Mutations in the kinesin-like protein Eg5 disrupting localization to the mitotic spindle. Proc Natl Acad Sci USA 92: 4289–4293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M, Lutum AS, Seufert W (1997) Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 90: 683–693 [DOI] [PubMed] [Google Scholar]
- Schuyler SC, Liu JY, Pellman D (2003) The molecular function of Ase1: evidence for a MAP-dependent midzone-specific spindle matrix. J Cell Biol 160: 517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen CS, Lukas C, Kramer ER, Peter J-M, Bartek J, Lucas J (2000) Nonperiodic activity of the human anaphase promoting complex-Cdh1 ubiquitin ligase results in continuous DNA synthesis uncoupled from mitosis. Mol Cel Biol 20: 7613–7623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkel CE, Glover DM (1988) Polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J Cell Sci 89: 25–38 [DOI] [PubMed] [Google Scholar]
- Surana U, Robitsch H, Price C, Schuster T, Fitch I, Futcher AB, Nasmyth K (1991) The role of Cdc28 and cyclins during mitosis in the budding yeast S. cerevisiae. Cell 65: 145–161 [DOI] [PubMed] [Google Scholar]
- Ubersax JS, Woodbury RL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO (2003) Targets of the cyclin-dependent kinase Cdk1. Nature 425: 859–864 [DOI] [PubMed] [Google Scholar]
- Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A (1998) The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell 2: 709–718 [DOI] [PubMed] [Google Scholar]
- Winey M, Mamay CL, O'Toole ET, Mastronarde DN, Giddings TH, McDonald KL, McIntosh JR (1995) Three-dimensional ultrastructural analysis of the Saccharomyces cerevisiae mitotic spindle. J Cell Biol 129: 1601–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeong FM, Lim HH, Wang Y, Surana U (2001) Early expressed Clb proteins allow accumulation of mitotic cyclin by inactivating proteolytic machinery during S phase. Mol Cell Biol 21: 5071–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae W, Nasmyth K (1999) Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev 13: 2039–2058 [DOI] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, Seufert W (1998) Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science 282: 1721–1724 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Table 1