

Abstract
ADAMs (a disintegrin and metalloproteinase) are sheddases possessing extracellular metalloproteinase/disintegrin/cysteine-rich (MDC) domains. ADAMs uniquely display both proteolytic and adhesive activities on the cell surface, however, most of their physiological targets and adhesion mechanisms remain unclear. Here for the first time, we reveal the ADAMs' MDC architecture and a potential target-binding site by solving crystal structures of VAP1, a snake venom homolog of mammalian ADAMs. The D-domain protrudes from the M-domain opposing the catalytic site and constituting a C-shaped arm with cores of Ca2+ ions. The disintegrin-loop, supposed to interact with integrins, is packed by the C-domain and inaccessible for protein binding. Instead, the hyper-variable region (HVR) in the C-domain, which has a novel fold stabilized by the strictly conserved disulfide bridges, constitutes a potential protein–protein adhesive interface. The HVR is located at the distal end of the arm and faces toward the catalytic site. The C-shaped structure implies interplay between the ADAMs' proteolytic and adhesive domains and suggests a molecular mechanism for ADAMs' target recognition for shedding.
Keywords: ADAM, MDC, protein–protein interaction, shedding, snake venom metalloproteinase
Introduction
ADAMs (a disintegrin and metalloproteinase) or MDC (metalloproteinase/disintegrin/cysteine-rich) proteins comprise an emerging class of mammalian metalloproteinases with potential regulatory roles in cell–cell and cell–matrix adhesion and signalling (Becherer and Blobel, 2003; Seals and Courtneidge, 2003; White, 2003; Blobel, 2005). To date, over 30 ADAMs have been identified in a variety of species from fission yeast to human. Roughly, half of these are believed to function as active metalloproteinases and thus to constitute major membrane-bound sheddase that can proteolytically release cell-surface-protein ectodomains including growth factors and cytokines, their receptors and cell adhesion molecules. For example, ADAM17 (TACE, TNF-α converting enzyme) releases many cell-surface proteins including TNF-α precursor (Black et al, 1997; Moss et al, 1997) and ADAM10 (kuzbanian), which dictates lateral inhibition of Drosophila neurogenesis (Rooke et al, 1996), releases Notch ligand Delta (Qi et al, 1999) and Notch itself (Pan and Rubin, 1997). With regard to cellular interactions, fertilin α and β (ADAM1 and ADAM2, respectively) have been identified as sperm surface molecules essential for fertilization (Primakoff et al, 1987; Blobel et al, 1990, 1992) and meltrin α (ADAM12) is implicated in myogenesis (Yagami-Hiromasa et al, 1995). ADAMs have been associated with numerous diseases including arthritis, Alzheimer's disease, and cancer (Duffy et al, 2003; Moss and Bartsch, 2004). ADAM33 has been genetically linked with asthma (Van Eerdewegh et al, 2002). ADAMs uniquely display both proteolytic and adhesive activities on the cell surface, however, most of their physiological targets and the adhesion mechanisms remain unclear.
Disintegrins are small proteins (40–90 aa) isolated from snake venom typically with an Arg-Gly-Asp (RGD) recognition sequence on an extended loop (disintegrin-loop) that inhibit platelet aggregation via integrin binding (Huang et al, 1987; Calvete et al, 2005). ADAMs are unique among cell surface proteins in possessing a disintegrin (D-) domain and thus it has been suggested that integrins might be common receptors for ADAMs (Blobel et al, 1992; Evans, 2001; White, 2003). However, the RGD sequence in the ADAMs' disintegrin-loop is usually replaced by XXCD and therefore, its adhesive potential has been controversial. Both the ADAMs' D- and cysteine-rich (C-) domains are involved in the protein–protein interactions (Myles et al, 1994; Almeida et al, 1995; Zolkiewska, 1999; Iba et al, 2000; Gaultier et al, 2002; Smith et al, 2002), however, the details of the interactions have remained elusive. This is because high-resolution structures have been available only for isolated domains (Maskos et al, 1998; Orth et al, 2004; Janes et al, 2005) and no structural information has been available for the C-domain of the canonical ADAMs. To clarify the molecular mechanisms of target recognition for shedding by and of cellular adhesion via ADAMs, elucidation of the atomic structure of the ADAMs' MDC domains is indispensable.
To obtain structural data on an ADAM family member, we exploited the fact that hemorrhagic P-III snake venom metalloproteinases (SVMPs) share the ADAMs' MDC architecture (Jia et al, 1996; Evans, 2001; Fox and Serrano, 2005). Most ADAMs possess additionally, EGF-like, transmembrane and cytoplasmic domains and therefore are primarily membrane-associated, whereas SVMPs are secreted. Vascular apoptosis-inducing protein-1 (VAP1) is a disulfide-bridged homodimer P-III SVMP isolated from Crotalus atrox venom (Masuda et al, 1998, 2000). VAP1's stability and intrinsic two-fold symmetry enabled us to solve the crystal structures at 2.5-Å resolution. The structure reveals the residues that are important for stabilizing the MDC architecture are strictly conserved throughout the primary structure among all the known ADAMs. Therefore, the present structure represents the general architecture of ADAMs' MDC domains and provides insights into the molecular mechanism of the ADAMs' target recognition.
Results
Structure determination
VAP1 yielded crystals readily, and initial phases were determined by molecular replacement method using the structure of P-I SVMP, acutolysin-C (1QUA) (Zhu et al, 1999) as a starting model. Although the initial model, with 99 identical residues out of 197, represented less than 50% of the total molecule, two distinct local noncrystallographic two-fold symmetry (NCS) operations (see below) allowed us to completely model the whole molecule. The native structures were determined from the crystals with two distinct space groups, P212121 and P41212, both at 2.5-Å resolution (Table I). Orthorhombic crystals were used for inhibitor soaking and the GM6001 ((3-(N-hydroxycarboxamido)-2-isobut yl-propanoyl-Trp-methylamide))-boun d structure was determined at 3.0-Å resolution (Table I). In either crystal forms, the asymmetric unit contained one dimer molecule. The four monomers in the two crystal forms have almost identical structures, except for slight variations in their domain orientations, terminal residues, surface loops and active-site GM6001-binding region.
Table 1.
Data collection and refinement statistics
| Native (orthorhombic) | Native (tetragonal) | GM6001-bound | |
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P41212 | P212121 |
| Cell dimensions | |||
| a, b, c (Å) | 86.7, 93.3, 137.7 | 93.9,93.9,244.8 | 86.3, 91.4, 136.0 |
| α, β, γ (deg) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50–2.50 (2.59–2.50) | 50–2.50 (2.59–2.50) | 50–2.95 (3.06–2.95) |
| Rmergea | 0.072 (0.369) | 0.084 (0.380) | 0.072 (0.367) |
| I/σI | 14.4 (2.9) | 18.7 (7.1) | 12.6 (4.3) |
| Completeness (%) | 99.4 (98.8) | 99.7 (99.6) | 99.9 (99.4) |
| Redundancy | 3.91 | 12.7 | 4.95 |
| Refinement | |||
| Resolution (Å) | 50–2.50 (2.59–2.50) | 50–2.50 (2.59–2.50) | 50–2.95 (3.06–2.95) |
| No. of reflections | 38874 | 38786 | 23295 |
| Rworkb/Rfreec | 0.212/0.258 | 0.229/0.269 | 0.208/0.264 |
| No. of atoms | |||
| Protein | 6558 | 6513 | 6558 |
| Zn2+ | 2 | 2 | 2 |
| Ca2+ | 4 | 4 | 4 |
| Co3+ | 1 | 1 | |
| N-acetyl glucosamine | 56 | 42 | 56 |
| GM6001 | 56 | ||
| Water | 205 | 165 | 35 |
| B-factors | |||
| Protein | 44.9 | 51.2 | 55.4 |
| Zn2+ | 40.9 | 41.6 | 46.4 |
| Ca2+ | 43.5 | 52.4 | 49.3 |
| Co3+ | 35.5 | 56.8 | |
| N-acetyl glucosamine | 69.8 | 65.1 | 75.8 |
| GM6001 | 78.6 | ||
| Water | 39.8 | 41.5 | 37.0 |
| R.m.s deviations | |||
| Bond lengths (Å) | 0.0052 | 0.0080 | 0.0038 |
| Bond angles (deg) | 1.18 | 1.39 | 0.92 |
| aRmerge=∑hkl∑I∣Ii(hkl)-<I (hlk)>∣ / ∑hkl∑iIi(hkl), where Ii(hkl) is the ith intensity measurement of reflection hkl and 〈I(hlk)〉 is its average. | |||
| bRwork=∑(∣∣Fobs∣−∣Fcalc∣∣/∑∣Fobs∣. | |||
| cRfree=R-value for a randomly selected subset (5%) of the data that were not used for minimization of the crystallographic residual. | |||
| Highest resolution shell is shown in parenthesis. | |||
| For each data set, single crystal was used for measurement. |
MDC architecture
The MDC architecture of VAP1 is shown in Figure 1A and B. The metalloproteinase (M-) domains in the dimer are related by NCS such that their active sites point in opposite directions and an intermolecular disulfide bridge is formed between symmetry-related Cys365 residues (Figure 1A). The M-domain is followed by a disintegrin (D-) domain that is further divided into Ds- and Da-domains (see below). The Ds-domain protrudes from the M-domain close to the Ca2+-binding site I (see below) opposing the catalytic site. The D-domain forms a C-shaped arm, together with the cysteine-rich (C-) domain, with its concave surface toward the M-domain. There are no direct interactions between the arm and the M-domain. Notably, the distal portion of the C-domain comes close to and faces toward the catalytic site in the M-domain. The C-terminus Tyr610 is located proximal to the boundary between the Da- and C-domains (Figure 1A and B). Aside from Cys365, each monomer contains 34 cysteinyl residues, all of which are involved in disulfide bonding, and their spacings are strictly conserved among ADAMs (Figure 2 and Supplementary Figure 1) except within the substrate-binding (between the helices H4 and H5) and the HVR (see below) regions. Figure 2 provides a selected subset of the sequence alignments and the entire alignments of VAP1 and 39 ADAM sequences, including all 23 human ADAMs so far available, can be found as Supplementary Figure 1.
Figure 1.
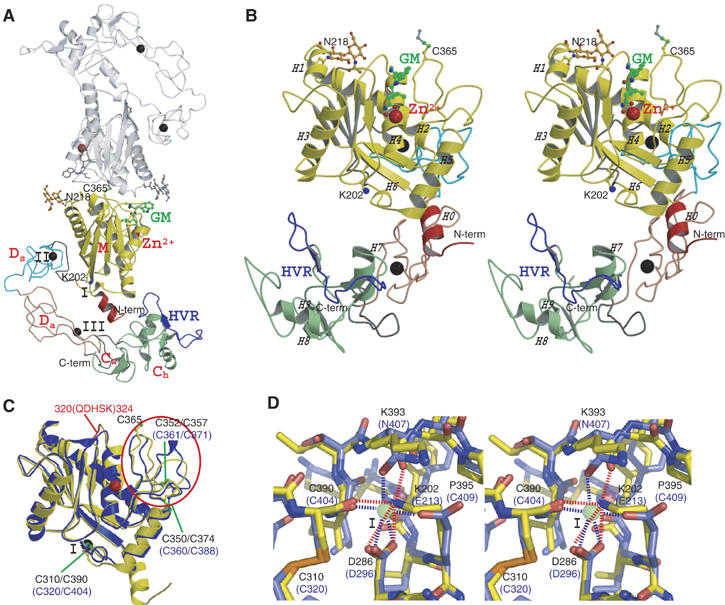
MDC architecture. (A) VAP1 dimer viewed from the NCS axis. The H0-helix, M-domain, linker, Ds-, Da-, Cw-, and Ch-domains and HVRs belonging to the one monomer are shown in red, yellow, gray, cyan, pink, gray, green and blue, respectively. The disulfide-linked counterpart is shown in gray. Zinc and calcium ions are represented as red and black spheres, respectively. The NAG (N-acetyl-glucosamine, in orange) moieties linked to Asn218, the calcium-mimetic Lys202 and the bound inhibitor GM6001 (GM, in green) are in ball-stick representations. (B) Stereo view of VAP1 monomer from the direction nearly perpendicular to (A). The helix numbers are labelled. (C) Superposition of the M-domains of ADAM33 (blue) and VAP1 (yellow). The calcium ion bound to site I and the zinc ion in ADAM33 are represented by black and red spheres, respectively. The disulfide bridges are indicated in black and blue letters for VAP1 and ADAM33, respectively. The QDHSK sequence for the dimer interface in VAP1 (residues 320–324) is in red. (D) Comparison of the calcium-binding site I structures of ADAM33 (blue) and VAP1 (yellow) in stereo. The residues in ADAM33 and in VAP1 are labelled in blue and black, respectively. A calcium ion and a water molecule bound to ADAM33 are represented as green and red spheres, respectively. The ammonium group of Lys202 in VAP1 occupies the position of the calcium ion in ADAM33. In ADAM33 (Orth et al, 2004), side-chain oxygen atoms of Glu213, Asp296 and Asn407, the carbonyl oxygen of Cys404 and a water molecule form the corners of a pentagonal bipyramid and ligand to the calcium ion.
Figure 2.
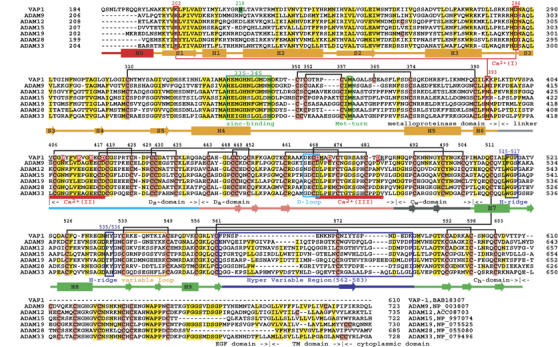
Sequence alignments of VAP1 and human ADAMs. The cysteinyl residues and the conserved residues are shaded in pink and yellow, respectively. Disulfide bridges, secondary structures and domains are drawn schematically. The HVR, calcium-binding site I, catalytic site and disintegrin-loop (D-loop) are boxed in blue, red, green and cyan, respectively. The hydrophobic ridges (H-ridges) are indicated. Calcium-binding sites II and III and the coordinating residues (shaded in red) are indicated. The NCBI accession numbers for the sequences are indicated.
M-domain
Each VAP1 M-domain corresponds to a very similar structure to that of ADAM33 (Orth et al, 2004), with a flat ellipsoidal shape having a central core made up of five stranded β-sheets and five α-helices and a conserved methionine (Met-turn) below the active site histidine residues, which bears the typical structural feature of metzincin family of metalloproteinases (Bode et al, 1993). However, they differ in the dimer interface and the loop structure around the substrate-binding site (Figure 1C) that corresponds to the variable region in the primary structure (between the helices H4 and H5, see Figure 2). The N-terminal helix (H0) is also unique in VAP1. The dimer interface is best characterized by the recognition sequence QDHSK (residues 320–324, see Figure 1C and Supplementary Figure 2A–C) and by Cys365, however these are not conserved among ADAMs; therefore, none of the ADAMs' M-domains are suggested to form a stable dimer as VAP1. A peptide-like hydroxamate inhibitor GM6001 binds to VAP1 (Figure 1A and B, and Supplementary Figure 2D and E) in exactly the same manner as in the marimastat-ADAM33 M-domain complex (Orth et al, 2004), suggesting that the catalytic sites of VAP1 and ADAM33 share a common substrate recognition mechanism. The ADAM33 M-domain structure suggests that most ADAMs have a Ca2+-binding site (designated Ca2+-binding site I) opposing the active-site cleft; however, in VAP1, the distal ammonium group of Lys202 substitutes for the Ca2+ ion (Figure 1D). Replacement of the calcium-coordinating glutamate residue with lysine also occurs in ADAM16, ADAM25 and ADAMs38–40 (Supplementary Figure 1).
C-shaped arm
The D-domain follows the M-domain, with a short linker that allows slightly variable domain orientations at V405 as a pivotal point (Figure 3C). The D-domain is further divided into two structural subdomains (Figure 3), the ‘shoulder' (Ds-domain, residues 396–440) and the ‘arm' (Da-domain, residues 441–487). The Ds- and Da-domains constitute a continuous C-shaped arm, together with the following N-terminus region of the C-domain which we designate the ‘wrist' (Cw-domain, residues 488–505). There are three disulfide bonds in the Ds-domain, three in the Da-domain and one in the Cw-domain. The subdomains are connected by single disulfide bridges (Figures 2 and 3A) with slightly variable angles (Figure 3B).
Figure 3.
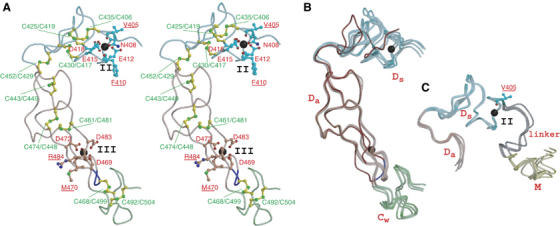
Arm structure. (A) Arm structure in stereo. The Ds-, Da-, and Cw-domains are in cyan, pink and light green, respectively. The calcium-coordinating residues and the disulfide bridges are shown in red and green, respectively. The residues with carbonyl oxygen atoms involved in calcium coordination are underlined. Calcium ions are represented as black spheres. The disintegrin-loop (DECD) is in blue. (B) Superimposition of the four Da-domains of VAP1 and trimestatin (1J2L). Trimestatin and its RGD loop are shown in red and blue, respectively. (C) Superimposition of the four Ds-domains. The linker between the M- and Ds-domains is shown in gray. Val405 at the pivotal point is indicated.
Both the Ds- and Da-domains contain structural calcium-binding sites. In the Ds-domain, the side-chain oxygen atoms in residues Asn408, Glu412, Glu415 and Asp418, and the carbonyl oxygen atoms of Val405 and Phe410 are involved in pentagonal bipyramidal coordination and constitute Ca2+-binding site II (Figures 2 and 3A). Notably, these residues are strictly conserved among all known ADAMs (Supplementary Figure 1). However, the side-chain oxygens of Asp469, Asp472 and Asp483, and carbonyl oxygens of Met470 and Arg484 form the corners of a pentagonal bipyramid to the calcium ligand and constitute the Da-domain Ca2+-binding site III (Figures 2 and 3A) and these residues are highly conserved among ADAMs except ADAM10 and ADAM17 (Supplementary Figure 1). Because of the few secondary-structural elements, bound calcium ions and the disulfide bridges are essential for the structural rigidity of ADAM's C-shaped arm. The RGD-containing disintegrin trimestatin (Fujii et al, 2003) has a similar structure with the Da-domain (r.m.s.d of 1.24 Å, Figure 3B); however, no disintegrins have been shown to bind Ca2+ ions.
Using isolated D-domains or portions thereof, numerous ADAMs and P-III SVMPs have been shown to interact specifically with particular integrins (Evans, 2001; White, 2003; Calvete et al, 2005). However, the disintegrin-loop is packed against the Cw-domain and a disulfide bridge (Cys468–Cys499) further stabilizes the continuous structure (Figure 3A). Therefore, the disintegrin-loop is inaccessible for protein binding.
Hand domain
The ‘hand' domain (Ch-domain, residues 505–610) follows the Cw-domain. The Ch-domain, together with the Cw-domain, constitutes a novel fold (Figure 4A). In either crystal form, VAP1 dimers interact with molecules of neighboring units through the Ch-domains such that the molecules form a handshake (Figure 4B). Each Ch-domain interacts with its counterpart through a relatively large complementary surface of 860 Å2 forming another NCS at the center, although VAP1 exists as dimers, not as oligomers, and is mono-dispersed in solution (data not shown).
Figure 4.

C-domain architecture and HVR. (A) The C-domain architecture in stereo. The Cw- and Ch-domains are in gray and light green, respectively. The disulfide bridges and the residues forming the hydrophobic ridges are indicated. The HVR and its NCS counterpart are shown in red and blue, respectively. The variable loop (residues 539–549), flanked by two adjacent cysteine residues, is in green. (B) Crystal packing in the orthorhombic crystal. The crystallographically equivalent molecules (HVRs) are in cyan (blue) and pink (red), respectively. The arrows indicate the directions of the HVR chains. Zinc and calcium ions are represented as red and black spheres, respectively. (C) Interactions between the HVRs (cyan and pink) in stereo. The molecular surface of the cyan molecule is shown with the electrochemical surface potential (red to blue). The residues constituting the hydrophobic ridges are in yellow. The residues are labelled in blue and red for cyan and pink, respectively. (D) Water-mediated hydrogen-bond network in the HVR. The HVR residues are in pink and cyan; non-HVR residues in the pink molecule are in gray.
HVR as a potential adhesive interface
Ch-domain residues 562–583 are predominantly involved in the handshake (Figure 4B). This is the region in which the ADAM sequences are most divergent and variable in length (16–55 aa) (Figure 2 and Supplementary Figure 1). We have designated this as the hyper-variable region (HVR). The HVR is subdivided into two structural elements. The N-terminal portion (residues 562–572) fits into an extended loop, filling the gap between the M-domain and the neighboring molecule's Ch-domain and thus fixing the position of the arm (Figure 4B). The variable structures and less-specific interactions suggest that this loop is stabilized by crystal packing. Some ADAMs possess a putative fusion peptide in this segment typical of viral fusion proteins (Blobel et al, 1992; Yagami-Hiromasa et al, 1995), although their role in the actual fusion process has not been demonstrated. However, the remainder of the HVR (residues 572–583) interacts extensively with its counterpart by forming an antiparallel β strand at the center (Figure 4C and D). Although the ability to form β strand is predictable from the sequence, this β strand is stabilized mainly by interchain interactions (Figure 4D). There are no intrachain hydrogen bonds between residues 574–577 and the remainder of the Ch-domain; however a water-mediated hydrogen-bond network stabilizes this segment (Figure 4D). Therefore, it appears, that this β strand might be formed by the induced-fit mechanism upon the association of the Ch-domains and that the conserved disulfide bond (Cys526–Cys572, see Figure 4D) may stabilize the structure when the HVRs are isolated in solution. In addition to the main-chain hydrogen bonds, side-chain atoms (particularly residues I574, Y575, Y576 and P578) in the HVR β strand contribute numerous von der Waals interactions with their counterparts. Aside from the HVR, aromatic residues located at both sides of the β strand in close proximity to the NCS axis create additional interaction surfaces: residues Phe515, Gly516, His535 and Tyr536 in the loop regions form hydrophobic ridges that fit complementarily into the NCS region (Figure 4C). The hydrophobic ridges are highly conserved among ADAMs (Figure 2 and Supplementary Figure 1), thus, in part, they may also constitute binding surfaces.
Discussion
The VAP1 structures reveal highly conserved structural calcium-binding sites and the numbers and the spacings of cysteinyl residues that are essential for maintaining structural rigidity and spatial arrangement of the ADAMs' MDC domains. The C-shaped MDC architecture implies meaningful interplay between the domains and their potential roles in physiological functions.
The HVR creates a novel interaction interface in collaboration with the conserved hydrophobic ridges. Different ADAMs have distinct HVR sequences, which result in distinct surface features, thus, they may function in specifying binding proteins. The HVR is at the distal end of the C-shaped arm and points toward the M-domain catalytic site, with a distance of ∼4 nm in between them. Collectively, these observations suggest that the HVR captures the target or associated protein that is processed by the catalytic site (Figure 5). The disintegrin portion is located opposit to and apart from the catalytic site and, thus, might play a primary role as a scaffold that allocates these two functional units spatially. The C-shaped structure also implies how the ADAMs' C-domains cooperate with their M-domains (Reddy et al, 2000; Smith et al, 2002). In membrane-bound ADAMs, the EGF-like domain (∼60 aa) follows the Ch-domain (Figure 2) and presumably works as a rigid spacer connecting the MDC-domains with and orientating against the membrane-spanning region (Figure 5A). Many ADAMs are proteolytically inactive (because of the defects in the catalytic HEXXHXXGXXHD sequence or the post-translational removal of the M-domain), and several of these are important developmentally. Therefore, the HVR may also work to modulate cell–cell and cell–matrix interactions. There is some experimental evidence for C-domain-mediated adhesion. Peptides encompassing the HVR and the hydrophobic ridge from P-III SVMPs interfere with platelet interaction and collagen binding (Kamiguti et al, 2003). A recombinant atrolysin-A C-domain specifically binds collagen I and von Willebrand factor (vWF) and blocks collagen–vWF interaction (Jia et al, 2000; Serrano et al, 2005). ADAM12 interacts with cell-surface syndecan through its C-domain and mediates integrin-dependent cell spreading (Iba et al, 2000). The D/C-domain portion of ADAM13 binds to the ECM proteins laminin and fibronectin (Gaultier et al, 2002). However, most of these studies do not assign specific regions of the C-domain to these interactions and the molecular recognition mechanisms are to be elucidated.
Figure 5.
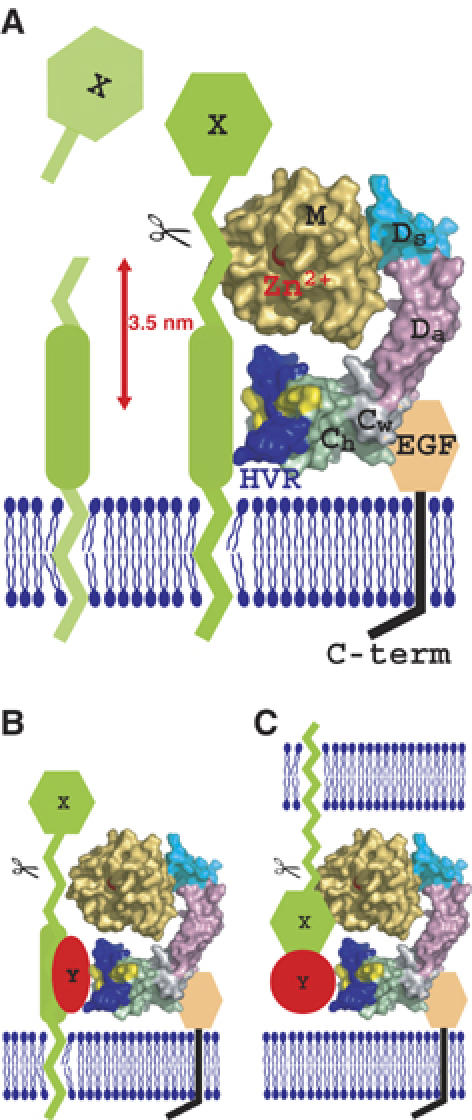
Models for ADAM's shedding. The molecular surface of the VAP1 monomer, without VAP1's unique H0-helix, are colored as in Figure 1A. Hydrophobic ridges are in yellow. EGF-like, transmembrane and cytoplasmic domains are represented schematically. (A) Membrane-anchored substrate molecule ‘X' is directly recognized and captured by the HVR on the membrane-bound ADAM molecule. The distance between the center of the HVR (Tyr575) and the catalytic zinc ion is about 3.5 nm. (B) Substrate ‘X' is recognized by the ADAM HVR via binding with an associated protein ‘Y'. (C) ADAM cleaves substrate ‘X' in trans via binding with an associated protein ‘Y'.
ADAM10 and ADAM17 lack the Ca2+-binding site III and show less sequence similarities in the C-domain with other canonical ADAMs (Supplementary Figure 1). Comparison of the recently solved ADAM10 D/C-domain partial structure (ADAM10D+C) (Janes et al, 2005) and that of VAP1 reveals that the atypical ADAM10 shares the continuous Da/Cw structure and the Ch-domain scaffold with VAP1; however, it has an disordered Ds-domain and an alternate HVR structure and a different orientation between Cw- and Ch-domains (Figure 6). The locations of four of the five disulfide bridges within the Ch-domain are conserved between VAP1 and ADAM10 (Figure 6B and C) and thus, they enabled us to align the two sequences (Figure 6E). Based on this alignment, we completed entire alignments (Supplementary Figure 1) including 38 sequences of mammalian ADAMs and Schizosaccharomyces pombe Mde10 (Nakamura et al, 2004), presumably the founding member of the ADAM family in evolutionary terms. The ADAM10D+C structure lacks the eight residues (583–590 in ADAM10) that may form a flexible loop. However, VAP1 (Figure 6E) and the canonical ADAMs except for ADAM8 (Supplementary Figure 1) have extra 16 residues in this segment that, in part, forms a variable loop, flanked by the adjacent cysteinyl residues (Cys539 and Cys549 in VAP1) and protrudes from the main body of the C-domain (Figures 4A and 6B). The variable loop has highest temperature factor in the molecule and resembles to the disintegrin-loop, thus can be an additional protein-binding interface. The six VAP1 monomer molecules represent almost the same Cw/Ch domain orientation (data not shown), however that is distinct from that of ADAM10 (Figure 5A). Thus, the possibility whether different ADAMs have distinct Cw/Ch domain orientation remains to be established. Janes et al (2005) have shown that the three glutamate residues outside of HVR are essential for ADAM10-mediated ephrin proteolysis in trans, however, roles of the ADAM10 HVR has not been examined. An extensive molecular surface of the elongated arm structure (12 000 Å2 for the VAP1 D/C-domains) might reveal additional protein–protein interaction interfaces other than the HVR. Multiple charged residues in the D-domain are essential for ADAM28 binding to α4β1 (Bridges et al, 2003) and the RX6DLPEF motif has been proposed for integrin α9β1 binding (Eto et al, 2002). However, the D-domain portion of the C-shaped scaffold is away from the catalytic site; thus, those additional sites might not directly serve as target recognition interfaces for catalysis.
Figure 6.

Comparison of the VAP1 and ADAM10 D/C domains. (A) Superimposition of the Da-domains of ADAM10 and VAP1. The Ds/Da/Cw-domains and the H7 helix of VAP1 and those of ADAM10 are shown in blue and red, respectively. The Ch-domains of VAP1 and ADAM10 are shown in cyan and pink, respectively. The arrow indicates the pivotal point between the Cw- and Ch-domains. Bound Ca2+ ions in VAP1 are shown as black spheres. (B) Ribbon representation of the Ch-domain of VAP1. The HVR is shown in blue. The common scaffold between the VAP1 and ADAM10 Ch-domains are shown in cyan and the segment lacking in ADAM10 is shown in light green. Disulfide bridges are indicated. (C) Ribbon representation of the Ch-domain of ADAM10. The HVR is shown in red. Disulfide brides are indicated. (D) Superimposition of the Ch-domains of VAP1 and ADAM10 in stereo with the colors as in (B, C). The N- and C-termini of the Ch-domains are indicated. (E) Structure-based alignments of VAP1, bovine ADAM10 (cADAM10), human ADAM17 (hADAM17) and S. pombe Mde10 (Mde10) Cw/Ch-domains. Secondary structures and the disulfide bridges are represented schematically. The HVR sequences and the missing segment in the ADAM10 structure are boxed in blue and green, respectively.
Uniquely among cell-surface proteins, ADAMs display both proteolytic and adhesive activities. The VAP1 structure reveals that these functions are spatially allocated to the ends of the unique C-shaped scaffold and face each other. This spatial allocation of the functional sites provide us insights into the molecular mechanism of ADAMs' target recognition, which ADAMs shed which key substrates in specific biological events. Since ADAMs are potential therapeutic targets, the distinct surface feature created by the HVR of the individual ADAMs might also provide insights into the future design of drugs with higher specificity for each member of ADAMs. We suggest that the HVR, not the disintegrin domain, should be the focus of searches for physiological targets of ADAMs.
Materials and methods
Protein preparation and crystallization
The details of the preparation, crystallization and preliminary X-ray analysis of VAP1 will be described elsewhere (T Igarashi et al, in preparation). VAP1 was isolated from the crude snake Crotalus atrox venom (Sigma-Aldrich, USA) and subjected to sitting- or hanging-drop vapor diffusion crystallization. Two distinct crystal forms (P212121 and P41212) were obtained with the reservoir solution containing 15% polyethyleneglycol 8000 and 100 mM sodium cacodylate at pH 6.5, with (orthorhombic form) or without (tetragonal form) 20 mM cobaltous chloride hexahydrate. GM6001-bound crystals were prepared by adding GM6001 (CALBIOCHEM) to the drop with the orthorhombic crystal at a final concentration qof 0.33 mM (twice the protein concentration) followed by a 12-h incubation. Crystals were flash-frozen under the nitrogen flow at 90 K.
Diffraction data collection
All the diffraction data were collected at SPring-8 beamlines using either ADSC quantum 310R CCD (for the inhibitor-bound crystal at the beamline BL41XU with λ=1 Å), Rigaku R-axis V imaging plate (for orthorhombic native crystal at the beamline BL45PX with λ=1 Å) or Jupitor CCD (for the tetragonal crystal at the beamline BL45PX with λ=0.98 Å) detectors at 90 K. The images were reduced using HKL2000. Both orthorhombic and tetragonal native data sets were collected to 2.5-Å resolution and inhibitor-bound crystal data sets were collected to 3.0 Å resolution (Table I).
Structural analysis
All structures were solved by the molecular replacement method by MOLREP in the CCP4 suite (CCP4, 1994) by using acutolysin-C (1QUA) (Zhu et al, 1999) as a starting model. Initially, the MR solution obtained from the orthorhombic crystal data set, assumed two M-domains in the asymmetric units. After manual rebuilding by TURBO-FRODO, the model was subjected to tortional molecular dynamic refinements with restrained NCS averaging of the M-domains using CNS (Brunger et al, 1998) and iterative refinements and manual rebuilding of the model improved the electron-density map and enabled us to extend the model. First, we found the electron densities associated with the pieces of helical segments of the molecules and modelled them as poly-alanine chains. After cycles of refinements, we assigned those segments as the parts of helices H7 and H8, where the secondary structures are predicted to be helices, judging from the electron densities associated with the side chains. At this stage, four tyrosine residues, Tyr575 and Try576 within the central β strands of the HVRs were clearly defined, and we noticed that there was another NCS-axis between the C-domains. After iterative rounds of refinements with restrained NCS averaging of the C-domains and manual model building, we completed modelling of the C-domains. From this stage onward, no NCS averaging was included in the refinements. Next, we modelled the D-domains with the help of automated chain tracing using the program ARP/wARP (Perrakis et al, 1999) and with the structural model of trimestatin (1J2L) as a guide. After completely modelling the polypeptide chains, we noticed that isolated lobes of high electron densities surrounded by oxygen atoms occurred both in the Ds- and Da-domains. For these sites, calcium ions fit optimally to the electron density with a refined occupancy of 100% and reasonably low B-values, thus, we included calcium ions in the model. We also assigned a cobalt ion, which was supplemented in the crystallization buffer for the orthorhombic crystal form, located between the M- and Ds-domains in the A molecule. The part of the carbohydrate chain linked to residue Asn218 (two N-acetyl-glocosamine (NAG) moieties) was modelled. Then, water molecules were assigned. The VAP1 cDNA encodes a protein with 610 amino-acid residues; however, the N-terminus is processed by post-translational modification (Masuda et al, 1998, 2000). Here, protein sequencing of the de-blocked VAP1 molecule clarified that the Glu184 side chain was modified into a pyro-form. The electron densities associated with almost the entire molecule except for the first pyroglutamic acid were defined in either monomer within the orthorhombic crystal. In the final model, 86.1% of the residues lay in the most favorable region, 13.3% in the additionally allowed region and 0.7% in the generously allowed region of the Ramachandran plot. The tetragonal crystal and inhibitor-bound crystal were solved by MR with the domains of the refined orthorhombic apo-form as a starting model. In the final model, 83.6% (80.6%) of the residues lay in the most favorable region, 15.7% (18.9%) in the additionally allowed region and 0.7% (0.5%) in the generously allowed region for tetragonal (inhibitor-bound) crystals in the Ramachandran plot. In either crystal form, the asymmetric unit contained one dimer molecule. All six monomers had almost identical structures. Refinement statistics are shown in Table I.
PDB accession codes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 2ERO, 2ERP and 2ERQ for the orthorhombic native, GM6001-bound form and tetragonal-form, respectively.
Supplementary Material
Supplementary Figures
Acknowledgments
We thank Yuko Oishi and staff in SPring-8 beamlines for assistance with data acquisition and Junichi Takagi for discussions and critical reading of the manuscript. This work was partly supported by Grant nano-001 for Research on Advanced Medical Technology from the Ministry of Health, Labor, and Welfare of Japan, and by grants from the Takeda Science Foundation, from the Kao Foundation for Arts and Science and from Senri Life Science Foundation. The authors declare no competing financial interests.
References
- Almeida EA, Huovila AP, Sutherland AE, Stephens LE, Calarco PG, Shaw LM, Mercurio AM, Sonnenberg A, Primakoff P, Myles DG, White JM (1995) Mouse egg integrin alpha 6 beta 1 functions as a sperm receptor. Cell 81: 1095–1104 [DOI] [PubMed] [Google Scholar]
- Becherer JD, Blobel CP (2003) Biochemical properties and functions of membrane-anchored metalloprotease-disintegrin proteins (ADAMs). Curr Top Dev Biol 54: 101–123 [DOI] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP (1997) A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385: 729–733 [DOI] [PubMed] [Google Scholar]
- Blobel CP (2005) ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol 6: 32–43 [DOI] [PubMed] [Google Scholar]
- Blobel CP, Myles DG, Primakoff P, White JM (1990) Proteolytic processing of a protein involved in sperm-egg fusion correlates with acquisition of fertilization competence. J Cell Biol 111: 69–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel CP, Wolfsberg TG, Turck CW, Myles DG, Primakoff P, White JM (1992) A potential fusion peptide and an integrin ligand domain in a protein active in sperm-egg fusion. Nature 356: 248–252 [DOI] [PubMed] [Google Scholar]
- Bode W, Gomis-Ruth FX, Stockler W (1993) Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the ‘metzincins'. FEBS Lett 331: 134–140 [DOI] [PubMed] [Google Scholar]
- Bridges LC, Hanson KR, Tani PH, Mather T, Bowditch RD (2003) Integrin alpha4beta1-dependent adhesion to ADAM 28 (MDC-L) requires an extended surface of the disintegrin domain. Biochemistry 42: 3734–3741 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54 (Part 5): 905–921 [DOI] [PubMed] [Google Scholar]
- Calvete JJ, Marcinkiewicz C, Monleon D, Esteve V, Celda B, Juarez P, Sanz L (2005) Snake venom disintegrins: evolution of structure and function. Toxicon 45: 1063–1074 [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Duffy MJ, Lynn DJ, Lloyd AT, O'Shea CM (2003) The ADAMs family of proteins: from basic studies to potential clinical applications. Thromb Haemost 89: 622–631 [PubMed] [Google Scholar]
- Eto K, Huet C, Tarui T, Kupriyanov S, Liu HZ, Puzon-McLaughlin W, Zhang XP, Sheppard D, Engvall E, Takada Y (2002) Functional classification of ADAMs based on a conserved motif for binding to integrin alpha 9beta 1: implications for sperm-egg binding and other cell interactions. J Biol Chem 277: 17804–17810 [DOI] [PubMed] [Google Scholar]
- Evans JP (2001) Fertilin beta and other ADAMs as integrin ligands: insights into cell adhesion and fertilization. BioEssays 23: 628–639 [DOI] [PubMed] [Google Scholar]
- Fox JW, Serrano SM (2005) Structural considerations of the snake venom metalloproteinases, key members of the M12 reprolysin family of metalloproteinases. Toxicon 45: 969–985 [DOI] [PubMed] [Google Scholar]
- Fujii Y, Okuda D, Fujimoto Z, Horii K, Morita T, Mizuno H (2003) Crystal structure of trimestatin, a disintegrin containing a cell adhesion recognition motif RGD. J Mol Biol 332: 1115–1122 [DOI] [PubMed] [Google Scholar]
- Gaultier A, Cousin H, Darribere T, Alfandari D (2002) ADAM13 disintegrin and cysteine-rich domains bind to the second heparin-binding domain of fibronectin. J Biol Chem 277: 23336–23344 [DOI] [PubMed] [Google Scholar]
- Huang TF, Holt JC, Lukasiewicz H, Niewiarowski S (1987) Trigramin. A low molecular weight peptide inhibiting fibrinogen interaction with platelet receptors expressed on glycoprotein Iib–IIIa complex. J Biol Chem 262: 16157–16163 [PubMed] [Google Scholar]
- Iba K, Albrechtsen R, Gilpin B, Frohlich C, Loechel F, Zolkiewska A, Ishiguro K, Kojima T, Liu W, Langford JK, Sanderson RD, Brakebusch C, Fassler R, Wewer UM (2000) The cysteine-rich domain of human ADAM 12 supports cell adhesion through syndecans and triggers signaling events that lead to beta1 integrin-dependent cell spreading. J Cell Biol 149: 1143–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB (2005) Adam meets Eph: an ADAM substrate recognition module acts as a molecular SWITCH for Ephrin cleavage in trans. Cell 123: 291–304 [DOI] [PubMed] [Google Scholar]
- Jia LG, Shimokawa K, Bjarnason JB, Fox JW (1996) Snake venom metalloproteinases: structure, function and relationship to the ADAMs family of proteins. Toxicon 34: 1269–1276 [DOI] [PubMed] [Google Scholar]
- Jia LG, Wang XM, Shannon JD, Bjarnason JB, Fox JW (2000) Inhibition of platelet aggregation by the recombinant cysteine-rich domain of the hemorrhagic snake venom metalloproteinase, atrolysin A. Arch Biochem Biophys 373: 281–286 [DOI] [PubMed] [Google Scholar]
- Kamiguti AS, Gallagher P, Marcinkiewicz C, Theakston RD, Zuzel M, Fox JW (2003) Identification of sites in the cysteine-rich domain of the class P-III snake venom metalloproteinases responsible for inhibition of platelet function. FEBS Lett 549: 129–134 [DOI] [PubMed] [Google Scholar]
- Maskos K, Fernandez-Catalan C, Huber R, Bourenkov GP, Bartunik H, Ellestad GA, Reddy P, Wolfson MF, Rauch CT, Castner BJ, Davis R, Clarke HR, Petersen M, Fitzner JN, Cerretti DP, March CJ, Paxton RJ, Black RA, Bode W (1998) Crystal structure of the catalytic domain of human tumor necrosis factor-alpha-converting enzyme. Proc Natl Acad Sci USA 95: 3408–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda S, Hayashi H, Araki S (1998) Two vascular apoptosis-inducing proteins from snake venom are members of the metalloprotease/disintegrin family. Eur J Biochem 253: 36–41 [DOI] [PubMed] [Google Scholar]
- Masuda S, Ohta T, Kaji K, Fox JW, Hayashi H, Araki S (2000) cDNA cloning and characterization of vascular apoptosis-inducing protein 1. Biochem Biophys Res Commun 278: 197–204 [DOI] [PubMed] [Google Scholar]
- Moss ML, Bartsch JW (2004) Therapeutic benefits from targeting of ADAM family members. Biochemistry 43: 7227–7235 [DOI] [PubMed] [Google Scholar]
- Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Warner J, Willard D, Becherer JD (1997) Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 385: 733–736 [DOI] [PubMed] [Google Scholar]
- Myles DG, Kimmel LH, Blobel CP, White JM, Primakoff P (1994) Identification of a binding site in the disintegrin domain of fertilin required for sperm-egg fusion. Proc Natl Acad Sci USA 91: 4195–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Abe H, Hirata A, Shimoda C (2004) ADAM family protein Mde10 is essential for development of spore envelopes in the fission yeast Schizosaccharomyces pombe. Eukaryot Cell 3: 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth P, Reichert P, Wang W, Prosise WW, Yarosh-Tomaine T, Hammond G, Ingram RN, Xiao L, Mirza UA, Zou J, Strickland C, Taremi SS, Le HV, Madison V (2004) Crystal structure of the catalytic domain of human ADAM33. J Mol Biol 335: 129–137 [DOI] [PubMed] [Google Scholar]
- Pan D, Rubin GM (1997) Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell 90: 271–280 [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Primakoff P, Hyatt H, Tredick-Kline J (1987) Identification and purification of a sperm surface protein with a potential role in sperm-egg membrane fusion. J Cell Biol 104: 141–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Rand MD, Wu X, Sestan N, Wang W, Rakic P, Xu T, Artavanis-Tsakonas S (1999) Processing of the notch ligand delta by the metalloprotease Kuzbanian. Science 283: 91–94 [DOI] [PubMed] [Google Scholar]
- Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA (2000) Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J Biol Chem 275: 14608–14614 [DOI] [PubMed] [Google Scholar]
- Rooke J, Pan D, Xu T, Rubin GM (1996) KUZ, a conserved metalloprotease-disintegrin protein with two roles in Drosophila neurogenesis. Science 273: 1227–1231 [DOI] [PubMed] [Google Scholar]
- Seals DF, Courtneidge SA (2003) The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev 17: 7–30 [DOI] [PubMed] [Google Scholar]
- Serrano SM, Jia LG, Wang D, Shannon JD, Fox JW (2005) Function of the cysteine-rich domain of the haemorrhagic metalloproteinase atrolysin A: targeting adhesion proteins collagen I and von Willebrand factor. Biochem J 391: 69–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KM, Gaultier A, Cousin H, Alfandari D, White JM, DeSimone DW (2002) The cysteine-rich domain regulates ADAM protease function in vivo. J Cell Biol 159: 893–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, Torrey D, Pandit S, McKenny J, Braunschweiger K, Walsh A, Liu Z, Hayward B, Folz C, Manning SP, Bawa A, Saracino L, Thackston M, Benchekroun Y, Capparell N, Wang M, Adair R, Feng Y, Dubois J, FitzGerald MG, Huang H, Gibson R, Allen KM, Pedan A, Danzig MR, Umland SP, Egan RW, Cuss FM, Rorke S, Clough JB, Holloway JW, Holgate ST, Keith TP (2002) Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature 418: 426–430 [DOI] [PubMed] [Google Scholar]
- White JM (2003) ADAMs: modulators of cell–cell and cell–matrix interactions. Curr Opin Cell Biol 15: 598–606 [DOI] [PubMed] [Google Scholar]
- Yagami-Hiromasa T, Sato T, Kurisaki T, Kamijo K, Nabeshima Y, Fujisawa-Sehara A (1995) A metalloprotease-disintegrin participating in myoblast fusion. Nature 377: 652–656 [DOI] [PubMed] [Google Scholar]
- Zhu X, Teng M, Niu L (1999) Structure of acutolysin-C, a haemorrhagic toxin from the venom of Agkistrodon acutus, providing further evidence for the mechanism of the pH-dependent proteolytic reaction of zinc metalloproteinases. Acta Crystallogr D 55: 1834–1841 [DOI] [PubMed] [Google Scholar]
- Zolkiewska A (1999) Disintegrin-like/cysteine-rich region of ADAM 12 is an active cell adhesion domain. Exp Cell Res 252: 423–431 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures