

Abstract
Steroid receptor coactivator 3 (SRC-3/ACTR/AIB-1/pCIP/RAC3/TRAM-1) is a member of the p160 family of nuclear receptor coactivators that plays an important role in mammary gland growth, development, and tumorigenesis. We show that deletion of SRC-3 gene decreases platelet and increases lymphocytes numbers, leading to the development of malignant B-cell lymphomas upon aging. The expansion of the lymphoid lineage in SRC-3−/− mice is cell autonomous, correlates with an induction of proliferative and antiapoptotic genes secondary to constitutive NF-κB activation, and can be reversed by restoration of SRC-3 expression. NF-κB activation is explained by the degradation of IκB, consequent to increases in free IκB kinase, which is no longer inhibited by SRC-3. These results demonstrate that SRC-3 regulates lymphopoiesis and in combination with previous studies indicate that SRC-3 has vastly diverging effects on cell proliferation depending on the cellular context, ranging from proliferative and tumorigenic (breast) to antiproliferative (lymphoid cells) effects.
Keywords: cofactors, hematopoiesis, lymphoma, NF-κB, transcription
Introduction
The steroid receptor coactivators (SRCs), also known as p160 proteins, were among the first cofactors identified that interact with nuclear receptors (NRs) to enhance their transactivation in a ligand-dependent manner (McKenna et al, 1999). Members of the SRC family, which include SRC-1, SRC-2/GRIP1/TIF-2, and SRC-3/ACTR/AIB-1/pCIP/RAC3/TRAM-1 (Onate et al, 1995; Voegel et al, 1996; Anzick et al, 1997; Li et al, 1997; Takeshita et al, 1997; Torchia et al, 1997), are all NR coactivators. SRC-1 and SRC-3 contain an intrinsic acetyltransferase activity and all p160 proteins can recruit histone acetyltranferases (Torchia et al, 1997) and histone methyltransferases (Chen et al, 1997), explaining their capacity to coactivate and interact with general transcription factors, such as TFIIB and TBP (Tsai and O'Malley, 1994; Takeshita et al, 1996). Although the three members of the SRC family have a great degree of overall sequence similarity and enhance the activity of many NRs in a similar manner, these proteins have distinct functions. The disruption of the SRC-1 gene in mice causes partial resistance to multiple hormones, including estrogen, progesterone, androgen, and thyroid hormone. The resistance results in a decreased response of certain target organs to hormone stimulation (Xu et al, 1998; Weiss et al, 1999). Inactivation of the TIF-2 gene revealed that it plays a critical role in mouse reproductive functions (Gehin et al, 2002; Mark et al, 2004). The relative levels of SRC-1 and TIF-2 were furthermore shown to influence energy homeostasis, since, SRC-1, but not TIF-2, was shown to enhance the thermogenic activity of the peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) (Picard et al, 2002). Ablation of the SRC-3 gene in mice results in growth retardation and SRC-3−/− mice are hence small in size, exhibit delayed puberty, reduced female reproductive function, and reduced mammary gland development (Wang et al, 2000; Xu et al, 2000; Wu et al, 2002). SRC-3 is suggested to be more promiscuous relative to other p160 proteins and to enhance the transcription of non-NR proteins including CREB (Torchia et al, 1997) and NF-κB (Wu et al, 2002, 2004).
SRC-3 activity has been linked with proliferative processes and the SRC-3 gene is amplified and strongly expressed in primary breast tumor cells (Anzick et al, 1997). SRC-3 is often overexpressed and sometimes amplified in other cancers, including those of the ovary (Anzick et al, 1997), endometrium (Glaeser et al, 2001), stomach (Sakakura et al, 2000), liver (Wang et al, 2002), and prostate (Gnanapragasam et al, 2001). The overexpression of SRC-3 in transgenic mice furthermore leads to an increased tumor incidence and activation of the PI3K/AKT pathway, defining SRC-3 as an oncogene (Torres-Arzayus et al, 2004). This adds SRC-3 to the list of individual components of the epigenetic machinery, including DNA methyltransferases, histone methyltransferases, and histone acetylase/deacetylases, which predispose to cancer development (Jones and Baylin, 2002; Lund and van Lohuizen, 2004).
In this study, the role and the function of SRC-3 in hematopoiesis was investigated using SRC-3−/− mice. SRC-3−/− mice have a hematological deficit, characterized by a decrease in platelets and a striking increase in lymphocyte numbers often resulting in lymphoma formation. The expansion of T and B cells in SRC-3−/− mice was at least in part cell autonomous, and correlated with an induction of both proliferative and antiapoptotic genes, secondary to constitutive NF-κB activation. These data, which are in apparent contradiction with previous results on the proliferative role of SRC-3 in epithelial cancers, show that the absence of SRC-3 also can lead to a cell-specific proliferative syndrome in lymphocytes, suggesting a selective antiproliferative action of SRC-3 in the hematopoietic system.
Results
Increased lymphocyte numbers in SRC-3−/− mice involves the activation of NF-κB
SRC-3−/− mice had increased white blood cell (WBC) counts, which was mainly due to an expansion of lymphocytes, and decreased platelet numbers (Figure 1A and C). These hematological disturbances were specific for SRC-3−/− mice since WBCs abnormalities were not observed in animals that were deficient for other p160 members, SRC-1−/− and TIF-2−/− mice (Figure 1B). Flow cytometry indicated a significant increase in the proportion of CD4+ and CD8+ T cells in bone marrow, spleen, lymph nodes, and thymus of SRC-3−/− mice (Figure 1D). Likewise, B cells were increased in the same organs of SRC-3−/− mice (Figure 1E). Precursor populations of T and B cells in thymus were normal (Supplementary Figure 1). These data indicate that SRC-3−/− lymphocytes differentiate normally but there is a selective disturbance in lymphoid proliferation.
Figure 1.
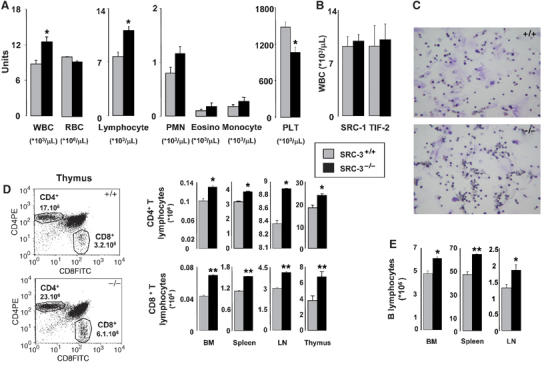
Increased number of mature lymphocytes in SRC-3−/− mice. (A) Quantification of circulating white (WBC) and red (RBC) blood cells and platelets (PLT) in SRC-3+/+ and SRC-3−/− mice (age 7 weeks, n=8). *P<0.05; (B) quantification of circulating WBCs in SRC-1+/+, SRC-1−/−, TIF-2+/+, and TIF-2−/− mice (age 7 weeks, n=8); (C) histology of SRC-3+/+ and SRC-3−/− bone marrow showing the increase in the lymphoid series (× 20); (D) quantification of the proportion of CD4+T and CD8+T lymphocytes populations in bone marrow (BM), spleen, lymph nodes (LN), and thymus (shown also at the left of the panel) of SRC-3+/+ and SRC-3−/− animals by flow cytometry; (E) quantification of the proportion of B lymphocytes in BM, spleen, and lymph nodes of SRC-3+/+ and SRC-3−/− animals by flow cytometry. *P<0.05, **P<0.01.
NF-κB plays a crucial role in hematopoiesis (Denk et al, 2000) and enhanced NF-κB activity can lead to lymphoproliferative disorders (Gilmore, 1999; Rayet and Gelinas, 1999; Hacker and Karin, 2002). Therefore, we quantified nuclear NF-κB p65 levels using a sensitive ELISA-based assay. In the thymus, bone marrow, and spleen, nuclear p65 levels were induced in SRC-3−/− mice (Figure 2A). In contrast, relative to wild-type mice, the nuclear p65 levels were decreased in breast and prostate biopsies of SRC-3−/− mice. Finally, the expression levels of p65 did not change between nuclei isolated from skin biopsies of SRC-3+/+ and SRC-3−/− mice. These findings indicate that the absence of SRC-3 selectively increased nuclear translocation of NF-κB in thymus, bone marrow, and spleen. Similar conclusions were derived from electrophoretic mobility shift assays (EMSA), which showed enhanced NF-κB DNA-binding activity in nuclear protein extracts of the thymus and bone marrow of SRC-3−/− mice (Figure 2B; lanes 4 and 5) relative to SRC-3+/+ littermates (lanes 2 and 3). The combined results of these ELISA and EMSA studies hence suggest that SRC-3 inhibits NF-κB nuclear translocation in vivo in thymus, bone marrow, and spleen.
Figure 2.
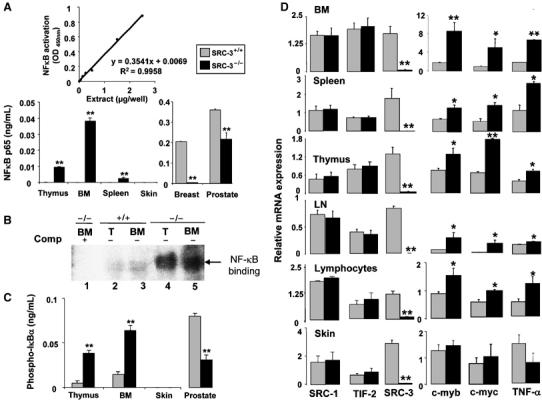
Increased NF-κB activity induces the expression of c-myc and c-myb and underpins the lymphoproliferation. (A) Quantification of the nuclear p65 NF-κB in SRC-3+/+ and SRC-3−/− thymus, bone marrow (BM), spleen, skin, breast, and prostate by TransAM® ELISA kit. **P<0.01; (B) NF-κB DNA-binding activity in nuclear protein extracts of BM and thymus of SRC-3−/− (lanes 4 and 5) and SRC-3+/+ mice (lanes 2 and 3) analyzed by EMSA. Binding in the presence of excess of unlabeled NF-κB-binding oligonucleotide (lane 1) is included as control; (C) quantification of phosphorylated IκB-α in SRC-3+/+ and SRC-3−/− thymus, BM, skin, and prostate by ELISA. **P<0.01; (D) mRNA levels of SRC-1, TIF-2, SRC-3, c-myb, c-myc, and TNF-α in BM, spleen, thymus, lymph nodes (LN), lymphocytes, and skin of SRC-3+/+ and SRC-3−/− animals (age 7 weeks, n=8) as determined by quantitative RT–PCR. *P<0.05, **P<0.01.
Since phosphorylation-induced IκB degradation enables NF-κB activation and its subsequent nuclear translocation (Hayden and Ghosh, 2004), we investigated the phosphorylation of IκB-α in thymus, bone marrow, skin, and prostate. Significantly more phosphorylated IκB-α was found in the thymus and bone marrow of SRC-3−/− mice (Figure 2C). In contrast, the phospho-IκB-α levels were decreased in prostate biopsies of SRC-3−/− mice. Finally, we did not detect phospho-IκB-α in skin biopsies from either SRC-3+/+ or SRC-3−/− mice. Consistent with these data, cytoplasmic IκB-α and IκB-β protein levels were decreased both in thymus and bone marrow of SRC-3−/− mice (Supplementary Figure 2). Moreover, no differences in mRNA levels of IκB-α were detected in thymus and bone marrow between SRC-3+/+ and SRC-3−/− mice, showing that SRC-3 does not affect IκB-α mRNA production (Supplementary Figure 2). These results suggest that SRC-3 stabilizes IκB in lymphoid tissues, which ultimately would lead to the activation of NF-κB.
SRC-3 controls lymphocyte proliferation and apoptosis in a cell autonomous manner
NF-κB influences cell division through the stimulation of the expression of c-myb (Zhang et al, 1998), and other cell cycle regulators such as the c-myc proto-oncogene (Evans et al, 1990; Zobel et al, 1991). As expected, c-myb and c-myc mRNA levels were significantly increased in bone marrow, spleen, thymus, lymph nodes, and isolated lymphocytes of SRC-3−/− mice compared to wild-type littermates (Figure 2D). No significant changes in c-myb and c-myc mRNA levels were, however, detected between skins of SRC-3−/− and SRC-3+/+ mice. The expression of TNF-α, a proinflammatory cytokine induced by NF-κB, was also selectively induced in lymphoid tissues of SRC-3−/− mice (Figure 2D). There was no compensatory increase in the expression of SRC-1 and TIF-2 in lymphoid tissues or skin (Figure 2D).
To further explore the mechanisms that account for high lymphocyte numbers in SRC-3−/− mice, in vitro cell proliferation assays were performed with the fluorescent dye CFSE. Loss of CFSE, as evidenced by a loss of fluorescence intensity, reflects cellular division. Interestingly, T cells of SRC-3−/− mice proliferated faster when stimulated with anti-CD3, either in the presence or absence of anti-CD28 (Figure 3A). Likewise, a more robust proliferative response after induction with anti-IgM and anti-CD40 was detected in B cells of SRC-3−/− mice (Figure 3B). B-cell proliferation seems to be slightly more enhanced when compared to that of T cells. Interestingly, there was no difference in the proliferation of SRC-3+/+ and SRC-3−/− mouse embryonic fibroblasts (MEFs) (Figure 3C). We hence conclude that increased lymphocyte proliferation in SRC-3−/− mice is specific to this cell lineage and caused by a cell autonomous process that may be the consequence of the NF-κB-mediated induction of proliferative genes such as c-myb and c-myc.
Figure 3.
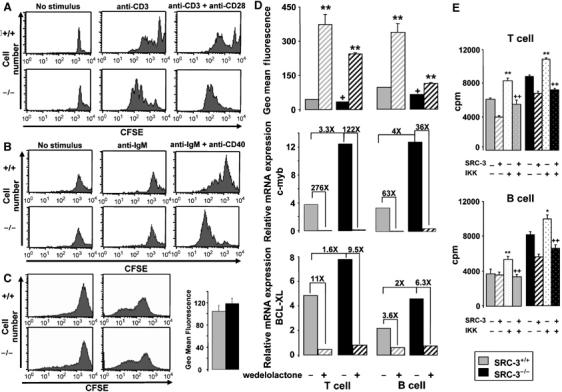
Cell autonomous increase in lymphocyte proliferation. (A) T cells of SRC-3+/+ and SRC-3−/− spleen mice were stimulated by anti-CD3 and/or anti-CD28 and analyzed by FACS. In vitro proliferation assays were performed with the fluorescent dye CFSE. Loss of CFSE reflects cellular division. CFSE staining is presented from a representative experiment (one mouse). Each experiment was repeated three times with similar results; (B) B cells of SRC-3+/+ and SRC-3−/− spleen mice were stimulated by anti-IgM and/or anti-CD40 and analyzed by FACS. CFSE staining is presented from a representative experiment (one mouse). Each experiment was repeated three times with similar results; (C) in vitro proliferation assays of SRC-3+/+ and SRC-3−/− MEFs using the fluorescent dye CFSE. The left histogram represents CFSE staining in MEFs at t=0. The right histogram shows MEFs stained with CFSE and then cultured for 2 days; (D) T and B cells isolated from the spleens of SRC-3+/+ and SRC-3−/− mice (age 7 weeks, n=8) were stimulated by anti-CD3+anti-CD28 and anti-IgM+anti-CD40, respectively, in the presence or absence of wedelolactone (20 μM). The geometric mean fluorescence is inversely proportional to proliferation rate. +P<0.05 indicates a significant difference compared with SRC-3+/+ cells without wedelolactone. **P<0.01 indicates a significant effect of wedelolactone. mRNA levels of c-myb and BCL-XL in T and B cells of SRC-3+/+ and SRC-3−/− mice (age 7 weeks, n=8) was determined by quantitative RT–PCR. Fold inductions are indicated; (E) SRC-3+/+ and SRC-3−/− T and B cells were transfected with pLXRN-SRC-3 or/and pFlag-CMV-IKK expression plasmids. T cells were stimulated with anti-CD3+anti-CD28 and B cells with anti-IgM+anti-CD40. Proliferation was assessed by [3H]thymidine incorporation 18 h later. Results are presented as mean c.p.m.±s.d. for cells from three individual mice for each condition. **P<0.01 indicates a significant difference compared with T or B cells transfected only with control plasmids.++P<0.01 indicates a significant difference compared with T or B cells transfected only with pFlag-CMV-IKK.
Recently, SRC-3 was found to be associated with the IKK complex (Wu et al, 2002). To evaluate the potential implication of this kinase in the antiproliferative effect of SRC-3, we studied the lymphocyte proliferation in the presence or absence of wedelolactone, a reportedly selective and irreversible inhibitor of IKK-α and IKK-β (Kobori et al, 2004). CFSE geometric mean fluorescence was lower in T cells and B cells of SRC-3−/− mice, indicating an increase in proliferation rate when grown, respectively, in the presence of anti-CD3+anti-CD28 and anti-IgM+anti-CD40 (Figure 3D). The addition of wedelolactone, which is reported to stabilize the interaction of NF-κB with IκB and hence decrease the transcriptional activity of NF-κB, diminished proliferation of T and B lymphocytes of SRC-3+/+ and SRC-3−/− mice. Wedelolactone also decreased c-myb and BCL-XL mRNA expression in T and B lymphocytes of SRC-3+/+ and SRC-3−/− mice (Figure 3D). In an independent experiment, we used aspirin to inhibit IKK-β activity, yielding qualitatively similar results as observed with wedelolactone (Supplementary Figure 3). Although we cannot exclude effects of wedelolactone and aspirin on other targets, these data together suggest an eventual involvement of IKK in the antiproliferative effect of SRC-3.
To demonstrate that IKK was involved in mediating the effects of SRC-3 on cell proliferation, we investigated the proliferation of T and B SRC-3+/+ and SRC-3−/− lymphocytes that were transfected with an IKK expression vector (pFlag-CMV-IKK) (Figure 3E). Relative to T and B lymphocytes of SRC-3+/+ and SRC-3−/− mice transfected with a control vector, the cells transfected with the IKK expression vector proliferated more after induction by anti-CD3+anti-CD28 or by anti-IgM+anti-CD40, respectively. However, following cotransfection with IKK together with SRC-3, proliferation rate of T and B lymphocytes of SRC-3+/+ and SRC-3−/− mice significantly decreased. These data indicate that inhibition of lymphocyte proliferation by SRC-3 is mediated by IKK, further underscoring the role of IKK/NF-κB in the enhanced lymphoproliferation observed in SRC-3−/− mice.
Since abnormalities in apoptosis contribute to the pathophysiology of lymphoproliferative disorders (Sanchez-Beato et al, 2003), we also studied apoptosis using annexin V–propidium iodide (PI) staining. In spleen cell suspensions of SRC-3−/− mice, populations of annexin V+PI− (early apoptotic cells) and annexin V+PI+ (late apoptotic cells) cells were decreased compared to SRC-3+/+, suggesting that SRC-3 favors apoptosis (Figure 4A). In SRC-3+/+ and SRC-3−/− MEFs, however, populations of annexin V+PI− and annexin V+PI+ were unchanged (Figure 4B).
Figure 4.
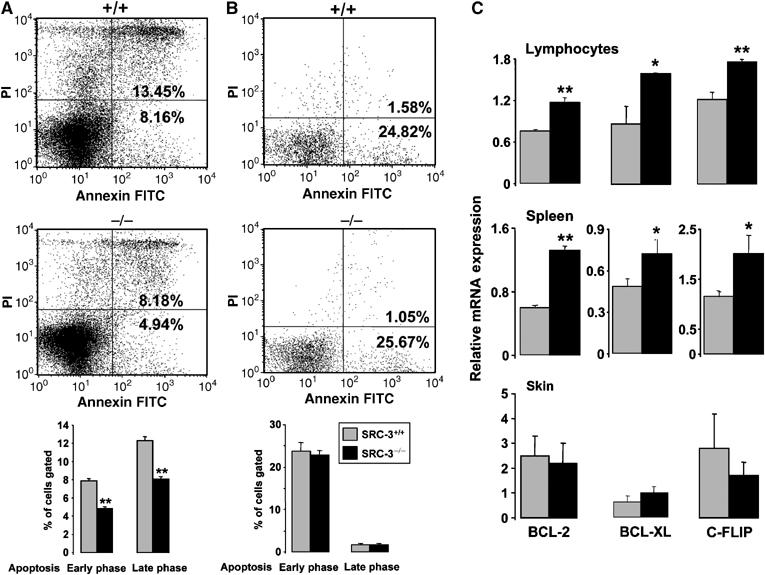
Decreased apoptotic activity in SRC-3−/− mice. Flow cytometric analysis of apoptotic SRC-3+/+ and SRC-3−/− spleen cell suspensions (A) and MEFs (B) after staining by annexin V FITC and propidium iodide (PI). The bar graphs at the bottom of (A) and (B) show the quantification of three independent experiments **P<0.01; (C) mRNA levels of BCL-2, BCL-XL, and c-FLIP in lymphocytes, spleen, and skin of SRC-3+/+ and SRC-3−/− animals (age 7 weeks, n=8) determined by quantitative RT–PCR. *P<0.05, **P<0.01.
We then investigated the expression of mRNAs encoding antiapoptotic NF-κB target genes. In isolated lymphocytes and spleen of SRC-3−/− mice, BCL-2, BCL-XL, and c-FLIP mRNA levels were all significantly increased compared to wild-type littermates (Figure 4C). In contrast, in other tissues of SRC-3−/− mice, such as skin and liver, the expression levels of antiapoptotic genes were not induced (Figure 4C, data not shown). Like the expression of the proliferative c-myb gene, inhibition of IKK by wedelolactone also reduced the expression of these antiapoptotic genes in lymphocytes, consistent with a commanding role of NF-κB in this process (Figure 3D; data not shown).
To unequivocally prove that the absence of SRC-3 leads to lymphoproliferation, we analyzed the proliferation of T and B lymphocytes of SRC-3+/+ and SRC-3−/− mice transfected with pLXRN-SRC-3 expression vector (Figure 5A and B). Relative to SRC-3+/+ mice, T and B lymphocytes of SRC-3−/−mice transfected with the control vector (pLXRN) proliferated more after induction by anti-CD3+anti-CD28 or by anti-IgM+anti-CD40, respectively. However, following transfection with SRC-3, the proliferation of T and B cells of SRC-3−/− mice decreased significantly. Likewise, the increased expression of antiapoptotic genes (BCL-XL, BCL-2, c-FLIP) was also reversed in SRC-3−/− lymphocytes upon SRC-3 transfection (Figure 5C). All these data together prove that SRC-3 directly controls both proliferation and apoptosis in T and B lymphocytes.
Figure 5.
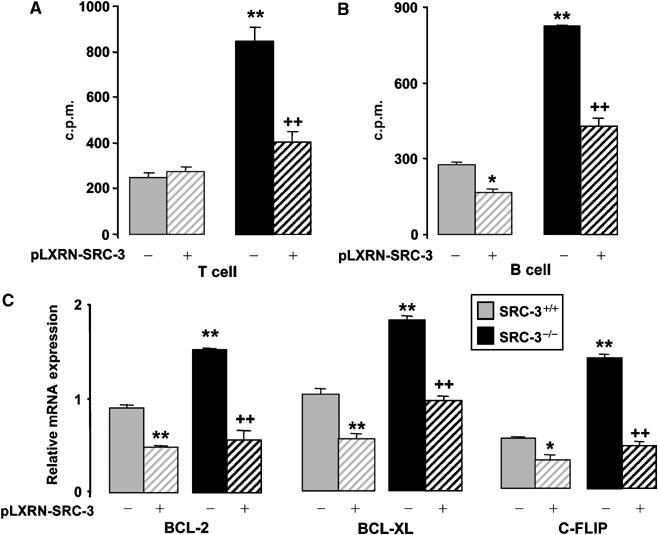
SRC-3 controls proliferation and apoptosis in T and B lymphocytes. SRC-3+/+ and SRC-3−/− T cells (A) and B cells (B) were transfected with pLXRN-SRC-3 or pLXRN control plasmid. T and B cells were stimulated, respectively, with anti-CD3+anti-CD28 and anti-IgM+anti-CD40, and then proliferation was assessed by [3H]thymidine incorporation 18 h later. Results in (A) and (B) are presented as mean c.p.m.±s.d. for cells from three individual mice plated in replicates of three for each condition. (C) BCL-2, BCL-XL, and c-FLIP mRNA levels were determined by quantitative RT–PCR in lymphocytes of SRC-3+/+ and SRC-3−/− animals transfected with pLXRN-SRC-3 or pLXRN plasmids. *P<0.05, and **P<0.01 indicate significant difference compared with SRC-3+/+ cells transfected with pLXRN.++P<0.01 indicates a significant difference compared with SRC-3−/− cells transfected with pLXRN.
Old SRC-3−/− mice develop lymphomas
As in lymphoproliferative disorders, the hematologic disturbance was in general amplified with age when white blood cell counts of young (6–8 weeks) and old mice (11–12 months) were compared (Figure 6A). Consistent with the progression of the lymphoproliferation, a gradual decrease in gain of body mass was observed in SRC-3−/− relative to SRC-3+/+ mice (Figure 6B). This could reflect the negative energy balance often seen in the context of proliferative syndromes and cancers. None of the SRC-3−/− animals were positive for anti-nuclear and anti-double-stranded DNA antibodies, indicating the absence of autoimmunity (data not shown). This suggests that the age-related increase in lymphocyte number and decrease in body weight is the consequence of the progression of the lymphoproliferative syndrome into malignant lymphoma. Consistent with this, the incidence of invasive disease (lymphoma) among mice of all ages was 40% higher in SRC-3−/− mice as compared to their wild-type littermates (Figure 6C). Moreover, when we evaluated the mortality at 14 months of age, survival was significantly decreased (by seven-fold) in SRC-3−/− mutant animals (Figure 6D).
Figure 6.
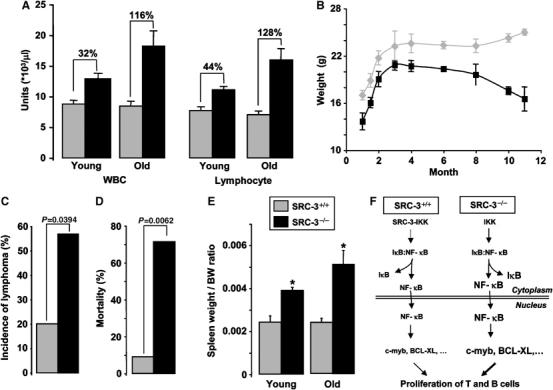
The lymphoproliferative syndrome in SRC-3−/− mice detoriates with age into malignant lymphoma. (A) WBC and lymphocyte counts of young (2–3 months, n=8) and old mice (11–14 months, n=7); (B) body weight gain in SRC-3+/+ and SRC-3−/− mice; (C) incidence of lymphoma in SRC-3+/+ (n=15) and SRC-3−/− (n=14) mice. Statistical significance (P<0.05; χ2 test) is indicated; (D) percentage of mortality of SRC-3+/+ (n=11) and SRC-3−/− (n=7) mice at 14 months of age. Statistical significance (P<0.01; χ2 test) is indicated; (E) spleen/body weight (BW) ratio of young (3 months, n=8) and old mice (14 months, n=7). *P<0.05; (F) scheme explaining the mechanism leading to the lymphoproliferative syndrome in SRC-3−/− mice. The size of symbols/arrows reflects the relative concentrations of signaling factors.
To chart the development of malignant lymphoma, we then performed systematic necropsy in groups of three SRC-3−/− and SRC-3+/+ mice at 3 and 11–14 months of age. The 3-month-old SRC-3−/− mutants did not display macroscopic defects at necropsy, but they showed a 1.6-fold increase in spleen weight/body weight ratio, a ratio which further increased upon aging (Figure 6E). Histological analyses of these young mutant mice consistently revealed hyperplastic germinal centers in both lymph nodes and spleen (white ovals, compare Figure 7A and B and data not shown; Taddesse-Heath et al, 2000). The cellularity of these hyperplastic germinal centers was, however, normal (compare Figure 7C and D). To further investigate the architecture of the SRC-3−/− spleen and lymph nodes, immunohistochemical analyses were performed with antibodies directed against CD3 and B220, which detect T and B cells, respectively. In the spleen, T-cell distribution was normal (data not shown). In contrast, B-cell distribution was disturbed: these cells invaded the marginal sinuses (S and brackets, Figure 7E and F) (Fredrickson et al, 1999), and their number was increased in the red pulp (R, compare Figure 7G and H). B- and T-cell distributions within lymph nodes were unaffected (data not shown).
Figure 7.
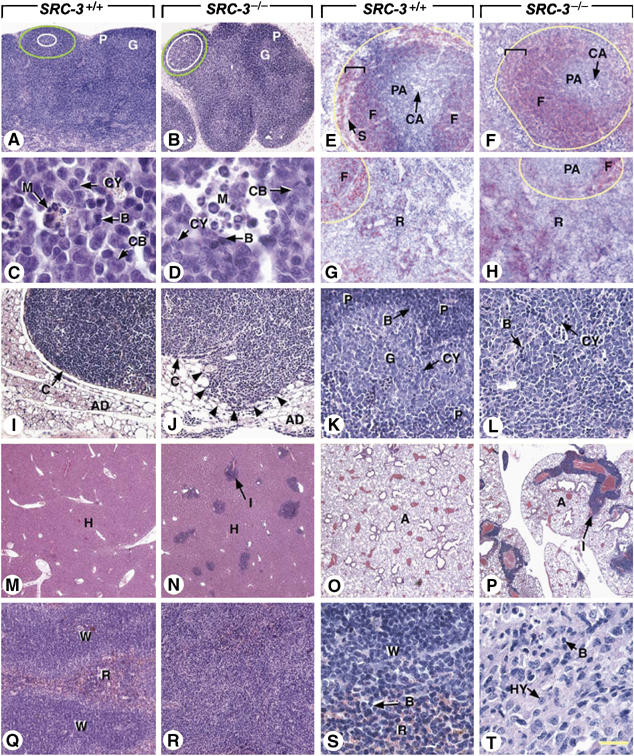
Ablation of SRC3 results in increased B-cell proliferation and predisposes to B-cell lymphomas. Histological sections through lymph nodes (A–D, I–L), spleen (E–H, Q–T), liver (M, N), and lung (O, P) of SRC3+/+ mice and SRC3−/− mice at 3 (A–H), 11 (I–P), and 14 (Q–T) months of age. (A–D) In young SRC3−/− and SRC3+/+ mice, the germinal centers (white ovals) of lymphoid follicles (green ovals) contain similar mixed populations of proliferative B lymphocyte precursors (CB and CY), mature lymphocytes (B), and macrophages (M). (E–H) Immunohistochemical detection of B cells in the spleen (red signal), showing an increase of their number in both the white pulp (yellow outlines) and the red pulp (R) of the mutants. (J, L, N, P) Follicular B-cell lymphoma with liver and lung metastases; the tumor (displayed in L) consists of a mixture of cells with scant amounts of slightly eosinophilic cytoplasm and large nuclei containing one or two nucleoli (CY), and of smaller basophilic cells with condensed chromatin and inconspicuous nucleoli (B). (I, K, M, O) Tissues from an SRC3+/+ littermate are displayed for comparaison. (R, T) Histiocyte-associated lymphoma consisting of a mixture of large cells (HY) with round nuclei, prominent nucleoli, and abundant eosinophilic cytoplasm, occasionally containing cellular debris, and of small cells with scant amounts of basophilic cytoplasm (B). (Q, S) The spleen of a SRC3+/+ littermate is displayed for comparative purposes. Abbreviations: A, lung alveoli; AD, adipose tissue; B, mature B cells; C, capsules of lymph nodes; CA, central arterioles; CB, centroblasts; CY, centrocytes; F, splenic lymphoid follicles; G, germinal centers of lymphoid follicles; H, hepatocytes; I, lymphoid infiltrates; M, macrophages; P, periphery of lymphoid follicles (mantle zone); PA, periarteriolar lymphoid sheath (main T-cell zone of the spleen); R, red pulp of the spleen; S, marginal sinus; W, white pulp of the spleen. White ovals, limits of germinal centers in lymph nodes; green ovals, limits of follicles in lymph nodes; yellow outlines, limits of the white pulp of the spleen; brackets, marginal zone of the white pulp; arrowheads, tumor cells invading adipose tissue. Hematoxylin and eosin stain (A–D and I–T) and computer generated bright field images of B220 immunodetection (red pseudocolor) with DAPI counterstain (E–H). Bar: 50 μm (A, B), 2.5 μm (C, D), 20 μm (E–L), 145 μm (M–P), 35 μm (Q, R), and 5 μm (S, T).
The malignant degeneration of the lymphoid lesions is clearly illustrated by the enlarged mediastinal lymph nodes with disrupted capsules (C and arrowheads in Figure 7I and J), and invasion of the surrounding adipose tissue by a mixture of centrocytes (CY compare Figure 7K and L) and mature B cells (B compare Figure 7K and L) in a 11-month-old SRC-3−/− mice. Infiltrates of centrocytes and mature B cells were also found in the liver, lungs, and kidneys (I, Figure 7N and P, compare with M and O, and data not shown). These aspects are indicative of a metastatic follicular B-cell lymphoma (Morse et al, 2002). In old SRC-3−/− mutant (14 months), the spleen was markedly enlarged, an abnormality that was present in all old SRC-3−/− mice (2.14-fold increase in spleen/body weights ratio; Figure 6E). Moreover, one of the mutant spleens exhibited several areas where its architecture was disorganized (Figure 7Q–T) due to invasion of the white and red pulps by abnormal sheets of cells consisting mainly in histiocytes (HY in Figure 7T), but also containing numerous, scattered, B lymphocytes (B in Figure 7T, compare with 7S). Infiltrates, probably formed by mature B cells, were present in the liver and lungs of this mutant (data not shown). These features are characteristic of a metastatic histiocyte-associated lymphoma (Morse et al, 2002). Hyperplastic germinal centers in lymph nodes and spleen, occurring as a consequence of aging, were observed in four other old SRC-3−/− mice (11 and 14 months of age) as well as in their SRC-3+/+ littermates, which otherwise displayed normal histological features. Altogether, these results indicate that ablation of SRC-3 results in increased B-cell proliferation and predisposes to B-cell lymphomas.
Discussion
The characterization of animals, which either lack or overexpress SRC-3, indicates that SRC-3 regulates somatic growth, affects steroid receptor function, and plays a role in the regulation of ovulation (Wang et al, 2000; Xu et al, 2000; Wu et al, 2002; Torres-Arzayus et al, 2004). SRC-3 was also shown to be involved in the hormonal regulation of cell proliferation, and its altered expression may play a role in the hormonal promotion of tumorigenesis (Liao et al, 2002; Torres-Arzayus et al, 2004). In this study, we provide evidence that the absence of SRC-3 induces thrombocytopenia and a lymphoproliferative disorder often evolving into lymphoma. Tumorigenesis in other organs of SRC-3−/− mice was not observed. The hematological disturbances were specific for SRC-3−/− mice since similar abnormalities were not observed in animals that were deficient in the other p160 members, that is, SRC-1−/− and TIF-2−/− mice. These results confirm that, despite sequence and functional similarities between the p160 family of coactivators, SRC-3−/− mice exhibit different phenotypes from those reported for SRC-1−/− and TIF-2−/− mice, proving that these coactivators play roles in distinct biological processes.
The most pronounced hematopoietic abnormality in SRC-3−/− mice concerns the lymphoid lineage, where SRC-3−/− mice show an expansion of both T and B cells. The lymphoid expansion in SRC-3−/− mice is not the consequence of an autoimmune disease and progresses with age into a B-cell lymphoma, as shown by the gradual increase in lymphocyte numbers, the progressive loss of body weight, the disorganization of hematopoietic tissues, and the presence of lymphoid tissue infiltrates (metastasis) in older mice. The effect of SRC-3 on the lymphoid lineage is cell autonomous as indicated by the stronger in vitro proliferative response observed after induction of T and B cells in SRC-3−/− mice and its normalization upon re-expression of SRC-3. Despite the increased proliferation in both T and B cells, it is unclear why mainly B-cell lymphomas develop. The absence of SRC-3 had, however, no effect on proliferation of MEFs showing that the effect is not present in all tissues but rather specific to cells of the lymphoid lineage.
Our data underscore that SRC-3 can act as a bona fide tumor suppressor in the hematopoietic system, whereas we detected no effects on cell proliferation and apoptosis in other tissues. The fact that SRC-3 acts as a tumor suppressor in the lymphoid system is in sharp contrast to the overexpression or amplification of the SRC-3 gene observed in breast (Anzick et al, 1997) and certain other cancers in humans (Sakakura et al, 2000; Glaeser et al, 2001). Furthermore, transgenic overexpression of SRC-3 favors the development of cancers of breast and other tissues (Torres-Arzayus et al, 2004). Other studies, however, have also documented a loss of heterozygosity of SRC-3 in breast cancers (Shibata et al, 2001), a feature characteristic of tumor suppressors, such as p53 and RB. Likewise, the loss of SRC-3 expression has been linked with pronounced vascular cell proliferation, indicative of a growth suppressive activity of SRC-3 in the vascular wall (Yuan et al, 2002). Altogether, these data indicate that SRC-3 could have both oncogenic and antioncogenic roles, depending on the particular cellular and molecular signaling pathway context. Several good examples exist of the impact of the cellular and molecular context on the proliferative/tumorigenic potential of proteins. For instance, depending on the presence of the retinoblastoma (Fajas et al, 2003) and adenomatous polyposis coli (APC) proteins (Girnun et al, 2002), PPARγ was shown to either induce or reduce cell proliferation. Such changes in the molecular milieu have been invoked to explain why treatment with PPARγ agonists inhibits tumor development in xenograft models of colon cancer (Sarraf et al, 1998), whereas it enhances tumorigenesis in the genetic APCMin/+ model of colon cancer (Lefebvre et al, 1998; Saez et al, 1998). Likewise, in an myc-inducible osteosarcoma mouse model, expression of the myc oncogene in immature osteoblasts induces tumors, but apoptosis ensues in differentiated osteocytes (Jain et al, 2002). The particular cellular context also may dictate whether SRC-3 will induce (breast epithelium) or reduce (lymphoid system) tumor formation.
NF-κB was recently shown to function as a tumor promotor (Aggarwal, 2004; Greten et al, 2004; Pikarsky et al, 2004). NF-κB furthermore plays a crucial role in hematopoiesis (Denk et al, 2000) and enhanced NF-κB activity leads to lymphoproliferative disorders and lymphomas (Rayet and Gelinas, 1999). This is often due to rearrangement and/or amplification of the NF-κB genes (Denk et al, 2000; Sanchez-Beato et al, 2003). The pivotal role of NF-κB in the development and function of hematopoietic cell types on one side and the reported association of SRC-3 with IKK (Wu et al, 2002) on the other side suggested that NF-κB could be implicated in the antiproliferative role of SRC-3. We show here that SRC-3 gene deletion results in constitutive NF-κB activation in vivo. This is attributed to the fact that IKK bound to SRC-3 (Wu et al, 2002), limits IκB phosphorylation and hence stabilizes IκB in hematopoietic tissues, inhibiting NF-κB nuclear accumulation and transcriptional activity. We excluded the possibility that the loss of SRC-3 affected IκB mRNA expression (Supplementary Figure 2). This mechanism may hence seem in contrast to previous studies which showed that SRC-3 can function as an NF-κB transcriptional coactivator in vitro (Werbajh et al, 2000; Wu et al, 2002; Gao et al, 2005). These studies, however, mainly tested the capacity of SRC-3 to activate NF-κB-mediated reporter gene expression in transfected breast and prostate cells and to control the expression of a single NF-κB target gene (IRF-1) in spleen (Wu et al, 2002). Our current results reflect a chronic hematopoetic selective effect in animals and substantiate gene-, cell- and signaling-specific actions of SRC-3. Although unexpected, this result is not without precedent. Multiple examples exist where SRC coactivators have been reported to enhance gene expression in certain tissues, while displaying gene repressive effects in others (Weiss et al, 1999; Takeuchi et al, 2002).
NF-κB, whose activity was enhanced in SRC-3−/− mice, was previously reported to influence cell growth through the stimulation of the c-myc or c-myb promoter (Zhang et al, 1998). Consistent with this, we show here that the expression of these two genes is elevated both in purified lymphocytes as well as in lymphoid tissues of SRC-3−/− mice, indicating that SRC-3 downregulates c-myb and c-myc expression hence controlling lymphoid proliferation. Interestingly, thrombopenia can also be attributed to elevated c-myb expression in SRC-3−/− mice. In fact, this inhibitory effect of c-myb on platelet production is in line with a recent study, which showed that the reduced expression of c-myb induces platelet production in the absence of thrombopoietin signaling (Carpinelli et al, 2004). The critical step by which thrombopoietin stimulates platelet production involves downregulation of c-myb expression or activity—a condition that is not met in the SRC-3−/− mice which have increased levels of c-myb in hematopoietic tissues.
Interference with apoptosis is another way how NF-κB could impact on lymphoproliferation. Raised threshold for apoptosis represents a central step in tumorigenesis (Adams and Cory, 1998), and antiapoptotic BCL-2 family members have oncogenic potential. For example, the BCL-2 gene is activated by chromosomal translocations in the majority of non-Hodgkin's lymphomas (Tsujimoto et al, 1985; Weiss et al, 1987). BCL-2 overexpression prolonges B-cell survival, and mice that overexpress BCL-2 are susceptible to autoimmune diseases and follicular hyperplasia that can progress to malignant large-cell lymphoma (McDonnell et al, 1989; McDonnell and Korsmeyer, 1991). NF-κB induces the expression of antiapoptotic factors such as the inhibitor of apoptosis proteins, c-FLIP, and TRAF1–2, as well as members of the BCL-2 family such as BCL-XL (Micheau et al, 2001; Karin and Lin, 2002). Consistent with this, we demonstrate here that lymphocytes, but not MEFs, of SRC-3−/− mice are resistant to apoptosis, and we correlate this with elevated BCL-2, BCL-XL, and c-FLIP mRNA levels. These data hence implicate SRC-3 also in the cell specific control of apoptosis. The increase in lymphocytes in SRC-3−/− mice is therefore the consequence of NF-κB-mediated induction of both proliferative and antiapoptotic genes.
In conclusion, the deletion of the SRC-3 gene leads to thrombocytopenia and cell autonomous induction of lymphocyte proliferation often culminating in malignant B lymphoma. The lymphoproliferation reflects an activation of NF-κB, which in turn induces the expression of both genes that stimulate proliferation and inhibit apoptosis of hematopoietic cells. Our results substantiate existing evidence linking SRC-3 and NF-κB signaling (Werbajh et al, 2000; Wu et al, 2002; Park et al, 2005) and fuel the hypothesis that NF-κB can also act as a potent tumor inducer in the hematopoietic system (Rayet and Gelinas, 1999; Greten et al, 2004; Pikarsky et al, 2004). Although, we cannot exclude the possibility that NF-κB-independent mechanisms contribute to the phenotype. In the lymphoid system, factors such as NF-κB, c-myb, or BCL-2 direct SRC-3's activity toward inhibition of tumor development. In the breast, other transcription factors (e.g. estrogen receptors) are most likely the predominant codeterminants of the stimulatory activity of SRC-3 on tumor development.
Materials and methods
Animal experiments
The generation of the SRC-1−/− (Xu et al, 1998), TIF-2−/− (Gehin et al, 2002), and SRC-3−/− (Xu et al, 2000) mouse lines were described. All mice were maintained on a pure C57BL/6J background. Only female, age-matched mice were used. Mice were maintained according to EU guidelines and had ad libitum access to water and chow (DO4, UAR, France). Mice were killed by decapitation and tissues were harvested, weighed, quickly frozen in liquid nitrogen, and stored at −80°C until further processing.
Hematological analysis and flow cytometry
Blood was collected from the retroorbital sinus and complete blood counts were performed with an ACT DIFF (Beckmann Coulter Fullerton, CA). The neutrophils, lymphocytes, monocytes, eosinophils, and basophils were determined by microscopic analysis. Cells were cytospun (105 cells/slide) onto glass slides, and stained with May–Grunwald followed by Giemsa. Lineage staining was performed using fluorescein isothiocyanate (FITC)-conjugated anti-CD8, phycoerythrin-conjugated anti-CD4, phycoerythrin-conjugated anti-IgM, Cy5-conjugated anti-CD19, and Cy5-conjugated anti-B220 antibodies (all from Pharmingen, San Diego, CA). All analyses were carried out on a FACScan (Becton Dickinson, San Jose, CA) using the CellQuest software (Becton Dickinson).
RNA extraction and RT–PCR
RNA preparation was as described (Rocchi et al, 2001). cDNA was synthesized by using the SuperScript System (Invitrogen, Carlsbad, USA) and random hexamer primers. Quantitative RT–PCR was performed by using LightCycler FastStart DNA Master SYBR Green I from Roche Diagnostics. The sequences of primers used are available at www.igbmc.ustrasbg.fr/Departments/Dep_V/Dep_VA/Publi/Paper.html. 18S rRNA was used as the invariant control.
Immunoblotting and ELISA for IκB-α
Tissue homogenization and preparation of cytosolic and nuclear fractions, SDS–PAGE, and electrotransfer were performed as described (Schoonjans et al, 1996). Filters were first incubated 4 h at 21°C with primary antibody against IκB-α and IκB-β (Santa Cruz Biotechnology, CA), and then for 1 h at 21°C with a peroxidase-conjugated secondary antibody. Phosphorylation of IκB-α was studied with the FunctionELISA® IκB-α kit (Active Motif, Rixensart, Belgium), which detects specifically phosphorylated IκB-α.
DNA-binding activity and EMSA
Tissue homogenization and nuclear proteins were prepared with the TransAM® nuclear extract kit (Active Motif). In brief, 20 μg of nuclear extract was added to each well of a 96-well plate into which an oligonucleotide with an NF-κB consensus-binding site had been immobilized. Linked NF-κB was detected with TransAM® ELISA kit using a specific p65 NF-κB antibody and a secondary HRP antibody. The final A450 was read on a microplate reader (Berthold Technologies, France).
Nuclear protein extraction and EMSA was carried out as described (Schoonjans et al, 1996) using a double-stranded 32P-ATP-labeled RelA NF-κB probe 5′-AGCTTGGGGACTTTCCACTAGTACG-3′. Specific binding was determined by adding 100-fold molar excess of unlabeled NF-κB-binding oligonucleotide to the reaction mixture.
Lymphocyte and MEF proliferation and apoptosis studies
Spleens were removed from 6- to 8-week-old mice and single-cell suspensions were prepared. MEFs were prepared from embryos at E13.5 from SRC-3+/+ and SRC-3−/− pregnant female mice and cells were self-immortalized (Picard et al, 2002). To determine cell division, cells were labeled with 20 μg/ml of 5-(and 6)-carboxyfluorescein diacetate succinimidyl ester (CFSE; Sigma, Saint Louis, MO) at 37°C for 15 min. T cells were cultured at 2 × 105 cells/well in 200 μl of 10% RPMI medium, precoated with 10 μg/ml anti-CD3 antibody (Becton Dickinson) and/or 25 μg/ml anti-CD28 antibody (BD Bioscience, Mountain View, CA). B lymphocytes were resuspended in 10% RPMI medium and supplemented with 10 μg/ml anti-IgM (Jackson Immunoresearch, Cambridge, UK) ±10 μg/ml anti-CD40 (Pharmingen, San Diego, CA). In certain experiments, cells were stimulated for 72 h in the presence or absence of wedelolactone (20 μM) (Calbiochem, Darmstadt, Germany) or aspirin (0.1 mM). For apoptosis analysis, cells were stained with fluorescein isothiocyanate (FITC)-conjugated annexin V and propidium iodide (PI) for 5 min at 21°C. Data for proliferation or apoptosis were collected in a FACS Calibur and analyzed by the CellQuest software. Cell culture experiments were repeated at least three times. Representative experiments are shown.
Transfection studies
Spleen cell suspensions were transfected with nucleofection (Amaxa, Koeln, Germany) using either the pLXRN control (BD Biosciences), the pLXRN-SRC-3 vector, which contains SRC-3 cDNA, and/or an IKK expression vector (pFlag-CMV-IKK). The pmaxGFP vector was cotransfected in all transfection studies enabling selection of GFP-positive cells, which were used for further culture. After transfection, T and B cells were cultured and stimulated as described above. Cell proliferation was analyzed by [3H]thymidine incorporation. RNA was extracted from parallel plates.
Histology and immunohistochemistry
Tissues were collected and prepared for hematoxylin and eosin staining and immunohistochemistry according to standard operating procedures available at http://www.eumorphia.org/. Antibodies directed against CD3 (KT3) and B220 (RA3.6B2) were produced in our laboratory. Sections were taken out from −80°C, dried at 20°C for 10 min, hydrated in PBS-0.5% Tween (PBST) incubated overnight at 4°C, with either the anti-CD3 or the anti-B220 antibody (diluted 1/1000 in PBST containing 5% normal goat serum), washed in PBST incubated with a secondary CY3-conjugated antibody (Jackson Laboratories) for 1 h at 21°C, washed and then counterstained with DAPI (Roche) and mounted with Vectashield® (Vector).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
This work was supported by grants of CNRS, INSERM, Hôpitaux Universitaires de Strasbourg, and NIH (DK59820, HD07857, and DK0677320). We thank Mohammed Selloum, Nassim Dali-Youcef, Kristina Schoonjans, Sami Heikkinen, Mei Li, and Marie-France Champy for helpful discussions, technical assistance, and gift of materials.
References
- Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281: 1322–1326 [DOI] [PubMed] [Google Scholar]
- Aggarwal BB (2004) Nuclear factor-kappaB: the enemy within. Cancer Cell 6: 203–208 [DOI] [PubMed] [Google Scholar]
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan X-Y, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS (1997) AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277: 965–968 [DOI] [PubMed] [Google Scholar]
- Carpinelli MR, Hilton DJ, Metcalf D, Antonchuk JL, Hyland CD, Mifsud SL, Di Rago L, Hilton AA, Willson TA, Roberts AW, Ramsay RG, Nicola NA, Alexander WS (2004) Suppressor screen in Mpl−/− mice: c-Myb mutation causes supraphysiological production of platelets in the absence of thrombopoietin signaling. Proc Natl Acad Sci USA 101: 6553–6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Schiltz L, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM (1997) Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms multimeric activation complexes with P/CAF and CBP/p300. Cell 90: 569–580 [DOI] [PubMed] [Google Scholar]
- Denk A, Wirth T, Baumann B (2000) NF-kappaB transcription factors: critical regulators of hematopoiesis and neuronal survival. Cytokine Growth Factor Rev 11: 303–320 [DOI] [PubMed] [Google Scholar]
- Evans JL, Moore TL, Kuehl WM, Bender T, Ting JP (1990) Functional analysis of c-Myb protein in T-lymphocytic cell lines shows that it trans-activates the c-myc promoter. Mol Cell Biol 10: 5747–5752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajas L, Egler V, Reiter R, Miard S, Lefebvre AM, Auwerx J (2003) PPARgamma controls cell proliferation and apoptosis in an RB-dependent manner. Oncogene 22: 4186–4193 [DOI] [PubMed] [Google Scholar]
- Fredrickson TN, Lennert K, Chattopadhyay SK, Morse HC III, Hartley JW (1999) Splenic marginal zone lymphomas of mice. Am J Pathol 154: 805–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Chiao P, Zhang X, Lazar MA, Seto E, Young HA, Ye J (2005) Coactivators and corepressors of NF-kappaB in IkappaB alpha gene promoter. J Biol Chem 280: 21091–21098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehin M, Mark M, Dennefeld C, Dierich A, Gronemeyer H, Chambon P (2002) The function of TIF2/GRIP1 in mouse reproduction is distinct from those of SRC-1 and p/CIP. Mol Cell Biol 22: 5923–5937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore TD (1999) Multiple mutations contribute to the oncogenicity of the retroviral oncoprotein v-Rel. Oncogene 18: 6925–6937 [DOI] [PubMed] [Google Scholar]
- Girnun GD, Smith WM, Drori S, Sarraf P, Mueller E, Eng C, Nambiar P, Rosenberg DW, Bronson RT, Edelmann W, Kucherlapati R, Gonzalez FJ, Spiegelman BM (2002) APC-dependent suppression of colon carcinogenesis by PPARgamma. Proc Natl Acad Sci USA 99: 13771–13776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaeser M, Floetotto T, Hanstein B, Beckmann MW, Niederacher D (2001) Gene amplification and expression of the steroid receptor coactivator SRC3 (AIB1) in sporadic breast and endometrial carcinomas. Horm Metab Res 33: 121–126 [DOI] [PubMed] [Google Scholar]
- Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN (2001) Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br J Cancer 85: 1928–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M (2004) IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118: 285–296 [DOI] [PubMed] [Google Scholar]
- Hacker H, Karin M (2002) Is NF-kappaB2/p100 a direct activator of programmed cell death? Cancer Cell 2: 431–433 [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S (2004) Signaling to NF-kappaB. Genes Dev 18: 2195–2224 [DOI] [PubMed] [Google Scholar]
- Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, Sundberg CD, Bishop JM, Felsher DW (2002) Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 297: 102–104 [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3: 415–428 [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A (2002) NF-kappaB at the crossroads of life and death. Nat Immunol 3: 221–227 [DOI] [PubMed] [Google Scholar]
- Kobori M, Yang Z, Gong D, Heissmeyer V, Zhu H, Jung YK, Gakidis MA, Rao A, Sekine T, Ikegami F, Yuan C, Yuan J (2004) Wedelolactone suppresses LPS-induced caspase-11 expression by directly inhibiting the IKK complex. Cell Death Differ 11: 123–130 [DOI] [PubMed] [Google Scholar]
- Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, Briggs M, Heyman R, Auwerx J (1998) Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med 4: 1053–1057 [DOI] [PubMed] [Google Scholar]
- Li H, Gomes PJ, Chen JD (1997) RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc Natl Acad Sci USA 94: 8479–8484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L, Kuang SQ, Yuan Y, Gonzalez SM, O'Malley BW, Xu J (2002) Molecular structure and biological function of the cancer-amplified nuclear receptor coactivator SRC-3/AIB1. J Steroid Biochem Mol Biol 83: 3–14 [DOI] [PubMed] [Google Scholar]
- Lund AH, van Lohuizen M (2004) Epigenetics and cancer. Genes Dev 18: 2315–2335 [DOI] [PubMed] [Google Scholar]
- Mark M, Yoshida-Komiya H, Gehin M, Liao L, Tsai MJ, O'Malley BW, Chambon P, Xu J (2004) Partially redundant functions of SRC-1 and TIF2 in postnatal survival and male reproduction. Proc Natl Acad Sci USA 101: 4453–4458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, Korsmeyer SJ (1989) bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 57: 79–88 [DOI] [PubMed] [Google Scholar]
- McDonnell TJ, Korsmeyer SJ (1991) Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature 349: 254–256 [DOI] [PubMed] [Google Scholar]
- McKenna NJ, Lanz RB, O'Malley BW (1999) Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev 20: 321–344 [DOI] [PubMed] [Google Scholar]
- Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J (2001) NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol 21: 5299–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse HC III, Anver MR, Fredrickson TN, Haines DC, Harris AW, Harris NL, Jaffe ES, Kogan SC, MacLennan IC, Pattengale PK, Ward JM (2002) Bethesda proposals for classification of lymphoid neoplasms in mice. Blood 100: 246–258 [DOI] [PubMed] [Google Scholar]
- Onate SA, Tsai SY, Tsai MJ, O'Malley BW (1995) Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 270: 1354–1357 [DOI] [PubMed] [Google Scholar]
- Park KJ, Krishnan V, O'Malley BW, Yamamoto Y, Gaynor RB (2005) Formation of an IKKalpha-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol Cell 18: 71–82 [DOI] [PubMed] [Google Scholar]
- Picard F, Gehin M, Annicotte J, Rocchi S, Champy MF, O'Malley BW, Chambon P, Auwerx J (2002) SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell 111: 931–941 [DOI] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y (2004) NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 431: 461–466 [DOI] [PubMed] [Google Scholar]
- Rayet B, Gelinas C (1999) Aberrant rel/nfkb genes and activity in human cancer. Oncogene 18: 6938–6947 [DOI] [PubMed] [Google Scholar]
- Rocchi S, Picard F, Vamecq J, Gelman L, Potier N, Zeyer D, Dubuquoy L, Bac P, Champy MF, Plunket KD, Leesnitzer LM, Blanchard SG, Desreumaux P, Moras D, Renaud JP, Auwerx J (2001) A unique PPARgamma ligand with potent insulin-sensitizing yet weak adipogenic activity. Mol Cell 8: 737–747 [DOI] [PubMed] [Google Scholar]
- Saez E, Tontonoz P, Nelson MC, Alvarez JG, Ming UT, Baird SM, Thomazy VA, Evans RM (1998) Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat Med 4: 1058–1061 [DOI] [PubMed] [Google Scholar]
- Sakakura C, Hagiwara A, Yasuoka R, Fujita Y, Nakanishi M, Masuda K, Kimura A, Nakamura Y, Inazawa J, Abe T, Yamagishi H (2000) Amplification and over-expression of the AIB1 nuclear receptor co-activator gene in primary gastric cancers. Int J Cancer 89: 217–223 [PubMed] [Google Scholar]
- Sanchez-Beato M, Sanchez-Aguilera A, Piris MA (2003) Cell cycle deregulation in B-cell lymphomas. Blood 101: 1220–1235 [DOI] [PubMed] [Google Scholar]
- Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, Holden SA, Chen LB, Singer S, Fletcher C, Spiegelman BM (1998) Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med 4: 1046–1052 [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Peinado-Onsurbe J, Lefebvre AM, Heyman RA, Briggs M, Deeb S, Staels B, Auwerx J (1996) PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J 15: 5336–5348 [PMC free article] [PubMed] [Google Scholar]
- Shibata A, Hayashi Y, Imai T, Funahashi H, Nakao A, Seo H (2001) Somatic gene alteration of AIB1 gene in patients with breast cancer. Endocr J 48: 199–204 [DOI] [PubMed] [Google Scholar]
- Taddesse-Heath L, Chattopadhyay SK, Dillehay DL, Lander MR, Nagashfar Z, Morse HC III, Hartley JW (2000) Lymphomas and high-level expression of murine leukemia viruses in CFW mice. J Virol 74: 6832–6837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita A, Cardona GR, Koibuchi N, Suen CS, Chin WW (1997) TRAM-1, a novel 160-kDa thyroid hormone receptor activator molecule, exhibits distinct properties from steroid receptor coactivator-1. J Biol Chem 272: 27629–27634 [DOI] [PubMed] [Google Scholar]
- Takeshita A, Yen PM, Misiti S, Cardona GR, Liu Y, Chin WW (1996) Molecular cloning and properties of a full-length putative thyroid hormone receptor coactivator. Endocrinology 137: 3594–3597 [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Murata Y, Sadow P, Hayashi Y, Seo H, Xu J, O'Malley BW, Weiss RE, Refetoff S (2002) Steroid receptor coactivator-1 deficiency causes variable alterations in the modulation of T(3)-regulated transcription of genes in vivo. Endocrinology 143: 1346–1352 [DOI] [PubMed] [Google Scholar]
- Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG (1997) The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function [see comments]. Nature 387: 677–684 [DOI] [PubMed] [Google Scholar]
- Torres-Arzayus MI, De Mora JF, Yuan J, Vazquez F, Bronson R, Rue M, Sellers WR, Brown M (2004) High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell 6: 263–274 [DOI] [PubMed] [Google Scholar]
- Tsai MJ, O'Malley BW (1994) Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem 63: 451–486 [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Cossman J, Jaffe E, Croce CM (1985) Involvement of the bcl-2 gene in human follicular lymphoma. Science 228: 1440–1443 [DOI] [PubMed] [Google Scholar]
- Voegel JJ, Heine MJS, Zechel C, Chambon P, Gronemeyer H (1996) TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J 15: 3667–3675 [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu MC, Sham JS, Zhang W, Wu WQ, Guan XY (2002) Prognostic significance of c-myc and AIB1 amplification in hepatocellular carcinoma. A broad survey using high-throughput tissue microarray. Cancer 95: 2346–2352 [DOI] [PubMed] [Google Scholar]
- Wang Z, Rose DW, Hermanson O, Liu F, Herman T, Wu W, Szeto D, Gleiberman A, Krones A, Pratt K, Rosenfeld R, Glass CK (2000) Regulation of sematic growth by the p160 coactivator p/CIP. Proc Natl Acad Sci USA 97: 13549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LM, Warnke RA, Sklar J, Cleary ML (1987) Molecular analysis of the t(14;18) chromosomal translocation in malignant lymphomas. N Engl J Med 317: 1185–1189 [DOI] [PubMed] [Google Scholar]
- Weiss RE, Xu J, Ning G, Pohlenz J, O'Malley BW, Refetoff S (1999) Mice deficient in the steroid receptor co-activator 1 (SRC-1) are resistant to thyroid hormone. EMBO J 18: 1900–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werbajh S, Nojek I, Lanz R, Costas MA (2000) RAC-3 is a NF-kappa B coactivator. FEBS Lett 485: 195–199 [DOI] [PubMed] [Google Scholar]
- Wu RC, Qin J, Hashimoto Y, Wong J, Xu J, Tsai SY, Tsai MJ, O'Malley BW (2002) Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) coactivator activity by I kappa B kinase. Mol Cell Biol 22: 3549–3561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O'Malley BW (2004) Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol Cell 15: 937–949 [DOI] [PubMed] [Google Scholar]
- Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, O'Malley BW (2000) The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci USA 97: 6379–6384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Qiu Y, DeMayo FJ, Tsai SY, Tsai MJ, O'Malley BW (1998) Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science 279: 1922–1925 [DOI] [PubMed] [Google Scholar]
- Yuan Y, Liao L, Tulis DA, Xu J (2002) Steroid receptor coactivator-3 is required for inhibition of neointima formation by estrogen. Circulation 105: 2653–2659 [DOI] [PubMed] [Google Scholar]
- Zhang MY, Sun SC, Bell L, Miller BA (1998) NF-kappaB transcription factors are involved in normal erythropoiesis. Blood 91: 4136–4144 [PubMed] [Google Scholar]
- Zobel A, Kalkbrenner F, Guehmann S, Nawrath M, Vorbrueggen G, Moelling K (1991) Interaction of the v-and c-Myb proteins with regulatory sequences of the human c-myc gene. Oncogene 6: 1397–1407 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3