

Abstract
Interferons (IFNs) regulate diverse cellular functions through activation of the Janus kinase–signal transducer and activator of transcription (JAK–STAT) pathway. Lack of Ubp43, an IFN-inducible ISG15 deconjugating enzyme, leads to IFN hypersensitivity in ubp43−/− mice, suggesting an important function of Ubp43 in downregulation of IFN responses. Here, we show that Ubp43 negatively regulates IFN signaling independent of its isopeptidase activity towards ISG15. Ubp43 functions specifically for type I IFN signaling by downregulating the JAK–STAT pathway at the level of the IFN receptor. Using molecular, biochemical, and genetic approaches, we demonstrate that Ubp43 specifically binds to the IFNAR2 receptor subunit and inhibits the activity of receptor-associated JAK1 by blocking the interaction between JAK and the IFN receptor. These data implicate Ubp43 as a novel in vivo inhibitor of signal transduction pathways that are specifically triggered by type I IFN.
Keywords: interferon (IFN), ISG15, JAK, STAT, UBP43 (USP18)
Introduction
Interferons (IFNs) are secreted pleiotropic cytokines that regulate diverse biological functions, including induction of the antiviral response, inhibition of cell proliferation, and immunomodulatory activities (Platanias and Fish, 1999; Biron, 2001; Sen, 2001; Chawla-Sarkar et al, 2003; Pestka et al, 2004). Type I IFNs signal by binding to a cognate receptor at the cell surface followed by activation of the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway (Darnell et al, 1994; Levy and Darnell, 2002; Shuai and Liu, 2003). JAK1 and TYK2, two members of the JAK family of nonreceptor tyrosine kinases, constitutively associate with the IFNAR2 and IFNAR1 subunits of the type I IFN receptor, respectively. Binding of IFN to the receptor triggers heterodimerization of the receptor chains, juxtaposing JAKs and initiating the phosphorylation cascade (Aaronson and Horvath, 2002). Activated JAK1 phosphorylates TYK2 and they further phosphorylate the cytoplasmic tails of IFNAR proteins, creating a docking site for STAT1/STAT2 binding and their subsequent phosphorylation. Once phosphorylated, STAT1/2 heterodimers disengage from the receptor, form a complex with p48 (IRF9), translocate to the nucleus and induce gene expression through binding to the ìnterferon stimulated response element (ISRE) within the promoters of interferon stimulated genes (ISGs) (Fu, 1995; Larner and Reich, 1996; Decker et al, 2002).
Stringent mechanisms of signal attenuation are essential for ensuring an appropriate and controlled cellular response. Several mechanisms of negative regulation have been implicated in the termination of IFN signaling (Yamada et al, 2003), including IFN receptor ubiquitination by SCFβ-TrCP/HOS E3 ubiquitin ligase and degradation by the lysosomal pathway, dephosphorylation of JAKs and the receptor by SHP-1 and SHP-2, dephosphorylation of STATs by TC45 and PTP1B, inhibition of STAT1 DNA binding by the family of PIAS proteins, and inhibition of JAK kinase activity and their subsequent degradation by SOCS proteins (Greenhalgh and Hilton, 2000; Yasukawa et al, 2000; Kumar et al, 2003; Shuai and Liu, 2005).
Along with other ISGs, IFN-α/β stimulation leads to upregulation of ISG15, a ubiquitin-like protein (Ubl) that conjugates to a number of cellular substrates (Narasimhan et al, 1995, 1996; Ritchie and Zhang, 2004). The conjugation involves an enzymatic cascade that includes an E1 activating enzyme (Ube1L) (Yuan and Krug, 2001), an E2 conjugating enzyme (Ubc8) (Kim et al, 2004; Zhao et al, 2004), and most likely some E3 ligases (Dao and Zhang, 2005; Zou and Zhang, 2006). The conjugation process is also reversible and controlled by an IFN-inducible cysteine protease of the ubiquitin-specific protease (USP) family of enzymes—Ubp43 (USP18) (Malakhov et al, 2002, 2003). Recently, we found that Ubp43 negatively regulates JAK–STAT signaling and may therefore represent a novel type of inhibitor in the IFN pathway (Malakhov et al, 2002, 2003). Ubp43-deficient cells exhibit high levels of ISG15 modified proteins (Ritchie et al, 2002). Furthermore, they are hypersensitive to type I IFN and undergo apoptosis upon IFN stimulation. Lack of Ubp43 results in enhanced and prolonged STAT1 phosphorylation, DNA binding, and increased induction of hundreds of ISGs as confirmed by gene expression microarray data (Malakhova et al, 2003; data in preparation for publication). As the consequence, loss of Ubp43 in mice results in greater resistance to the cytopathic effects caused by a number of viruses including lymphocytic choriomeningitis virus (LCMV), vesicular stomatitis virus (VSV), and Sindbis virus (SNV) (Ritchie et al, 2004). Although Ubp43 apparently inhibits JAK–STAT signaling, the molecular mechanisms of such inhibition are yet to be determined. As isopeptidase activity toward removal of ISG15 from its substrate is the only currently known function of Ubp43 (Malakhov et al, 2002), it would be plausible that the negative regulation of IFN signaling by Ubp43 is mediated by de-conjugation of ISG15. However, ablation of ISG15 or Ube1L in mice does not reverse the phenotype of the Ubp43 knockout (Knobeloch et al, 2005; Osiak et al, 2005; Kim et al, 2006).
In this report, we investigated the functional mechanism of Ubp43 action within the IFN signaling pathway and showed that Ubp43 negatively regulates JAK–STAT signaling independently of its isopeptidase activity. Ubp43 action is strictly specific to type I IFN responses and achieved through a direct interaction between Ubp43 and the IFNAR2 subunit of the receptor. Binding of exogenous and endogenous Ubp43 to IFNAR2 in vivo interferes with the JAK-receptor interaction and leads to inhibition of the downstream phosphorylation cascade and other signaling events. These data point to a mechanistically novel nonenzymatic function of Ubp43 as a specific in vivo inhibitor of cellular responses to type I IFN.
Results
Neither ISG15 conjugation nor Ubp43 isopeptidase activity is required for the negative regulation of IFN signaling by Ubp43
Genetic ablation of Ubp43 in mice causes hypersensitivity to IFN and subsequent hyperactivation of ISGs, suggesting that the Ubp43 protein is a negative regulator of the JAK–STAT signaling pathway (Malakhova et al, 2003). The only reported function of Ubp43 is its enzymatic activity against ISG15 conjugates. Indeed, ubp43-deficient mice show high levels of ISG15-conjugated proteins in most tissues in response to IFN treatment. However, it remains to be determined whether sustained ISG15 conjugation may lead to IFN hypersensitivity in ubp43-null mice or if such hypersensitivity might be mediated by other mechanisms that are independent of Ubp43 enzymatic activity. To address both possibilities, we performed a set of experiments to separate the impact of the ISG15 conjugation process from any ISG15-independent function of Ubp43. For this purpose, we generated MEFs that included parental ubp43+/+ and ubp43−/− MEFs or stably transfected pools of ubp43−/− MEFs expressing Ube1L siRNA, as well as either wild-type Ubp43(wt) or an active site cysteine mutant (c61s) of Ubp43, Ubp43C61S, that is no longer enzymatically active. First, we confirmed that reconstitution of ubp43−/− MEFs with Ube1L siRNA noticeably decreased the degree of ISG15 conjugation, bringing ISGylation to a level comparable with the ubp43+/+ cells upon IFN-β stimulation (Figure 1A, top panel). Reconstitution of ubp43−/− MEFs with constitutively expressed ubp43 (Figure 1A, second panel) resulted in a lower level of ISGylation than that of ubp43+/+ (Figure 1A, top panel). Unexpectedly, expression of the enzymatically inactive mutant of Ubp43 in ubp43−/− MEFs also resulted in lower levels of ISG15 conjugates although the ratio between conjugated and free ISG15 was higher in these cells than in cells expressing wt Ubp43. Given that both wt and mutant forms of Ubp43 were expressed at similar levels (Figure 1A, top two panels), it is likely that the decrease levels of ISG15 conjugation in cells transduced with catalytically inactive Ubp43 reflects an impaired IFN induced expression of free ISG15 and enzymes responsible for ISG15 conjugation and, therefore, might be indicative of inhibition of the JAK–STAT pathway in an isopeptidase-independent manner.
Figure 1.

Negative regulation of IFN signaling by Ubp43 is independent of its enzymatic activity. (A) Total protein extracts were prepared from ubp43+/+ and ubp43−/− MEFs or ubp43−/− MEFs stably expressing Ube1L siRNA, wt murine Ubp43, or Ubp43C61S after mIFN-β (1000 U/ml) treatment for 0′, 30′, and 10 h and analyzed by Western blotting with the respective antibodies. (B) 293T cells were transiently transfected with vector-control (lane 1) or plasmids containing 6xHis-ISG15, HA-Ube1L, and Flag-Ubc8 in the absence (lane 2) or the presence of wt (lane 3) or mutant (c61s) Ubp43-V5 (lane 4). The level of ISG15 conjugation was determined by Western blotting with anti-mISG15 antibodies. Blots were stripped and reprobed with anti-HA, anti-Flag, or anti-V5 antibodies to ensure equal levels of protein expression.
Indeed, reconstitution of ubp43−/− cells with either wt or mutant Ubp43 resulted in a significant inhibition of STAT1 tyrosine phosphorylation on the Tyr701 residue to a level similar to ubp43+/+ cells (Figure 1A, third panel), suggesting that catalytic activity of Ubp43 toward ISG15 conjugates is not required for this inhibition. This conclusion was further corroborated by data showing that downregulation of the total level of ISG15 conjugates in ubp43-deficient cells by inhibiting Ube1L function with specific siRNA did not affect the level of STAT1 phosphorylation compared to the parental ubp43−/− MEFs (Figure 1A). STAT3 and tubulin blots were performed to ensure equal protein loading in this experiment (Figure 1A, fifth and sixth panels).
We also confirmed that Ubp43C61S is enzymatically inactive in vivo. ISG15 conjugation can be artificially induced in 293T cells by overexpression of ISG15 and enzymes involved in its conjugation (Ube1L (E1), and Ubc8 (E2)) in the absence of IFN (Figure 1B, lane 2) (Kim et al, 2004; Zhao et al, 2004). When wt Ubp43 was co-expressed in 293T cells with this artificial ISG15-conjugation system, it significantly lowered the amount of ISG15 conjugates in vivo (Figure 1B, lane 3). In contrast, co-expression of Ubp43C61S did not have any effect on the level of ISG15 conjugation (Figure 1B, lane 4). These data confirmed that the active site cysteine 61 to serine substitution completely abolished the enzymatic activity of Ubp43 towards ISG15.
To determine whether Ubp43 can function as a negative regulator of JAK–STAT signaling independent of the level of ISG15 conjugation at the physiological level, we performed a VSV protection assay (Wong et al, 2001) using MEF pools that included parental ubp43+/+ and ubp43−/− MEFs, as well as ubp43−/− MEFs expressing Ube1L siRNA, wt Ubp43, or Ubp43C61S. Consistent with previous observations (Ritchie et al, 2004), MEF cells deficient for Ubp43 resisted significantly higher VSV titers as compared to ubp43+/+ cells (Figure 2) (105 versus 104 PFUs in untreated control and >1010 versus 108 PFUs in IFN-β treated samples). Reconstitution of ubp43−/− cells with either wt or active site mutated Ubp43 reduced the resistance level back to that of ubp43+/+ cells. However, downregulation of ISG15 conjugation by knocking down Ube1L did not significantly affect cellular resistance to VSV in ubp43−/− cells. These results, together with similar activities exhibited by wt Ubp43 and Ubp43C61S in this assay, indicate that the lack of Ubp43 is important for the IFN-induced resistance of these cells to viral infections whereas ISG15 conjugation mediated by Ube1L is not essential.
Figure 2.
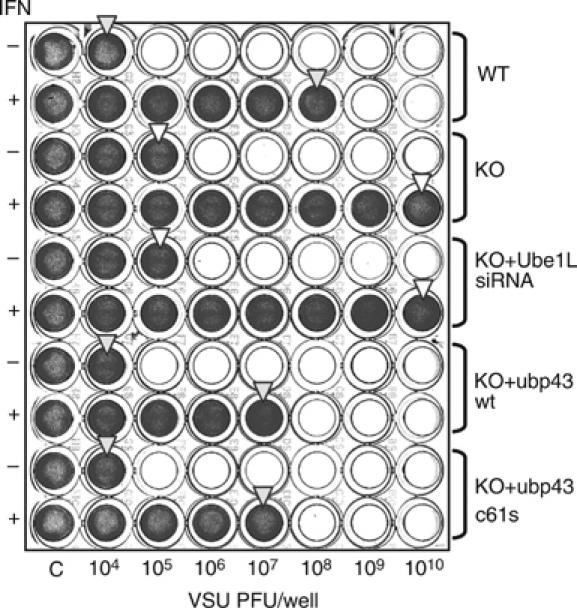
VSV protection assay. ubp43+/+, ubp43−/−, ubp43−/− MEFs with stable expression of Ube1L siRNA, wt Ubp43, or Ubp43C61S were left untreated or treated with 1000 U/ml of mIFN-β for 24 h, followed by mock or VSV infection ranging from 104 to 1010 PFU/well for additional 24 h. Cell viability was assessed by crystal violet staining. The arrows indicate the level of protection.
Ectopic expression of wt or inactive mutant of Ubp43 blocks STAT1 phosphorylation and IFN-mediated gene induction
To determine the effect of forced Ubp43 expression on JAK–STAT signaling, we also generated stably transfected human U3A cells (STAT1-deficient cells, which were selected for this study based on their low basal level of endogenous UBP43) and KT-1 cell pools with different levels of Ubp43 expression. U3A cells constitutively expressing wt Ubp43 or Ubp43C61S (Malakhov et al, 2002) were transiently transfected with STAT1 in combination with an ISRE-driven promoter-luciferase reporter. As shown in Figure 3A, left panel, the expression of either the enzyme active or inactive form of Ubp43 decreased the induction of STAT1 phosphorylation in response to IFN-α in U3A cells. In accordance with this observation, the level of IFN-specific promoter activation was found to be significantly lower in cells expressing wt or mutant Ubp43 (Figure 3A, right panel). Similar data were also obtained from KT-1 cells that stably expressed the wt or mutant Ubp43 (data not shown). Both cell lines showed a substantially reduced IFN response as judged by the level of STAT1 phosphorylation. In contrast, the knockdown of Ubp43 by specific siRNA in KT-1 cells extended the duration of IFN signaling (Figure 3B) mimicking the IFN hypersensitive phenotype observed in ubp43-knockout mouse cells (Figure 1A; Malakhova et al, 2003). These results provide additional genetic evidence in support of our conclusion that Ubp43 negatively regulates JAK–STAT signaling and subsequent activation of ISGs in response to IFN in both murine and human cells in a manner that is independent of Ubp43 enzyme activity toward ISG15 conjugates.
Figure 3.
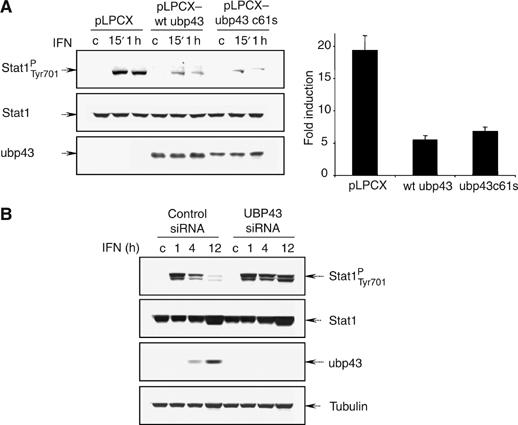
Ectopic expression of Ubp43 blocks STAT1 phosphorylation and IFN-mediated gene induction. (A) STAT1-deficient U3A stable cell lines expressing vector-control, wt mUbp43, or mUbp43C61S were transiently co-transfected by STAT1 and ISRE-driven luciferase reporter plasmid. At 24 h post-transfection, cells were either left untreated or treated with hIFN-α for 15′, 1 h, and 16 h. Level of STAT1 phosphorylation and expression was assessed by Western blotting with the respective antibodies (left). Luciferase activities were measured, normalized, and presented as fold increase of relative luciferase activity in IFN treated cells (at 16 h point) over the untreated controls (right). The error bars indicate the s.d. of the mean. (B) KT-1 cells were stably transfected with control siRNA, hUBP43-specific siRNA. After 1 week of drug selection, cells were either left untreated or treated with hIFN-α for 1, 4 or 12 h respectively. STAT1 phosphorylation and expression was assessed by Western blotting with the respective antibodies. Specific inhibition of endogenous hUBP43 by siRNA in the respective stable lines was confirmed by Western blotting with anti-hUBP43-specific antibodies.
Cell surface expression and rate of degradation of IFN receptor are not altered in Ubp43 deficient cells
Given that de-conjugation of ISG15 does not play a role in the inhibition of JAK–STAT signaling by Ubp43, it is plausible that expression of Ubp43 may either affect IFN receptor downregulation or be involved in the activation/deactivation of JAK. We next tested whether Ubp43 affects ligand-induced downregulation of IFN receptor, which plays a primary role in restricting the extent and duration of IFN signaling. Treatment of cells with IFN was shown to trigger rapid IFNAR1 ubiquitination (Kumar et al, 2003) and degradation (Constantinescu et al, 1994); furthermore, stable IFNAR1 mutants that bear deletions in their C-terminus mediate enhanced responses to IFN-α (Gibbs et al, 1996; Basu et al, 1998).
We first investigated whether Ubp43 could affect the level of the endogenous IFNAR1 and IFNAR2 subunits of the receptor at the cell surface. Since antibodies against extracellular domains of IFN receptor subunits are only available for human proteins, we performed flow cytometry analysis of the cell surface level of both IFNAR1 and IFNAR2 using human KT-1 cells expressing either control siRNA or Ubp43-specific siRNA. Despite highly efficient UBP43 knockdown (Figure 4A), no apparent differences in steady state or IFN-induced surface levels of either IFN receptor subunit were detected in UBP43-defficient KT-1 cells as compared with control cells (Figure 4A), suggesting that Ubp43 is not involved in regulating the cellular surface level of IFN receptors.
Figure 4.
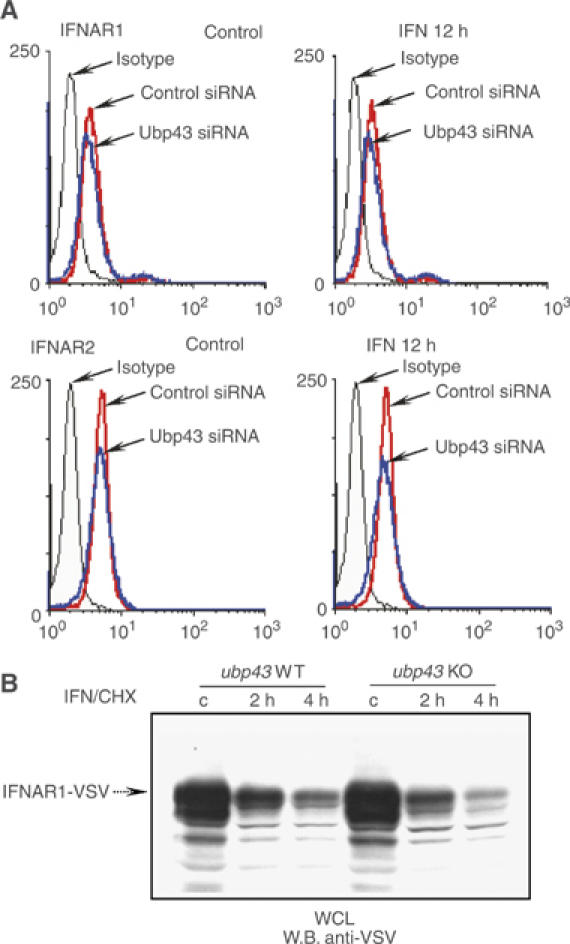
IFN receptor cell surface expression and degradation are not altered in Ubp43 deficient cells. (A) The steady-state or IFN-induced level of the endogenous IFNAR1 (upper left panels) and IFNAR2 (lower left panels) chains was determined by flow cytometry analysis of KT-1 cells, expressing control or UBP43-siRNA by using polyclonal anti-IFNAR1 and monoclonal MMHAR-2 antibodies, respectively. Graphs, corresponding to the isotype-matched control or IFNAR-specific staining in KT-1 control and UBP43-siRNA cells are marked by arrows. (B) Protein extracts were prepared from ubp43+/+ and ubp43−/− MEFs with stable expression of VSV-tagged mIFNAR1 that were either left untreated or treated with IFN and cycloheximide (CHX, to inhibit the protein synthesis) for 2 and 4 h respectively. The rate of IFNAR1 degradation was assessed by Western blotting.
We then analyzed the rate of receptor ubiquitination and degradation in MEF cells that express VSV-tagged murine IFNAR1 (as analysis of endogenous murine IFNAR1 is hindered due to the unavailability of appropriate mIFNAR1-specific antibodies). Neither the half-life of mIFNAR1-VSV (treated with IFN-β and cycloheximide, Figure 4B) nor ubiquitination of this receptor (data not shown) differed between ubp43−/− and ubp43+/+ cells. These results indicate that Ubp43 is not involved in the control of either IFNAR1 ubiquitination or IFNAR1 proteolysis.
Ubp43 inhibits the phosphorylation of JAK
The magnitude and duration of IFN signaling is negatively controlled by phosphatases at different levels such as dephosphorylation of STATs, JAKs, and the receptor. On the other hand, given that JAK activation is the initial event in activation of the JAK–STAT signaling pathway, a sustained activation of JAKs could also positively affect components of this pathway. No difference in dephosphorylation regulation was detected between wt and ubp43−/− cells (data not shown). Therefore, we examined the phosphorylation rate of JAK1 in the presence of IFN and the phosphatase inhibitor sodium orthovanadate. Upon this treatment, we observed that re-expression of Ubp43 in ubp43-deficient MEF cells decreases the extent of JAK1 phosphorylation (Figure 5A). An increase in the magnitude and duration of IFN-induced JAK1 phosphorylation was also observed in the bone marrow cells derived from Ubp43 knockout mice as compared with cells from wt animals (Figure 5B). We further examined the role of Ubp43 in downregulation of the JAK–STAT signaling pathway by assessing JAK activation in human KT-1 cells (Figure 5C). The inhibition of endogenous UBP43 in KT-1 cells by specific siRNA significantly extended the phosphorylation of both JAK1 and TYK2 kinases upon IFN-α stimulation (phosphorylation was still detectable at 4 h of IFN stimulation versus 1 h in control cells). Conversely, KT-1 cells constitutively expressing either the wt or mutant form of Ubp43 exhibited reduced levels of JAK1 and TYK2 phosphorylation. Together, the results suggest that Ubp43 inhibits the tyrosine kinase activity of these JAKs in a manner that does not rely on the protease activity of Ubp43.
Figure 5.
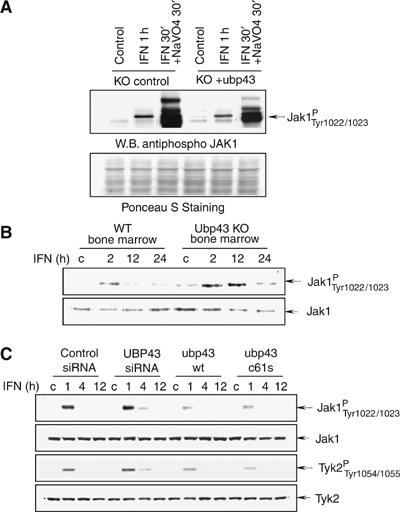
Ubp43 inhibits the activation of JAK-kinases. (A) Whole-cell lysates (WCL) were prepared from ubp43−/− MEFs or ubp43−/− MEFs reconstituted with wt mUbp43 after treatment with mIFN-β alone or mIFN-β+sodium orthovanadate for the indicated period of time. Level of JAK1 phosphorylation was determined by Western blotting using phospho-specific antibodies against JAK1 (pYpY1022/1023) (upper panel). After SDS–PAGE membrane was stained with Ponceau S solution to assure equal protein loading (lower panel). (B) Bone marrow cells were prepared from ubp43+/+ or ubp43−/− mice and incubated with 100 U/ml of mIFN-β for various periods of time as indicated in the figure. JAK1 was immunoprecipitated from WCL and subjected to an in vitro kinase assay, followed by immunoblotting with anti-phosphospecific JAK1 antibodies. Blots were stripped and re-probed with anti-JAK1 antibodies. (C) KT-1 cells stably expressing control siRNA, human UBP43-specific siRNA, plasmids encoding wt Ubp43 or Ubp43C61S mutant protein were stimulated with hIFN-α (1000 U/ml) for the indicated periods of time. WCL were subjected to immunoblotting with antiphosphospecific JAK1 and TYK2 antibodies. Blots were stripped and re-probed with anti-JAK1 and TYK2 antibodies respectively to assure equal protein loading.
Ubp43 attenuates IFN signaling through the specific interaction with IFNAR2 subunit of the receptor
Ubp43 deficient cells are hypersensitive to type I IFN, but not to type II IFN (Malakhov et al, 2002, 2003). In line with these findings, we did not observe any effects of Ubp43 knockdown in human cells on the activation of the JAK–STAT pathway in response to IFNγ, IL-6, or IL-12 (data not shown). Furthermore, forced expression of Ubp43 did not affect the autophosphorylation of either Jak1 or Tyk2 induced by overexpression of the respective kinases in 293T cells in the absence of IFN stimulation (data not shown). The ability of Ubp43 to attenuate the activity of JAK in a type I IFN dependent manner suggested that it may be associated with the type I IFN specific component of the IFN-receptor complex and that both IFNAR1/IFNAR2 receptor chains represent potential targets for Ubp43 action. To determine whether these proteins are in a complex with Ubp43, we performed reciprocal co-precipitation assays for the assessment of Ubp43-receptor interactions. Specific binding of the IFNAR2 subunit of the type I IFN receptor to Ubp43 was detected (Figure 6A, middle). Neither IFNAR1 (Figure 6A, left) nor IFNGR1 (type II IFN) receptor subunits (Figure 6A, right) were able to interact with Ubp43. Furthermore, both wt Ubp43 and Ubp43C61S were found to bind to IFNAR2 to a similar extent (Supplementary Figure S1).
Figure 6.

Ubp43 interacts with IFNAR2 receptor subunit. (A) 293T cells were transiently transfected with Flag-IFNAR1, HA-IFNAR2, or Flag-IFNGR1 and either GST control or GST-Ubp43. Reciprocal immunoprecipitations (I.P.) were performed using anti-Flag/HA or anti-GST antibodies. Whole-cell lysates (WCL) or immunoprecipitated complexes were subjected to immunoblotting with anti-HA antibodies (top middle panel), anti-Flag (top left & right panels) or anti-GST (bottom panel) antibodies, respectively. (B) Transient co-transfections of 293T cells were performed using HA-tagged IFNAR2 and various deletion mutants of Ubp43 (a.a. positions are indicated in the figure). Ubp43 deletion constructs used for this study are schematically represented in the top panel. WCL or immunoprecipitated (anti-HA) complexes were subjected to immunoblotting with anti-HA antibodies (middle panel) or anti-GST (bottom panel) antibodies, respectively. (C) wt Ubp43 and Ubp43 mutants: D331K340-AA, R350R352R354-AAA, K364-A (positions of a.a. substitutions are graphically shown on the top panel of the figure) were co-expressed with GST-IFNAR2 (a.a. 265–515) in 293T cells. I.P. were performed using anti-HA antibodies followed by immunoblotting with anti-HA antibodies (top) to verify equal loading and anti-GST-antibodies (bottom). (D) U3A cells were transiently co-transfected by the combination of ISRE-driven luciferase reporter plasmid, STAT1, and either vector-control, wt Ubp43, or Ubp43 mutants: D331K340-AA (DK), R350R352R354-AAA (RRR) and K364-A (K). At 24 h post-transfection, cells were either left untreated or treated with hIFN-α for 24 h. Luciferase activities were measured, normalized, and presented as fold increase of relative luciferase activity in IFN treated cells over the untreated controls (average of 4 independent experiments). The error bars indicate the s.d. of the mean.
To further delineate the region of Ubp43 responsible for the interaction with IFNAR2, a panel of GST fusion proteins encoding portions of Ubp43 was used for binding studies (Figure 6B, top). Following co-expression/co-immunoprecipitation assays, we found that the Ubp43–IFNAR2 interaction was mediated by the C-terminus of Ubp43, since the deletion of the N-terminal portion of Ubp43 up to amino acid (a.a.) 217 did not affect its interaction with IFNAR2 (Figure 6B, middle panel). Meanwhile, GST-fusions expressing various fragments encompassing the N-terminal two-thirds of Ubp43 protein (up to a.a. 243) or GST alone were not able to bind IFNAR2 (Figure 6B, middle panel, lanes 1, 4 and 5). Further truncation from the amino terminus revealed that a 56-a.a. fragment (312–368) of Ubp43 binds to IFNAR2, as efficiently as the full-length protein (Figure 6B, bottom panel, lane 5). From these data we concluded that the very C-terminus of Ubp43 provides the main interaction motif for the association with IFNAR2 and residues in the region of a.a.312–368 might be critical for the interaction.
To further characterize the Ubp43–IFNAR2 interaction domain and its role in IFN signaling, several charged residues potentially present on the surface of the Ubp43 312–368 fragment (based on the alignment with HAUSP deubiquitinating enzyme, for which the crystal structure is available (Hu et al, 2002)) were substituted to alanines. As a result, three Ubp43 mutants were made (i.e. D331K340-AA, R350R352R354-AAA and K364-A) within the IFNAR2 interaction region and these mutants were tested for their ability to interact with IFNAR2. Co-immunoprecipitation assays revealed that mutations at the positions 331–340 or 364 of Ubp43 did not affect its ability to interact with IFNAR2; however, binding of Ubp43 was diminished when amino-acid residues at positions 350–354 were substituted for alanines (Figure 6C, right panel, compare lanes 2 and 4), indicating that charged residues at positions 350–354 of Ubp43 were critical for its association with IFNAR2 in vivo. These data prompted us to examine if mutation of arginines within the 350–354 a.a. span would also reduce the potency of Ubp43 in downregulation of IFN response. To test this, U3A cells constitutively expressing wt or charged amino acids mutants of Ubp43 within the 312–368 region described above, were co-transfected with STAT1 in combination with an ISRE-driven promoter-luciferase reporter. As shown in Figure 6D, expression of either wt or D331K340-AA and K364-A mutants of Ubp43 noticeably decreased the level of IFN-specific promoter activation in response to IFN in U3A cells. However, the R350R352R354-AAA mutant of Ubp43 (that exhibited lower IFNAR2 binding capacity) showed substantially lower efficiency in inhibiting IFN signaling. These data provided further evidence that Ubp43 attenuates IFN signaling through the specific interaction with the IFNAR2 subunit of the receptor.
Ubp43 binds to the IFNAR2 subunit within the Box1–Box2 region and interferes with JAK1-receptor interaction
In order to determine the functional consequences of Ubp43–IFNAR2 interaction, we further identified the region of IFNAR2 that is required for Ubp43 binding. We generated constructs that expressed various fragments of the cytoplasmic portion of IFNAR2 as GST-fusions (Figure 7A, top panel). Deletion of either Box1 alone or the Box1–Box2 regions of IFNAR2 abrogated its ability to interact with Ubp43 in co-immunoprecipitation assays (Figure 7A, bottom), indicating that the Box1–Box2 domain of the receptor is required for Ubp43 binding. Interestingly, the same domain of IFNAR2 has been reported to be a critical interaction surface for JAK1 binding (Domanski et al, 1997; Usacheva et al, 2002), suggesting that Ubp43–IFNAR2 complex formation could compete with JAK1 receptor binding, consequently inhibiting activation and the downstream signaling cascade. To test this hypothesis, we co-expressed JAK1 and IFNAR2 in 293T cells in the absence or presence of increasing amounts of Ubp43 and performed a set of reciprocal pull down assays either for JAK1 (Figure 7B, left bottom panel) or IFNAR2 (Figure 7B, right bottom panel). As shown in the top panels (left and right) of Figure 7B, all proteins were expressed at the expected levels. We then confirmed that both JAK1 (Figure 7B, left bottom panel, lane 1) and IFNAR2 (Figure 7B, right bottom panel, lane 1) were able to interact with their respective partners in the absence of Ubp43 by co-immunoprecipitation experiments. Ubp43, however, was capable of interfering with JAK1–IFNAR2 complex formation in a dose-dependent manner (Figure 7B, left and right bottom panels, lanes 2–4). Furthermore, forced expression of Ubp43 had no effect on the interaction between JAK1 and IFNGR1 (data not shown). Given that modulation of Ubp43 level did not affect the extent of Jak–Stat signaling triggered by IFNγ (data not shown), these data are consistent with our hypothesis that Ubp43 specifically regulates type I IFN signaling via the interaction with a cognate receptor.
Figure 7.
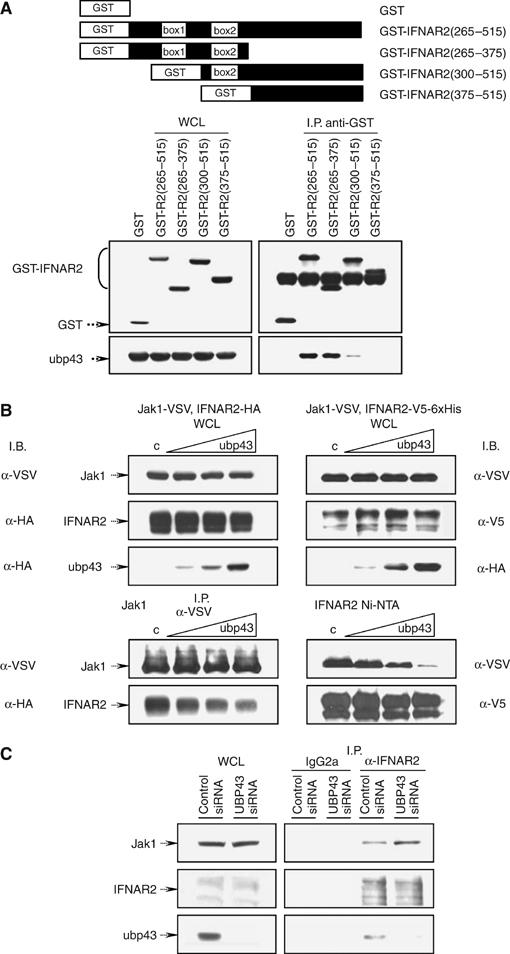
Ubp43 competes with JAK1 for receptor binding. (A) 293T cells were transiently co-transfected with HA-Ubp43 and GST or GST-IFNAR2 truncation constructs schematically presented in the top panel of the figure. Whole cell lysate (WCL) (left panels) or immunoprecipitated complexes (right panels) were subjected to immunoblotting against GST or HA-tag, respectively. (B) 293T cells were transiently co-transfected with JAK1-VSV and IFNAR2-HA (left) or JAK1-VSV and IFNAR2-V5-6xHis (right) in the absence or presence of increasing concentration of HA-Ubp43 followed by immunoprecipitation using antibodies against VSV-tagged JAK1 (left, bottom panels) or Ni-NTA purification of 6xHis-tagged IFNAR2 (right, bottom panels). WCL (top panels) or immunoprecipitated complexes (bottom panels) were subjected to immunoblotting with antibodies indicated in the figure. (C) Protein extracts from stable KT-1 transfectants expressing either control siRNA or UBP43 specific siRNA and treated with hIFN-α for 4 h were used for the immunoprecipitations with control IgG2a antibodies or with anti-IFNAR2 antibodies. WCL and immunoprecipitates were subjected to immunoblotting with anti-JAK1 (top panel), anti-IFNAR2 (middle panel), and anti-Ubp43 antibodies (bottom panel).
In order to determine whether Ubp43 confers this mode of regulation under physiological conditions, we used the RNAi approach followed by the assessment of interaction between endogenous IFNAR2 and JAK1. First, we confirmed that endogenous Ubp43 specifically interacts with endogenous IFNAR2 upon IFN stimulation in human KT-1 cells (Figure 7C, anti-IFNAR2 versus IgG2a-control immunoprecipitaion). Furthermore, knockdown of Ubp43 in KT-1 cells by Ubp43-specific siRNA increased the association between endogenous IFNAR2 and JAK1 (Figure 7C), indicating that the endogenous Ubp43 is capable of competing with JAK1 for IFN receptor binding. These data provide the genetic evidence to support the hypothesis that Ubp43-mediated titration of JAKs away from the receptor may inhibit downstream phosphorylation cascade events.
Discussion
IFNs regulate diverse cellular processes involved in cell growth, differentiation, and host defense by recruiting JAK kinases and STAT transcription factors to the specific receptors present at the cell surface (Ivashkiv and Hu, 2004; Rawlings et al, 2004). In order to achieve an appropriate biological response to a diverse range of extracellular stimuli, cells acquired a sophisticated and strictly controlled network involving a great variety of proteins that negatively regulate cytokine signal transduction by various means (Greenhalgh and Hilton, 2001; Yamada et al, 2003). Our previous studies demonstrated that ubp43-deficient cells are hypersensitive to type I IFN treatment, indicating that Ubp43 is a novel negative regulator of IFN signaling (Malakhova et al, 2003). Furthermore, Ubp43 is a specific ISG15 protease that removes the ubiquitin-like modifier ISG15 from targeted proteins and therefore controls the level of ISG15 conjugated proteins in cells (Malakhov et al, 2002). Indeed, ubp43-deficient cells have higher basal and IFN induced levels of ISGylated proteins. Naturally, the level of protein ISG15 modification was thought to be linked to the hypersensitivity of these cells to IFN. However, one of the first questions raised was whether sustained ISG15 conjugation is the cause of IFN hypersensitivity or whether the negative regulation of JAK–STAT signaling by Ubp43 is mediated by an ISG15-independent mechanism. Analyses of stable ubp43−/− MEF cell lines reconstituted with either ISG15 activating enzyme Ube1L siRNA or Ubp43 demonstrated that the inhibition of ISG15 conjugation by knocking down Ube1L in ubp43−/− cells cannot reverse the IFN hypersensitive phenotype of ubp43-deficient cells. On the other hand, the reintroduction of wt or isopeptidase active site mutated Ubp43 protein into ubp43−/− MEFs can effectively inhibit IFN sensitivity to a level comparable with wt cells. Such an observation is also confirmed in two different human cell lines used in this study. When either wt or the enzyme active site mutated Ubp43 is ectopically expressed in U3A or KT-1 cells, a specific inhibition of STAT1 phosphorylation and subsequent activation of ISGs in response to IFN is observed. These results clearly demonstrate that Ubp43 downregulates the cellular response to IFN independently of its enzymatic activity towards ISG15. These data were also confirmed in ubp43−/− ube1L−/− double knockout mice, where MEF cells deficient for both Ubp43 and ISG15 conjugation retained the same level of IFN sensitivity as was observed in ubp43-knockout cells (Kim et al, 2006).
In recent years, several proteins within the JAK–STAT pathway were found to be targets for ISG15 modification, including JAKs and all STAT family members (Malakhov et al, 2003 and our unpublished data), implying that modification of these proteins by ISG15 may influence their structure, function, and overall signaling in general. Our current data suggest that high accumulation of ISG15 conjugates in ubp43-deficient cells is most likely a result but not a main cause of the IFN hypersensitivity observed in these cells.
Since the duration of IFN signaling is known to be controlled at multiple levels, including receptor proteolysis and activity of kinases and phosphatases, we further examined which particular step of the IFN signaling cascade involves Ubp43. We found that Ubp43 inhibits JAK–STAT signaling at the level of the IFN receptor, but is not involved in the control of receptor proteolysis or in sustaining the level of the IFN receptor at the cellular surface. Importantly, Ubp43 is capable of inhibiting the activation of the receptor-associated kinases—JAK1 and TYK2. While Ubp43 deficiency results in sustained phosphorylation of these JAKs in response to IFN, the forced expression of either wt or active site mutated Ubp43 reduces the phosphorylation level of these kinases and their downstream phosphorylation and transcriptional events as well as resistance to viral infection.
The ability of Ubp43 to attenuate the activity of JAKs suggests that it may associate with one or more components of the type I IFN-receptor complex and both JAKs and the receptor subunits represent potential targets for Ubp43 action. It is tempting to hypothesize that the direct binding of Ubp43 to JAKs may cause inhibition of its tyrosine kinase activity. However, further investigation did not support this theory since Ubp43 was not able to inhibit JAK1 or TYK2 activities that were auto-induced by constitutive overexpression of the respective kinases (data not shown). In addition, Ubp43 was found to act specifically for type I IFN signaling and did not inhibit the IFN-γ, IL-6, or IL-12 signaling pathways; even though all tested cytokines required JAK1 or TYK2 for signal transduction. These results suggested that inhibition of JAK phosphorylation by Ubp43 depended on the molecular specifics of type I IFN signaling and was likely to be achieved through an interaction between Ubp43 and type I IFN specific components of the IFN-receptor complex. Indeed, further investigation revealed that Ubp43 specifically interacted with the IFNAR2 chain of the type I IFN receptor, but not with IFNAR1 or IFNGR1. The physiologic effects of disrupting the Ubp43–IFNAR2 interaction were also investigated. The most important result of these studies is the identification of an Ubp43 mutant that cannot efficiently bind IFNAR2 and impair IFN-dependent induction of an ISRE-linked reporter gene. This result strongly suggests that the Ubp43–IFNAR2 interaction is critical for Ubp43-dependent inhibition of JAK–STAT signaling. Additional studies will be required to delineate the structure-function relationships of this interaction.
In order to determine the functional consequences of the Ubp43–IFNAR2 interaction, we identified the region of IFNAR2 required for Ubp43 binding. We found that the Ubp43 binding site is located in the membrane-proximal region of IFNAR2, covering the Box1–Box2 motifs of the receptor. The same domain of IFNAR2 is essential for the interaction with JAK1 (Domanski et al, 1997; Usacheva et al, 2002), suggesting that Ubp43 could compete with JAK1 for receptor binding, consequently inhibiting its activation and the downstream intracellular signaling. Indeed, our experimental data from co-expression experiments demonstrate that Ubp43 inhibits the formation of the JAK1–IFNAR2 complex in a dose-dependent manner, suggesting that titration of JAKs away from the receptor may account for the observed inhibition of the downstream phosphorylation cascade and therefore represent the putative mode of Ubp43 action in the negative regulation of IFN signaling. This hypothesis has been corroborated by genetic evidence showing that knockdown of endogenous Ubp43 promotes the interaction between endogenous JAK1 and endogenous receptor subunit. It is also possible that, as in the case of SOCS proteins (Greenhalgh and Hilton, 2000; Yasukawa et al, 2000; Kumar et al, 2003; Shuai and Liu, 2005), there are multiple roles for Ubp43 in regulating signal transduction, including its function in regulating protein ISGylation. Indeed, recent report clearly point to the role of ISG15 in the antiviral defense (Lenschow et al, 2005). Further studies, such as knockin of the enzymatically inactive ubp43 into the ubp43 locus, will provide additional information about different aspects of Ubp43 in cellular innate immune responses.
The ability of Ubp43 to inhibit signal transduction and ultimately the biological response to IFN suggests that, like other known negative regulators of signal transduction, such as phosphatases (SHP-1, SHP-2, TC45, and PTP1B), SOCS and PIAS proteins, Ubp43 plays an important role in controlling the magnitude and/or duration of cellular responses to type I IFN. The evidence provided here, as well as our previous findings, suggests that Ubp43 acts in a classic negative feedback loop for IFN signal transduction. Like other genes such as SOCS, Ubp43 is induced by IFNs through the activation of the JAK–STAT pathway (Li et al, 2000; Kang et al, 2001). Once induced and expressed, Ubp43 can directly bind to the IFN receptor and inhibit the receptor interaction with JAK, thereby reducing the phosphorylation of the receptor and STATs and suppressing signal transduction and downstream biological responses. Ubp43 deficient mice are highly resistant to viral/bacterial infection and cancer development in several experimental mouse models (Ritchie et al, 2004; Kim et al, 2005;Yan and Zhang, data in preparation). It is of added importance that, unlike phosphatases, SOCS and PIAS that affect cellular responses to many cytokines, Ubp43-mediated inhibition is specific for the type I IFN signaling. Given that specificity and the fact that type I IFN are widely used as antiviral, anticancer, and immunomodulatory agents in therapy of chronic viral infections, malignancies, and relapsing-remitting multiple sclerosis, Ubp43 appears to be a promising therapeutic target for improving the benefits of treatment.
Materials and methods
Details provided as Supplementary data.
Supplementary Material
Supplementary Figure S1
Acknowledgments
We thank Drs Ernest Borden, James Darnell, Ueli Gubler, John Krolewski, Sandra Pellegrini, Lawrence Pfeffer, George Stark, and Gilles Uze for antibodies, cell lines, and DNA constructs, and members of Zhang lab for valuable discussions. This work was supported by research funding from National Institutes of Health (CA079849 to DEZ and CA092900 to SYF) and Department of Defense (DAMD17-03-1-0269 to DEZ). The Stein Endowment Fund has partially supported the departmental molecular biology service laboratory for DNA sequencing and oligonucleotide synthesis. This is manuscript 17422-MEM from The Scripps Research Institute.
References
- Aaronson DS, Horvath CM (2002) A road map for those who don't know JAK–STAT. Science 296: 1653–1655 [DOI] [PubMed] [Google Scholar]
- Basu L, Yang CH, Murti A, Garcia JV, Croze E, Constantinescu SN, Mullersman JE, Pfeffer LM (1998) The antiviral action of interferon is potentiated by removal of the conserved IRTAM domain of the IFNAR1 chain of the interferon alpha/beta receptor: effects on JAK–STAT activation and receptor down-regulation. Virology 242: 14–21 [DOI] [PubMed] [Google Scholar]
- Biron CA (2001) Interferons alpha and beta as immune regulators—a new look. Immunity 14: 661–664 [DOI] [PubMed] [Google Scholar]
- Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC (2003) Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 8: 237–249 [DOI] [PubMed] [Google Scholar]
- Constantinescu SN, Croze E, Wang C, Murti A, Basu L, Mullersman JE, Pfeffer LM (1994) Role of interferon-alpha/beta receptor chain-1 in the structure and transmembrane signaling of the interferon-alpha/beta receptor complex. Proc Natl Acad Sci USA 91: 9602–9606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao CT, Zhang DE (2005) ISG15: a ubiquitin-like enigma. Front Biosci 10: 2701–2722 [DOI] [PubMed] [Google Scholar]
- Darnell JE Jr, Kerr IM, Stark GR (1994) Jak–STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264: 1415–1421 [DOI] [PubMed] [Google Scholar]
- Decker T, Stockinger S, Karaghiosoff M, Muller M, Kovarik P (2002) IFNs and STATs in innate immunity to microorganisms. J Clin Invest 109: 1271–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanski P, Fish E, Nadeau OW, Witte M, Platanias LC, Yan H, Krolewski J, Pitha P, Colamonici OR (1997) A region of the beta subunit of the interferon alpha receptor different from box 1 interacts with Jak1 and is sufficient to activate the Jak–Stat pathway and induce an antiviral state. J Biol Chem 272: 26388–26393 [DOI] [PubMed] [Google Scholar]
- Fu XY (1995) A direct signaling pathway through tyrosine kinase activation of SH2 domain-containing transcription factors. J Leukoc Biol 57: 529–535 [DOI] [PubMed] [Google Scholar]
- Gibbs VC, Takahashi M, Aguet M, Chuntharapai A (1996) A negative regulatory region in the intracellular domain of the human interferon-alpha receptor. J Biol Chem 271: 28710–28716 [DOI] [PubMed] [Google Scholar]
- Greenhalgh CJ, Hilton DJ (2000) The regulation of cytokine signaling by SOCS proteins. Immunologist 8: 45–47 [Google Scholar]
- Greenhalgh CJ, Hilton DJ (2001) Negative regulation of cytokine signaling. J Leukocyte Biol 70: 348–356 [PubMed] [Google Scholar]
- Hu M, Li P, Li M, Li W, Yao T, Wu JW, Gu W, Cohen RE, Shi Y (2002) Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 111: 1041–1054 [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB, Hu XY (2004) Signaling by STATs. Arthritis Res Therapy 6: 159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Jiang H, Wu Q, Pestka S, Fisher PB (2001) Cloning and characterization of human ubiquitin-processing protease-43 from terminally differentiated human melanoma cells using a rapid subtraction hybridization protocol RaSH. Gene 267: 233–242 [DOI] [PubMed] [Google Scholar]
- Kim KI, Giannakopoulos NV, Virgin HW, Zhang DE (2004) Interferon-inducible ubiquitin E2, Ubc8, is a conjugating enzyme for protein ISGylation. Mol Cell Biol 24: 9592–9600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KI, Malakhova OA, Hoebe K, Yan M, Beutler B, Zhang DE (2005) Enhanced antibacterial potential in UBP43-deficient mice against salmonella typhimurium infection by up-regulating type I IFN signaling. J Immunol 175: 847–854 [DOI] [PubMed] [Google Scholar]
- Kim KI, Yan M, Malakhova O, Luo JK, Shen MF, Zou W, de la Torre JC, Zhang DE (2006) Ube1L and protein ISGylation are not essential for alpha/beta interferon signaling. Mol Cell Biol 26: 472–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobeloch KP, Utermohlen O, Kisser A, Prinz M, Horak I (2005) Reexamination of the role of ubiquitin-like modifier ISG15 in the phenotype of UBP43-deficient mice. Mol Cell Biol 25: 11030–11034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KGS, Tang W, Ravindranath AK, Clark WA, Croze E, Fuchs SY (2003) SCFHOS ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. EMBO J 22: 5480–5490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larner A, Reich NC (1996) Interferon signal transduction. Biotherapy 8: 175–181 [DOI] [PubMed] [Google Scholar]
- Lenschow DJ, Giannakopoulos NV, Gunn LJ, Johnston C, O'Guin AK, Schmidt RE, Levine B, Virgin HW (2005) Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus infection in vivo. J Virol 79: 13974–13983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Darnell JE Jr (2002) Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3: 651–662 [DOI] [PubMed] [Google Scholar]
- Li XL, Blackford JA, Judge CS, Liu M, Xiao W, Kalvakolanu DV, Hassel BA (2000) RNase-L-dependent destabilization of interferon-induced mRNAs. A role for the 2-5A system in attenuation of the interferon response. J Biol Chem 275: 8880–8888 [DOI] [PubMed] [Google Scholar]
- Malakhov MP, Kim KI, Malakhova OA, Jacobs BS, Borden EC, Zhang DE (2003) High-throughput immunoblotting—ubiquitin-like protein ISG15 modifies key regulators of signal transduction. J Biol Chem 278: 16608–16613 [DOI] [PubMed] [Google Scholar]
- Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE (2002) UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem 277: 9976–9981 [DOI] [PubMed] [Google Scholar]
- Malakhova OA, Yan M, Malakhov MP, Yuan YZ, Ritchie KJ, Kim KI, Peterson LF, Shuai K, Zhang DE (2003) Protein ISGylation modulates the JAK–STAT signaling pathway. Genes Dev 17: 455–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan J, Potter JL, Haas AL (1996) Conjugation of the 15-kDa interferon-induced ubiquitin homolog is distinct from that of ubiquitin. J Biol Chem 271: 324–330 [DOI] [PubMed] [Google Scholar]
- Narasimhan J, Rasmussen JL, Haas AL (1995) Conjugation of Ucrp (Isg15) to cellular proteins. FASEB J 9: A1472 [Google Scholar]
- Osiak A, Utermohlen O, Niendorf S, Horak I, Knobeloch KP (2005) ISG15, an interferon-stimulated ubiquitin-like protein, is not essential for STAT1 signaling and responses against vesicular stomatitis and lymphocytic choriomeningitis virus. Mol Cell Biol 25: 6338–6345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S, Krause CD, Walter MR (2004) Interferons, interferon-like cytokines, and their receptors. Immunol Rev 202: 8–32 [DOI] [PubMed] [Google Scholar]
- Platanias LC, Fish EN (1999) Signaling pathways activated by interferons. Exp Hematol 27: 1583–1592 [DOI] [PubMed] [Google Scholar]
- Rawlings JS, Rosler KM, Harrison DA (2004) The JAK/STAT signaling pathway. J Cell Sci 117: 1281–1283 [DOI] [PubMed] [Google Scholar]
- Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, de la Torre JC, Zhang DE (2004) Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat Med 10: 1374–1378 [DOI] [PubMed] [Google Scholar]
- Ritchie KJ, Malakhov MP, Hetherington CJ, Zhou L, Little MT, Malakhova OA, Sipe JC, Orkin SH, Zhang DE (2002) Dysregulation of protein modification by ISG15 results in brain cell injury. Genes Dev 16: 2207–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie KJ, Zhang DE (2004) ISG15: the immunological kin of ubiquitin. Semin Cell Dev Biol 15: 237–246 [DOI] [PubMed] [Google Scholar]
- Sen GC (2001) Viruses and interferons. Annu Rev Microbiol 55: 255–281 [DOI] [PubMed] [Google Scholar]
- Shuai K, Liu B (2003) Regulation of JAK–STAT signalling in the immune system. Nat Rev Immunol 3: 900–911 [DOI] [PubMed] [Google Scholar]
- Shuai K, Liu B (2005) Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol 5: 593–605 [DOI] [PubMed] [Google Scholar]
- Usacheva A, Sandoval R, Domanski P, Kotenko SV, Nelms K, Goldsmith MA, Colamonici OR (2002) Contribution of the Box 1 and Box 2 motifs of cytokine receptors to Jak1 association and activation. J Biol Chem 277: 48220–48226 [DOI] [PubMed] [Google Scholar]
- Wong AHT, Durbin JE, Li SY, Dever TE, Decker T, Koromilas AE (2001) Enhanced antiviral and antiproliferative properties of a STAT1 mutant unable to interact with the protein kinase PKR. J Biol Chem 276: 13727–13737 [DOI] [PubMed] [Google Scholar]
- Yamada S, Shiono S, Joo A, Yoshimura A (2003) Control mechanism of JAK/STAT signal transduction pathway. Febs Lett 534: 190–196 [DOI] [PubMed] [Google Scholar]
- Yasukawa H, Sasaki A, Yoshimura A (2000) Negative regulation of cytokine signaling pathways. Annu Rev Immunol 18: 143–164 [DOI] [PubMed] [Google Scholar]
- Yuan WM, Krug RM (2001) Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J 20: 362–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Beaudenon SL, Kelley ML, Waddell MB, Yuan WM, Schulman BA, Huibregtse JM, Krug RM (2004) The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-alpha/beta-induced ubiquitin-like protein. ProcNatl Acad Sci USA 101: 7578–7582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Zhang DE (2006) The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J Biol Chem 281: 3989–3994 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1