

Abstract
Gfi1b is a 37 kDa nuclear protein with six C2H2 zinc-finger domains that can silence transcription upon binding to specific target gene promoters. Here we show by using a chromatin immunoprecipitation and cloning protocol that Gfi1b also binds to γ-satellite sequences that mainly occur in pericentric heterochromatin. Immuno-FISH experiments demonstrated that Gfi1b is localized at foci of pericentric heterochromatin identified by DAPI staining. Elevated levels of Gfi1b correlated with increased histone H3 lysine 9 dimethylation at sites neighboring γ-satellite sequences but also at Gfi1b target gene promoters. In Gfi1b-deficient cells, however, a decrease of histone H3 lysine 9 trimethylation and a loss of heterochromatic structures was observed. Strikingly, we found that Gfi1b binds to both SUV39H1 and G9A histone methyl transferases, which provides a direct link between histone methylation and Gfi1b at heterochromatic and euchromatic sites. We propose that Gfi1b functions in heterochromatin formation and silencing of euchromatic transcription by recruiting histone methyl transferases to either γ-satellite sequences or specific target gene promoters.
Keywords: Gfi1 proteins, heterochromatin, histone methylation, satellite sequences, transcriptional silencing
Introduction
Gfi1 and Gfi1b are two very similar transcription factors that are encoded by two different single-copy genes. Both proteins are exclusively localized in the nucleus and can act as transcriptional repressors (for a review see Duan and Horwitz, 2003a; Möröy, 2005). Two clearly identifiable domains are present in Gfi1 and Gfi1b: six C-terminal C2H2 type zinc-finger domains and a characteristic stretch of 20 amino acids, called SNAG domain, at their N-termini. The sequences of both domain types are almost entirely conserved, suggesting a close functional relationship between both transcription factors. However, the amino-acid stretch that separates both the SNAG and zinc-finger domains comprises 234 amino acids in Gfi1 and 144 amino acids in Gfi1b where virtually no sequence similarity exists. Therefore, this area might confer some functional difference possibly by providing a protein–protein interaction domain. Binding site selection from oligonucleotide libraries has revealed that both proteins can recognize the same AATC core DNA sequence (Grimes et al, 1996; Zweidler-McKay et al, 1996; Tong et al, 1998). Gfi1 and Gfi1b do efficiently repress reporter genes appended to this DNA binding site and the repression depends on the presence of the SNAG domain and an intact zinc-finger domain (Grimes et al, 1996; Tong et al, 1998).
Infection of lymphoid cells in culture or mice with the non-acute transforming Moloney murine leukemia virus led to the discovery of the Gfi1 gene, as it is one of the major target sites for proviral integration (Schmidt et al, 1996; Zörnig et al, 1996; Scheijen et al, 1997). Subsequently, by virtue of sequence homology, the Gfi1b gene was found in the murine and human genome (Rödel et al, 1998; Tong et al, 1998). Whereas Gfi1 is expressed in T cells, monocytes and granulocytes, Gfi1b is most prominently expressed in cells of the fetal and adult erythroid lineage (Osawa et al, 2002; Saleque et al, 2002). In the B-cell lineage, there is some overlap of the expression of both proteins in early developmental stages, but both Gfi1 and Gfi1b are absent in peripheral mature B cells and only re-expression of Gfi1 is seen after induction with antigen or LPS (Yücel et al, 2004). Loss-of-function mutants in mice revealed a complex phenotype for both Gfi1- and Gfi1b-deficient animals, which reflected their cell-type-specific expression pattern. The loss of Gfi1b leads to early embryonic death at about stage 13.5–14.5 dpc, mainly owing to the lack of definitive erythropoiesis and the absence of megakaryocytes (Saleque et al, 2002). Hence, the consequences of Gfi1b deletion in adult organ development is still an unresolved question and can only be assessed by conditional gene targeting. By contrast, Gfi1-deficient mice are viable but lack granulocytes and have a defect in the development of early pre-T cells (Karsunky et al, 2002; Hock et al, 2003; Yücel et al, 2003). In addition, Gfi1 is also expressed in the nervous system, most prominently in hair cells of the inner ear, which leads to deafness in Gfi1 knockout mice (Wallis et al, 2003; Hertzano et al, 2004).
A number of target gene candidates that harbor promoter sequences with an ‘AATC' core containing binding site have been determined for Gfi1 (Duan and Horwitz, 2003b; Yücel et al, 2003) and Gfi1b (Jegalian and Wu, 2002). In the present study, we have used chromatin immunoprecipitation (Ch-IP) to identify direct binding sites of Gfi1b within the genome. We found that Gfi1b can bind to γ-satellite sequences similar to the zinc-finger transcription factor Ikaros. We have assessed the consequences of this new activity of Gfi1b and present evidence that Gfi1b might be involved in transcriptional silencing by altering histone methylation patterns. We present evidence that Gfi1b binds to two important histone methyltransferases: G9A, which can induce histone H3K9 dimethylation on euchromatic genes (Tachibana et al, 2002; Rice et al, 2003), and SUV39H1, which maintains histone methylation at pericentric heterochromatin (Jenuwein and Allis, 2001; Peters et al, 2001, 2003; Lachner et al, 2003). Moreover, we find that loss of Gfi1b correlates with a decreased histone methylation and heterochromatinization in nuclei of erythroid precursor cells, which critically depend on Gfi1b to maturate into definitive erythrocytes.
Results
Chromatin immunoprecipitation reveals γ-satellite sequences as direct Gfi1b targets
Wild-type (wt) murine splenocytes and splenocytes from vav-Gfi1b transgenic mice that constitutively express high levels of Gfi1b (Vassen et al, 2005; Supplementary Figure S1) were used as chromatin sources (Figure 1A). After the precipitation of protein–DNA complexes with an anti-Gfi1b antibody and reverse crosslinking, linkers were added to the precipitated DNA and the ligation products were amplified by PCR and subcloned (Figure 1A). The obtained ‘primary clones' were sequenced and 12 out of 60 primary clones (20%) were found to contain γ-satellite type A or B sequences (Figure 1A–C). Nine primary clones (15%) contained sequences with tg/tc or tgc repeats and 39 (65%) contained other unidentified sequences (Figure 1C). To enrich for directly Gfi1b-bound sequences and to exclude false positives, we incubated the precipitated and purified DNA that was used to generate the primary clones with in vitro-translated, Myc epitope-tagged Gfi1b and precipitated with anti-Myc antibodies. The obtained DNA was purified, re-amplified by PCR and subcloned. These ‘secondary clones' were sequenced and now 24 out of 50 (48%) contained γ-satellite sequences, seven clones (14%) contained tg/tc or tgc repeats and 19 clones (38%) contained other unidentified sequences. This indicated that by affinity purification of Ch-IP reactions, γ-satellite DNA sequences could be significantly enriched as would be expected for directly Gfi1b-bound sequences. As a positive control, we found that sequences containing the Gfi1b promoter itself, a known Gfi1b target gene, could be amplified from the primary and secondary IPs (data not shown).
Figure 1.
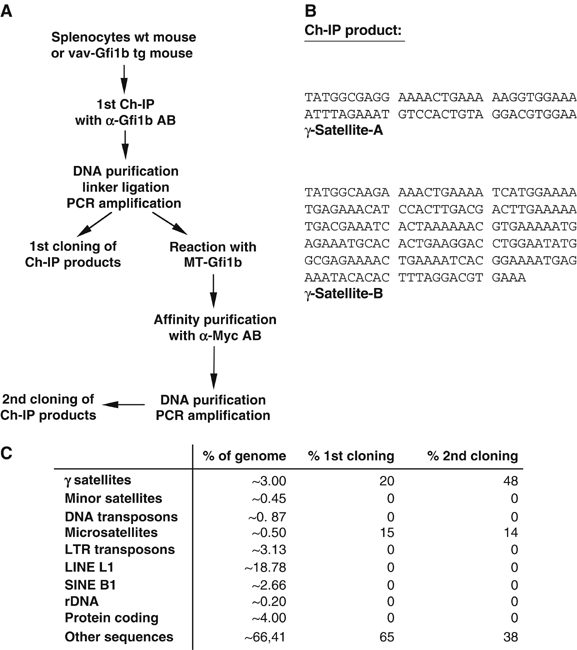
Ch-IP and cloning procedure. (A) Work flow protocol for the Ch-IP cloning procedure described in detail in Materials and methods. MT-Gfi1b: Myc-tagged Gfi1b. (B) Sequence of the γ-satellite repeat with its two parts, γ-satellite A (SAT-A) and γ-satellite B (SAT-B). (C) Overview of Ch-IP products and their representation after affinity purification in comparison to the frequency of these and other sequences in the mouse genome. There is a clear over-representation of γ-satellites and microsatellites after the Ch-IP and affinity purification procedure.
We compared the genome-wide frequency of the γ-satellite element, and other types of sequences with the frequency by which they appeared in our Ch-IP cloning procedure. Whereas the frequency of γ-satellite sequences accounts for about 3% of the entire murine genome, this sequence element was found in 20 and 48% of the sequences isolated after the first or second step of our Ch-IP cloning procedure, respectively (Figure 1C). This indicated that γ-satellite sequences are a bona fide Gfi1b target sequence and excluded that they represent a randomly generated artifact of the Ch-IP/cloning procedure.
Gfi1b binds to γ-satellite-B but not to γ-satellite-A sequences
Three putative Gfi1b binding sites with an ‘AATC' core could be found in the SAT-B part of the γ-satellite sequence and were termed SAT-B1, SAT-B2 and SAT-B3, but no such site was present in the SAT-A part (Figure 2A). Sequence comparison between an ideal Gfi1/Gfi1b binding site (‘ideal site'; Grimes et al, 1996; Zweidler-McKay et al, 1996), a previously determined bona fide Gfi1b binding site (Gfi1b-BS) from the Gfi1b promoter (Vassen et al, 2005) and the sequences surrounding the SAT-B1-3 putative binding sites revealed similarities at several positions 5′ and 3′ of the AATC core (Figure 2B, lower panel). To test their relative binding affinities, oligonucleotides covering the potential binding sites were used as unlabeled competitors in EMSA experiments with the radiolabeled Gfi1b-BS and in vitro-translated Myc-tagged Gfi1b (MT-Gfi1b) (Figure 2B). As expected, a strong complex with Gfi1b was observed, which could be supershifted with an anti Myc-tag antibody (Figure 2B, lanes 4 and 5). The complex could be disrupted with the ideal site and with SAT-B3 (lanes 8 and 9) as competitors but not with the unrelated CREB oligonucleotide (lane 10) and less efficiently with SAT-B1 or SAT-B2 sequences (lanes 6 and 7). When radiolabeled SAT-B1, SAT-B2, SAT-B3 and SAT-A oligonucleotides were used in EMSA experiments with in vitro-translated Gfi1b, SAT-B1 and SAT-B3 sequences but not SAT-B2 or SAT-A sequences were able to form complexes with Gfi1b that could be supershifted with an anti-Myc tag antibody (see Supplementary Figure S2).
Figure 2.
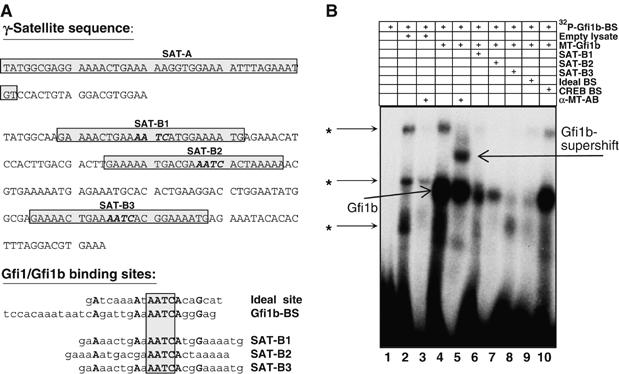
Presence of Gfi1/Gfi1b binding sites in the SAT-B part of the γ-satellite sequence. (A) Upper panel: Representation of DNA sequences in the SAT-A and SAT-B part of the γ-satellite repeat. ‘AATC' core sequences found in the SAT-B part are indicated by bold face and italicized letters. Oligonucleotides used for EMSA experiments are boxed. Lower panel: Sequence comparison between the ideal Gfi1/Gfi1b binding site (ideal site), a Gfi1b binding site from the Gfi1b promoter (Gfi1b-BS) and three putative Gfi1b binding sites from the SAT-B part of the γ-satellite sequence repeat (termed SAT-B1, SAT-B2 and SAT-B3). Note that several bases outside the ‘AATC' core are conserved between the ideal site, the Gfi1b-BS and the SAT-B1, SAT-B2 and SAT-B3 sequences. (B) Electrophoretic mobility shift assay (EMSA) with a radiolabeled Gfi1b-BS oligonucleotide and in vitro-translated Myc-tagged Gfi1b (MT-Gfi1b). Incubation of the labeled probe with empty (unprogrammed lysate) resulted in three nonspecific signals marked with an asterisk (*, lane 2). In the presence of MT-Gfi1b, the formation of a complex with the labeled probe is observed (lane 4), which can be supershifted by anti-Myc tag antibodies (α-MT-AB, lane 5) and is disrupted in the presence of unlabeled SAT-B3 or the ‘ideal site' oligonucleotide (lanes 8 and 9). Competition is less efficient with SAT-B1 or SAT-B2 oligonucleotides (lanes 6 and 7) and ineffective with an unrelated CREB oligonucleotide (lane 10).
To test whether γ-satellite sequences could represent a functional target site of Gfi1b in living cells, one or two copies of the SAT-A or SAT-B1 sequences were inserted 5′ of a luciferase reporter gene that was under the control of the SV40 promoter (Supplementary Figure S3A). As a control, a similar reporter gene construct with the Gfi1b-BS from the Gfi1b promoter was used (Vassen et al, 2005). As expected and described before (Vassen et al, 2005), Gfi1b was able to repress and a Gfi1b-VP16 fusion could transactivate transcription from the reporter constructs containing the Gfi1b-BS (Supplementary Figure S3A). However, reporter genes containing one or two copies of the SAT-A sequence could not be repressed by Gfi1b and were only marginally activated by Gfi1b-VP16 (Supplementary Figure S3A). In contrast, Gfi1b could repress and a Gfi1b-VP16 fusion protein could noticeably activate transcription from reporter genes containing SAT-B1 sequences but did not reach the levels obtained with the Gfi1b-BS-driven reporter (Supplementary Figure S3A). The activation by Gfi1b-VP16 increased with the numbers of SAT-B1 sequences inserted upstream of the SV40 promoter (Supplementary Figure S3A). This suggests that SAT-B but not SAT-A sequences are bona fide Gfi1b targets.
Gfi1b and Ikaros bind to different sites in the SAT-B part of the γ-satellite sequence but both colocalize at foci of pericentric heterochromatin
The zinc-finger transcription factor Ikaros has also been shown to bind to γ-satellite sequences (Cobb et al, 2000) and we decided to test whether Gfi1b and Ikaros have overlapping binding sites. To this end, the SAT-A and SAT-B1 oligonucleotides (Supplementary Figure S3B) were radiolabeled and used in an EMSA with in vitro-synthesized Myc-tagged Gfi1b (MT-Gfi1b) and the Myc-tagged Ikaros protein (MT-Ikaros). The results are documented in Supplementary Figure S3B and show that Ikaros and Gfi1b bind independently to the SAT-B1 sequence.
To visualize and control the binding of Gfi1b to the γ-satellite DNA sequence in cells, NIH 3T3 cells were transiently transfected with an expression vector for a Gfi1b-GFP fusion protein and were treated with an Immuno-FISH probe for γ-satellite sequences. A perfect colocalization of Gfi1b with DAPI-positive areas of heterochromatin and the γ-satellite sequence was observed in the transfected cells (Figure 3A). The previously described colocalization of Ikaros and γ-satellite sequences (Cobb et al, 2000) was also reproduced in our experimental system (Figure 3B), demonstrating the specificity of our immuno-FISH reaction. In addition, Gfi1b also showed a perfect colocalization with the transcription factor Ikaros in experiments where an Ikaros-GFP fusion protein was coexpressed in NIH 3T3 cells along with Gfi1b fused to the ds-Red protein (Figure 3C).
Figure 3.
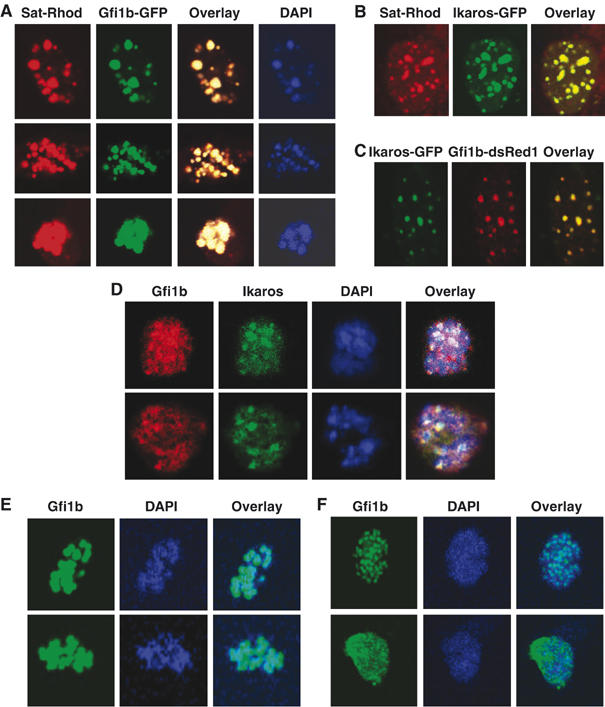
Colocalization of Gfi1b and Ikaros to foci of pericentric heterochromatin. (A) NIH 3T3 cells were transiently transfected with expression vectors for a fusion protein between Gfi1b and GFP, stained with DAPI and subjected to an immuno-FISH reaction with the rhodamine-labeled γ-satellite sequence as a probe. (B) Sat-Rhodamin immuno-FISH as in (A) but in the presence of an expression vector for a fusion protein between Ikaros and GFP. (C) Transient transfections of NIH 3T3 cells as in (A) with expression vectors for fusion protein between the transcription factor Ikaros and GFP or Gfi1b and ds-Red. (A–C) Shown are representative pictures from several independent experiments representative for virtually all transfected cells. (D) MEL75Acl.19 cells were stained with antibodies against endogenous Gfi1b, Ikaros and DAPI. The overlay shows colocalization of Gfi1b and Ikaros and intense DAPI stain (white spots). Pictures are representative of those cells that showed a nuclear localization of Ikaros, which shuttles in and out of the nucleus during the cell cycle. (E) Staining of Ter119+/CD71+-positive erythrocyte precursor cells sorted by flow cytometry from murine bone marrow with an anti-Gfi1b antibody and DAPI. The overlay of both pictures suggests localization of endogenous murine Gfi1b at foci of heterochromatin. Shown are two examples of cells with these features that represent about 10% of all analyzed cells. (F) Staining of Ter119+/CD71+ cells with anti-Gfi1b antibodies and DAPI. Shown are cells with more evenly distributed nuclear Gfi1b and heterochromatin that represent the majority in this population.
Cells from the Gfi1b-expressing murine erythroleukemia cell line MEL75Acl.19 with DAPI and antibodies against Gfi1b and Ikaros showed a colocalization of both proteins (red and green signals) with DAPI (blue signals) as white spots in all cells that coexpressed Gfi1b and Ikaros (Figure 3D). As the MEL75Acl.19 line comprises cells of different developmental stages, not all cells coexpressed Gfi1b and Ikaros. Hence, we also tested murine primary Ter119+/CD71+ erythroid precursor cells that express high levels of Gfi1b. In these cells, nuclear staining of Gfi1b varied from a clear localization at DAPI-positive foci of pericentric heterochromatin (Figure 3E) to a more even nuclear distribution not associated with a subnuclear structure (Figure 3F), which very likely reflected again different maturation or cell cycle stages of the cells. Deletion of the zinc-finger domains abrogated the localization of Gfi1b at DAPI-positive foci of nuclear chromatin (Supplementary Figure S4) indicating that the zinc binding domains mediate the association of Gfi1b with areas of heterochromatin.
Gfi1b alters histone methylation
B220+ B cells were purified from spleens of wt mice that are Gfi1b negative or from spleens of vav-Gfi1b transgenic mice that express elevated levels of Gfi1b (Vassen et al, 2005; Supplementary Figure S1). Nuclei from these cells were prepared and Ch-IP was performed with antibodies against histone H3 dimethylated at lysine 9 and histone H4 dimethylated at lysine 20. To detect whether Gfi1b can induce changes in chromatin regions adjacent to γ-satellite sequences, regions 5′ and 3′ of a γ-satellite sequence on chromosome 16 that had been pulled down in the Ch-IP cloning procedure and that were unique and free of repetitive sequences (hence ‘usable' as primer sequences) were identified. These regions were around 1 kb upstream and 5 kb downstream of this γ-satellite sequence, and primers from this region were designed for amplification after Ch-IP with the respective antibodies (Figure 4A). A significantly increased amount of histone H3 methylated at lysine 9 could be detected bound at the region 5′ of the γ-satellite sequence of chromosome 16 but not at the more distant 3′ region in B cells from vav-Gfi1b animals over wt B cells (Figure 4A). Unexpectedly, histone H4 lysine 20 dimethylation was decreased in regions both 5′ and 3′ of γ-satellite DNA sequences in cells from mice harboring a vav-Gfi1b transgene as opposed to wt cells (Figure 4A).
Figure 4.
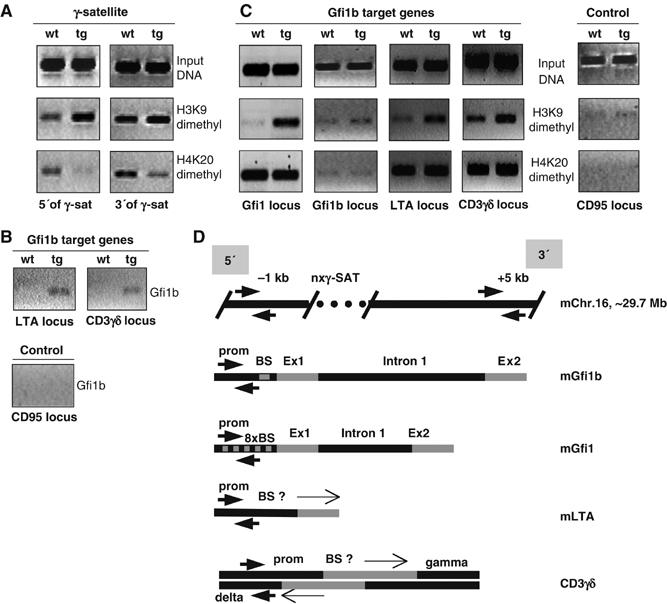
Elevated levels of Gfi1b expression are associated with altered histone methylation patterns. (A) Purified B cells (B220+) from spleens of either wt or vav-Gfi1b transgenic (tg) mice were fixed and the chromatin was subjected to IP (Ch-IP) with antibodies against dimethylated histone H3K9 or H4K20 as indicated. The precipitates were amplified with primer pairs covering the 5′ or 3′ adjacent regions of a γ-satellite sequence on chromosome 16 (see panel D). The chromatin preparation and input DNA was as described by Vassen et al (2005). Shown is a representative of three independent experiments. (B) As in (A), but precipitation was performed using an antibody against Gfi1b and PCR amplification was carried out with primer pairs specific for direct Gfi1b target genes (upper panel) or as a control for an actively transcribed gene that is not a Gfi1b target (CD95, lower panel) as indicated. Shown is a representative of three independent experiments. (C) As in (A) with primer pairs specific for the promoters of the indicated gene loci. (D) Schematic diagram of the primer pair positions on the Gfi1b occupied gene loci as indicated.
This prompted us to test whether altered histone methylation is also seen on Gfi1b target gene loci as for instance at the Gfi1b and Gfi1 promoter regions that have been confirmed as direct targets by a number of independent studies (Doan et al, 2004; Yücel et al, 2004; Vassen et al, 2005). In addition to this, by using comparative DNA microarray analysis of B220+ cells from wt and vav-Gfi1b transgenic mice, we could identify two additional Gfi1b target genes, namely lymphotoxin-A and CD3γ. Real-time PCR with cDNA from B-cell lines that had been previously established from vav-Gfi1b transgenic mice (Vassen et al, 2005) demonstrated that indeed the expression of these loci was downregulated in the presence of Gfi1b (Supplementary Figure S5). In addition, we could show by Ch-IP with an antibody against Gfi1b that the promoter regions of both genes are occupied by Gfi1b (Figure 4B) confirming them as bona fide Gfi1b target genes. We found a very strong enhancement of H3K9 dimethylation but no difference in histone H4K20 dimethylation at the CD3γ, LTA and Gfi1 loci and to a lesser extent at the Gfi1b locus in B cells from vav-Gfi1b transgenic mice compared to cells from wt mice (Figure 4C and D). As a control, we did not observe any alteration of H3K9 dimethylation at the CD95 locus, which is not a direct target of Gfi1b (Figure 4B and C).
Gfi1b binds histone methyltransferases G9A and SUV39H1
As Gfi1b correlated with increased histone H3K9 dimethylation, we hypothesized that Gfi1b may be able to recruit histone methyl transferases to target gene promoters or γ-satellite sequences and we tested whether Gfi1b can interact with the major histone methyltransferases G9A or SUV39H1. Both methyltransferases are expressed at detectable levels in nuclear extracts of MEL75Acl.19 cells, where Gfi1b was also detected by immunoblot and IP/Western as two forms of 37 and 48 kDa (Figure 5A; Supplementary Figure S6) as previously described (Rodriguez et al, 2005). Both G9A and SUV39H1 could be precipitated with an anti-Gfi1b antibody but not with an irrelevant anti-GST antibody from whole MEL cell extracts (Figure 5B), strongly suggesting a direct physical interaction with Gfi1b and both G9a and SUV39H1 histone methyltransferases at endogenous expression levels. In addition, in the same cells, endogenous Gfi1b could be precipitated with both anti-G9a and anti-SUV39H1 antibodies but not with an irrelevant control antibody (anti-GFP) nor in a second control reaction where only protein A/G beads were present confirming the existence of Gfi1b/G9a and Gfi1b/SUV39H1 complexes at endogenous expression levels in erythroid cells (Figure 5C).
Figure 5.
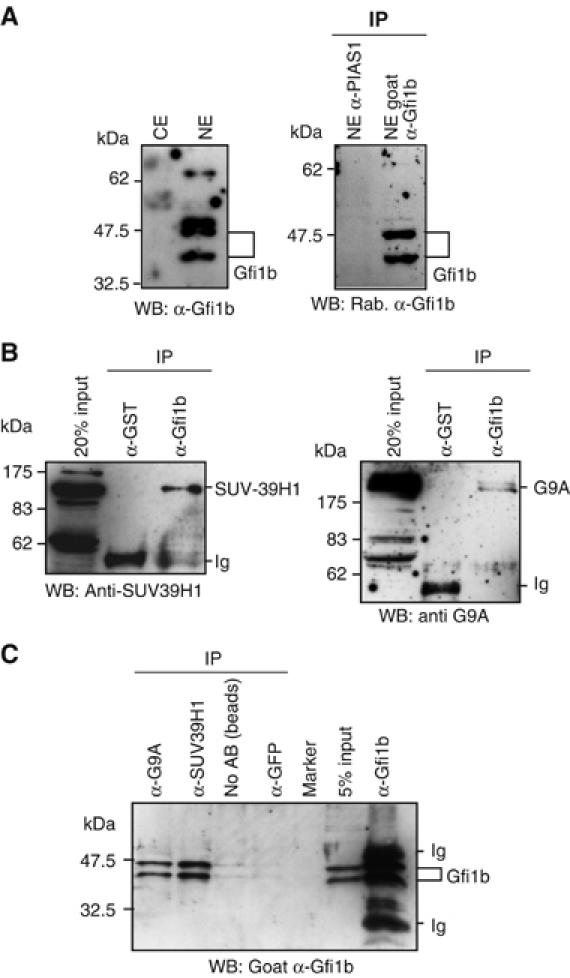
Gfi1b can bind to the histone methylases G9A and SUV39H1. (A) Western blots using an anti-Gfi1b antibody on either cytoplasmic (CE) or nuclear (NE) extracts from MEL75Acl.19 cells (left panel) or immunoprecipitates from these extracts using an anti-Gfi1b or an unrelated anti-PIAS1 antibody as indicated (right panel) showing an exclusively nuclear distribution and specific IP of Gfi1b. Detected are two forms of Gfi1b in these cells (37 and 48 kDa) as described (Rodriguez et al, 2005). (B) Extracts of MEL75Acl.19 cells were immunoprecipitated with either anti-Gfi1b antibodies or with an unrelated anti-GST antibody. The precipitates were separated by SDS–PAGE, transferred onto a solid support and probed with either an anti-SUV-39H1 antibody (left panel) or an anti-G9a antibody (right panel). (C) Extracts of MEL75Acl.19 cells were immunoprecipitated with anti-G9a, anti-SUV39H1, anti-Gfi1b or unrelated anti-GFP antibodies or were treated with protein-A/G beads alone. The precipitates were separated by SDS–PAGE, transferred onto a solid support and probed with anti-Gfi1b antibodies.
Gfi1b-deficient erythroid precursor cells show a defect in heterochromatinization
To test whether Gfi1b plays a role in heterochromatinization, we used fetal liver cells from a Gfi1-deficient mouse that we generated by replacing the Gfi1b coding exons with the GFP cDNA (Vassen and Möröy, unpublished). Whereas Gfi1b+/− animals are normal and indistinguishable from wt mice, Gfi1b−/− animals do not develop to term and arrest at embryonal stage E13.5 d.p.c. owing to a block in erythroid maturation (Figure 6A) as was previously described (Saleque et al, 2002). Fetal liver cells from Gfi1b−/− and Gfi1b+/− were isolated and stained with DAPI. Whereas Gfi1b+/− cells clearly showed distinct spots of intense DAPI stain, typical for foci of heterochromatin, Gfi1b−/− cells lacked this subnuclear pattern and displayed a rather even distribution of DAPI staining (Figure 6B–E). Cell counts estimated that this loss of DAPI staining pattern associated with heterochromatin was typical for over 80% of Gfi1b−/− cells (not shown). Further, Gfi1b-deficient cells showed a noticeable loss of the characteristic punctate staining with anti-trimethyl H3K9 antibodies that in wt or Gfi1b+/− cells is indicative of pericentric heterochromatin (Figure 6C–E), suggesting that Gfi1b is required to maintain or support H3K9 trimethylation at sites of pericentric heterochromatin.
Figure 6.
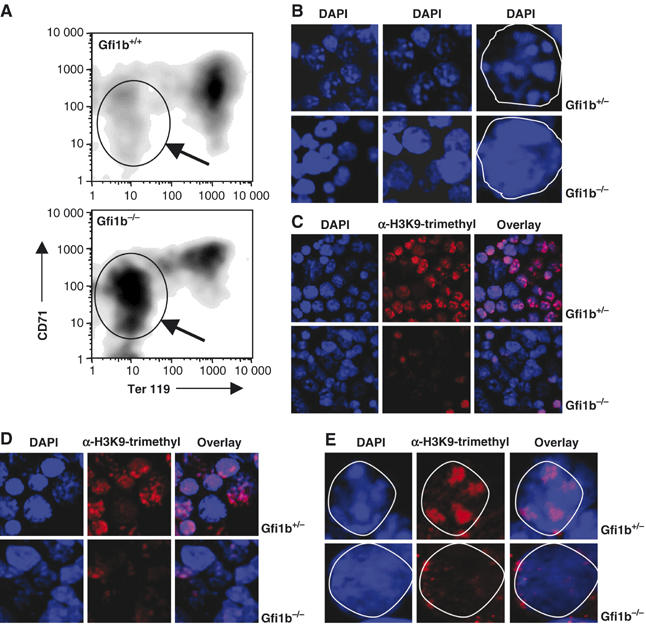
Heterochromatinization defect in Gfi1b-deficient erythroid precursor cells. (A) FACS analysis of fetal liver cells from wt or Gfi1b-deficient mice at day E13.5 stained with antibodies against CD71 and Ter119. Gfi1b-deficient cells from fetal liver show a block in maturation and accumulate cells in an immature state (see gated population and arrow). (B) Confocal image analysis of cytospins from fetal liver cells derived from heterozygous Gfi1b:GFP knock-in (Gfi1b+/−) or homozygous Gfi1b:GFP knock-in (Gfi1b−/−) embryos. Cells were stained with DAPI and foci of heterochromatin are clearly visible in Gfi1b+/− cells but not in Gfi1b−/− cells. Rightmost pictures: single cell nucleus is displayed and is marked by white line. (C, D) Fetal liver cells from heterozygous Gfi1b:GFP knock-in (Gfi1b+/−) or homozygous Gfi1b:GFP knock-in (Gfi1b−/−) embryos stained with DAPI or with antibodies against trimethylated H3K9. Foci of heterochromatin are visible as blue (DAPI positive) regions. Distinct areas of H3K9 trimethylation are also visible in the cell nuclei of Gfi1b+/− cells (red staining). In Gfi1b−/− cell nuclei, foci of heterochromatin (DAPI positive, blue) regions are not readily seen and distinct areas of H3K9 trimethylation (red) are almost absent. (E) As in (C) and (D), but a single fetal liver cell is displayed and delineated by a white margin. Distinct areas of H3K9 trimethylation (red) as well as heterochromatic foci (DAPI positive, blue) are visible in the Gfi1b+/− cell nucleus, but in Gfi1b−/− cells the typical DAPI-positive foci are not readily observed and distinct areas of H3K9 trimethylation (red areas) are completely lacking.
Discussion
Gfi1b is a small nuclear protein, which is able to repress the transcription of specific target genes by binding to cognate sites in their promoter sequences (Tong et al, 1998; Jegalian and Wu, 2002; Doan et al, 2004; Vassen et al, 2005). DNA binding of Gfi1b is mediated by C-terminal zinc-finger domains that recognize stretches of DNA with the core sequence AATC. Our data presented here reveal a novel function of Gfi1b and show that besides occupying specific target gene promoter sequences, Gfi1b is also able to bind to γ-satellite sequences that are a constituent of pericentric heterochromatin but are also found outside this area (Grewal and Elgin, 2002). Several lines of evidence support the notion that Gfi1b can bind to γ-satellite sequences and specifically to the SAT-B1 and SAT-B3 parts: First, γ-satellite sequences were isolated by a Ch-IP and cloning procedure with an anti-Gfi1b antibody and were significantly enriched in a affinity selection with a tagged Gfi1b protein. Second, a direct binding of Gfi1b to γ-satellite sequences in vitro could be shown by gel EMSAs. Third, a Gfi1b binding site from the SAT-B parts of the isolated γ-satellite sequence was able to recruit Gfi1b or Gfi1b-VP16 fusion proteins to repress or stimulate the transcription of an appended reporter gene. Fourth, Gfi1b sequences were found to be localized at sites identified by a γ-satellite-specific immuno-FISH probe and, fifth, Gfi1b colocalized with Ikaros, a transcription factor known to reside at γ-satellite sequences.
Our findings that ectopically expressed Gfi1b is enriched in subnuclear regions of DNA that stained positive with DAPI, with a γ-satellite immuno-FISH probe and with antibodies against the zinc-finger protein Ikaros further confirm that Gfi1b is localized at foci of pericentric heterochromatin and suggest that it is involved in its formation or maintenance. Consistent with this view is our observation that the association of Gfi1b with heterochromatin strictly depended on an intact zinc-finger DNA binding domain, as the deletion of the last of the six zinc-finger domains in Gfi1b abrogated the binding to the SAT-B part of the γ-satellite sequence as well as association with DAPI-stained areas of heterochromatin. This behavior is very similar to the Ikaros protein, which also binds to pericentric heterochromatin sequences via its zinc-finger domains. However, our experiments with primary cells or MEL cells also suggested that the association of Gfi1b at endogenous expression levels at foci of heterochromatin may depend on the particular status of an individual cell, as in contrast to the situation found in transfected cells, endogenous Gfi1b was found in patterns of subnuclear structures that varied from clear punctate structures typical for heterochromatin to a more homogeneous nuclear distribution. This may be due to the fact that not all cells express the same endogenous levels of Gfi1b or that the cells represent even in culture different differentiation stages or the cells are at different positions in the cell cycle, as it has been described before for Ikaros (Dovat et al, 2002).
Heterochromatin contains few genes, is replicated late in the cell cycle and is rich in repetitive sequences, in particular satellite repeats. In the mouse, pericentric heterochromatin is almost exclusively composed of the typical 234 bp γ-satellite repeats to which Gfi1b can bind according to our data. In contrast, euchromatin replicates early in cell cycle, is poor of repetitive sequences and comprises stretches of DNA dense with single-copy genes (for a review, see Dillon and Festenstein, 2002; Li et al, 2002; Fahrner and Baylin, 2003). The most prominent differences between hetero- and euchromatin are the degree of nucleosome packaging regulated by histone tail modifications. The methylation of histone H3 at lysine 9 has recently been recognized as a key feature in maintaining pericentric heterochromatin and in silencing of euchromatic genes (for a review see Lachner et al, 2003). Two histone methyl transferases, Suv39H1 and G9a, were identified as the responsible enzymes mediating the methylation of histone H3K9 at pericentric heterochromatin or at euchromatic gene loci, respectively (Peters et al, 2001; Tachibana et al, 2002; Lehnertz et al, 2003; Rice et al, 2003). Whereas G9a mediates histone H3K9 mono- and dimethylation at euchromatic regions associated with gene silencing (Tachibana et al, 2002, 2005), Suv39H1 appears to be the major methylase to direct histone H3K9 trimethylation in pericentric heterochromatin (Peters et al, 2003).
Our observation that Gfi1b resides at γ-satellite repeats and at foci of pericentric heterochromatin and the well-documented fact that Gfi1b is a transcriptional repressor led to the hypothesis that Gfi1b is involved in the methylation of histone H3 at lysine 9 or even exerts its function through histone methylation. Our IP experiments with erythroleukemia cells suggest that at least a fraction of Gfi1b physically interacts with both histone methylases, G9a and Suv39H1, at endogenous expression levels. As we also found that elevated levels of Gfi1b expression correlated with increased histone H3K9 dimethylation at four independent, direct Gfi1b target genes and taking into account that G9a mainly directs H3K9 dimethylation at euchromatic regions, it is very likely that G9a methylates H3K9 at Gfi1b target gene promoters through interaction with Gfi1b. This would offer an explanation of the biochemical mechanism by which Gfi1b represses transcription of target genes. The dimethylation of histone H4 at lysine 20, which is broadly distributed over euchromatic regions and is mediated by methylases other than G9a (Sanders et al, 2004; Schotta et al, 2004), was not affected by Gfi1, indicating the specificity of the interaction and the Gfi1b-dependent methylation at target gene promoters.
Moreover, H3K9 dimethylation was also increased by elevated Gfi1b levels at the 5′ region that neighbors a γ-satellite sequence on chromosome 16 pointing to the possibility that Gfi1b is able to direct G9a-dependent H3K9 dimethylation at euchromatic regions outside of direct target genes, which may reflect a role of Gfi1b in the maintenance of overall genomic stability. By contrast, our observation that dimethylation of H4K20 is decreased at sites neighboring γ-satellite sequences on chromosome 16 at 3′ and 5′ flanking regions when Gfi1b is overexpressed is not readily explained. It is not unlikely that in these regions H4K20 trimethylation is predominant over H4K20 dimethylation, which would suggest that higher Gfi1b levels should correlate with increased H4K20 trimethylation at these sites. This would indicate that these sites belong to an area of heterochromatin outside the pericentromeric region, as H4K20 trimethylation has been associated with constitutive heterochromatin (Schotta et al, 2004). As neither G9a or Suv39H1 is involved in H4K20 methylation (Sanders et al, 2004; Schotta et al, 2004), this effect may only be indirectly related to Gfi1b and would require further experimentation to be clarified.
In contrast to G9a, Suv39H1 has been shown to be the major methyl transferase to direct H3K9 trimethylation to pericentric heterochromatin (Peters et al, 2001, 2003; Lehnertz et al, 2003). Our findings that Gfi1b on the one hand interacts with Suv39H1 and on the other binds to γ-satellite sequences in pericentric heterochromatin would therefore be consistent with a model in which Gfi1b participates in the formation or maintenance of pericentric heterochromatin by recruiting Suv39H1 to γ-satellite sequences, enabling it to direct trimethylation of H3K9 in this area. One critical test for this hypothesis and for the significance of Gfi1b in such a process would be the analysis of H3K9 trimethylation at pericentric heterochromatin in the absence of Gfi1b. Our experiments with erythroid precursor cells from Gfi1b-deficient mice demonstrated that, indeed, the ablation of Gfi1b correlated with a noticeable decrease of DAPI-positive foci of heterochromatin and H3K9 trimethylation and thus clearly supports this hypothesis.
Furthermore, from the very critical role of Gfi1b in erythrocyte maturation, which becomes apparent in Gfi1b-deficient mice (Saleque et al, 2002; Vassen and Möröy, this study and unpublished data), and our findings that suggest a role of Gfi1b in heterochromatin maintenance or formation, a more general role of Gfi1b in chromatin condensation can be inferred that goes beyond transcriptional silencing. Erythrocyte precursors develop into definitive erythrocytes by expelling their nucleus and prior to this enucleation, nuclear chromatin condenses, and the nucleus reduces in size and moves adjacent to the plasma membrane (Rothmann et al, 1997). When Gfi1b is deleted from the mouse germ line, the most striking features are the absence of definitive enucleated erythrocytes (Saleque et al, 2002). If the enucleation of erythrocyte precursors, which is a prerequisite for the development of red blood cells in mammals, requires the compaction of chromatin, the formation of heterochromatin is very likely to be involved in this process. Our observation that Gfi1b deficiency was accompanied by a loss of heterochromatic foci identified by DAPI and H3K9 trimethylation points to the possibility that Gfi1b is required for chromatin condensation in erythroblasts before enucleation can take place. Our findings may thus provide a basis to explain the role of Gfi1b in erythrocyte development and its specific activity as an inducer or executer of heterochromatin formation by promoting the recruitment of Suv39H1 histone methyl transferases to γ-satellite sequences and also support the hypothesis that Gfi1b is both a conventional transcriptional repressor binding to upstream regulatory sequences and at the same time a constituent of heterochromatin by recognizing γ-satellite sequences as a functional target.
Materials and methods
Chromatin immunoprecipitation and cloning (Ch-IP cloning)
Isolation of nuclei: Single-cell suspensions of 108 splenocytes from two wt and two vav-Gfi1b transgenic mice were washed in ice-cold PBS, resuspended in 25 ml homogenization buffer (300 mM sucrose, 60 mM KCl, 15 mM NaCl, 50 mM Tris–HCl pH 7.5, 15 mM MnCl2 (add freshly), 2 mM EDTA, 0.5 mM spermine, 0.15 mM spermidine, 0.2 mM AEBSF) and lysed by addition of 25 ml homogenization buffer plus 1% NP-40. After incubation for 5 min on ice, nuclei were spun down (1500 r.p.m., 4°C, 5 min, Heraeus Megafuge) and washed twice with homogenization buffer. DNAse digestion: Nuclei were resuspended in 500 μl homogenization buffer and 10 or 20 μl DNAseI (20 mg/ml Grade II; Roche) respectively was added. After incubation for 15 min on ice, the reaction was stopped by transferring the nuclei to 45 ml ice-cold PBS/ED(G)TA (50 mM EDTA/50 mM EGTA). Crosslinking: A 5 ml portion of fixation solution (100 mM NaCl, 50 mM Tris–HCl pH 8.0, 11% formaldehyde) was added immediately and nuclei were stirred on ice for 30 min. The reaction was stopped by successive addition of 5 g solid glycine until complete solution. Nuclei were spun down (1500 r.p.m., 4°C, 5 min, Heraeus Megafuge) and washed in ice-cold PBS, wash-buffer-A (10% Triton X-100, 10 mM Tris–HCl pH 8.0, 0.5 mM EDTA, 1.0 mM EGTA) and wash-buffer-B (200 mM NaCl, 10 mM Tris–HCl pH 8.0, 0.5 mM EDTA, 1.0 mM EGTA) successively. Immunoprecipitation: The pellets were resuspended in 2 ml of IP buffer/0.1% NP-40 and 50 μl of the solution was saved for reverse crosslink as an input control at −80°C. About 5 μg of affinity-purified rabbit anti-Gfi1b antibody was added and the samples were incubated overnight on a roller incubator at 4°C. A 25 μl portion of protein-A-Sepharose beads (preincubated IP buffer containing 1% BSA and washed with IP buffer) was added and after incubation at 4°C for 2 h, beads were washed five times with IP buffer/0.1% NP-40, twice with high-salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.0, 500 mM NaCl), twice with low-salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.0, 150 mM NaCl) and twice with IP buffer. The beads were spun down and as much supernatant as possible was removed. Immune complexes were eluted by two-fold extraction with 50 μl of elution buffer (15 mM Tris–HCl pH 7.5, 0.5 mM EDTA, 1% SDS) by shaking vigorously at 30°C for 15 min. The beads were spun down and the supernatant was used for reverse crosslink. Reverse crosslink: The eluates and the input controls were diluted with 300 μl TE and 10 μg RNAse A was added. After 20 min of incubation at 37°C, SDS (0.5% final) and Proteinase K (500 μg/ml final) were added and samples were incubated at 37°C overnight. Reverse crosslink was carried out by incubation at 65°C for an additional 6 h. Samples were extracted with 400 μl phenol/chloroform/isoamylalcohol (25:24:1) and the aqueous phase was extracted with chloroform/isoamylalcohol (24:1) again. The DNA containing aqueous phase was ethanol precipitated, the precipitate washed with 70% ethanol and dried down at 37°C for 15 min. The DNA was redissolved in 50 μl of TE overnight.
Linker ligation and PCR amplification
To each sample, 6 μl ligation buffer, 100 pM annealed unphosphorylated linker (US: 5′-gatccagtacatcgtcgaattct-3′, LS: 5′-agaattcgacgatgtactg-3′) and 3 μl T4 DNA ligase (3 U; Roche). After ligation at 16°C overnight, DNA was purified (and excess linker removed) using a PCR purification kit (Qiagen, Hilden, Germany). The ligation products were amplified by PCR (24 cycles) using the linker-lower-strand oligonucleotide. The PCR products were purified using a PCR purification kit and 4 μl of the 50 μl eluate was directly used for cloning using a TOPO cloning kit (Invitrogen). The inserts of the derived clones were sequenced.
FISH labeling and confocal image analysis
The procedures were performed following modified protocols (Brown et al, 2001). Briefly, cells grown on glass coverslips were treated with fixation and permeabilization buffer (0.5% glutaraldehyde, 1% Triton X-100 in PBS without Ca, Mg) for 30 min and washed three times with PBS (15 min each). After washing twice with 1 mg/ml borohydride solution, cells were incubated sequentially with blocking solution (5% FCS/5% goat serum), specific antiserum and a fluorescent dye-labeled secondary antiserum (diluted in blocking solution) for 30 min at 37°C each. Thereafter, cells were kept in the dark, washed with PBS three times and incubated with 50 mM EGS in PBS for 30 min at 37°C for post-fixation crosslinking. After washing twice with PBS, cells were incubated with 100 μg/ml RNAse-A in 2 × SSC at 37°C, washed in PBS and incubated with 100 μl drops of 0.1 N NaOH for 2 min. Cells were rinsed immediately in ice-cold PBS and tetramethylrhodamine-5′-dUTP (Roche) PCR-labeled, heat-denatured DNA probes comprising γ-satellite sequences dissolved in hybridization buffer (50% formamide, 4 × SSC, 100 mM NaPO4 pH 7.0, 0.1% Tween 20) were applied. The cells were hybridized overnight at 38–40°C in humid chambers. Then, cells were washed with hybridization buffer (2 × SSC, without Tween) containing 40, 30, 20, 10 and 0% formamide successively. Then, the cells were washed twice with PBS and once with distilled water for 10 s. Coverslips were embedded using the ‘ProLong' Antifade Kit (Molecular Probes, Oregon) and fluorescence was analyzed using a laser scanning microscope.
Gel-shift analysis
The Myc-tagged Gfi1b or Gfi1 proteins were produced in the TNT™ Rabbit Reticulocyte Lysate System (Promega). A 1 μl portion of lysate was incubated for 20 min at room temperature with 7 μl of gel-shift buffer (40 mM KCl, 30 mM HEPES–KOH pH 7.6, 1 mM MgCl2, 5% glycerol, 15% Ficoll, 5 mM DTT, 0.25 μg/μl poly-dIdC) with or without 32P-labeled double-stranded oligonucleotides. Where indicated, for test of specificity, 50 pM unlabeled target or non-target oligonucleotide was added to compete for the protein of interest. After preincubation for 20 min, anti-Myc antibody was added to the protein:DNA complex for 10 min to further retard its mobility (supershift) during electrophoresis. Protein:DNA complexes were separated using a native acrylamide gel (4% PAA, 0.5 × TBE) and visualized by autoradiography.
Immunoprecipitation
A 200 μg portion of MEL cell whole-cell extract was diluted in 500 μl IP buffer (150 mM KCl, 0.1 mM EDTA, 3 mM MgCl2, 10% glycerol, 0.1% NP-40, 10 mM Tris–HCl pH 8), incubated with the primary antibodies as indicated overnight and 30 μl of protein-A/protein-G agarose mixture was added for an additional 3 h. The beads were washed six times with IP buffer and purified proteins were denatured in SDS sample buffer. Probes were separated on a 10% PAA gel and subjected to Western blot procedure. Blots were probed with a goat anti-Gfi1b antibody (SANTA-CRUZ SC8559).
Supplementary Material
Supplementary Text
Supplementary Figure S1
{kind=link}
Supplementary Figure S2
{kind=link}
Supplementary Figure S3
{kind=link}
Supplementary Figure S4
{kind=link}
Supplementary Figure S5
{kind=link}
Supplementary Figure S6
{kind=link}
Acknowledgments
We are indebted to Angelika Warda, Wojciech Wegrzyn, Adriane Parchatka, Nadine Esser and Inge Spratte for technical assistance and Petra Plessow and Tomas Civela for excellent animal care. We particularly thank Marshall Horwitz and Zhi-Jun Duan for sharing results prior to publication. This work was supported by the Deutsche Forschungsgemeinschaft, DFG (grant Mo 435/18-1), the ‘Fonds der chemischen Industrie' and the ‘IFORES Program' of the University of Essen Medical School.
References
- Brown KE, Amoils S, Horn JM, Buckle VJ, Higgs DR, Merkenschlager M, Fisher AG (2001) Expression of alpha- and beta-globin genes occurs within different nuclear domains in haemopoietic cells. Nat Cell Biol 3: 602–606 [DOI] [PubMed] [Google Scholar]
- Cobb BS, Morales-Alcelay S, Kleiger G, Brown KE, Fisher AG, Smale ST (2000) Targeting of Ikaros to pericentromeric heterochromatin by direct DNA binding. Genes Dev 14: 2146–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon N, Festenstein R (2002) Unravelling heterochromatin: competition between positive and negative factors regulates accessibility. Trends Genet 18: 252–258 [DOI] [PubMed] [Google Scholar]
- Doan LL, Porter SD, Duan Z, Flubacher MM, Montoya D, Tsichlis PN, Horwitz M, Gilks CB, Grimes HL (2004) Targeted transcriptional repression of Gfi1 by GFI1 and Gfi1b in lymphoid cells. Nucleic Acids Res 32: 2508–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovat S, Ronni T, Russell D, Ferrini R, Cobb BS, Smale ST (2002) A common mechanism for mitotic inactivation of C2H2 zinc finger DNA-binding domains. Genes Dev 16: 2985–2990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z, Horwitz M (2003a) Gfi-1 oncoproteins in hematopoiesis. Hematology 5: 339–344 [DOI] [PubMed] [Google Scholar]
- Duan Z, Horwitz M (2003b) Targets of the transcriptional repressor oncoprotein Gf11. Proc Natl Acad Sci USA 100: 5932–5937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrner JA, Baylin SB (2003) Heterochromatin: stable and unstable invasions at home and abroad. Genes Dev 17: 1805–1812 [DOI] [PubMed] [Google Scholar]
- Grewal SI, Elgin SC (2002) Heterochromatin: new possibilities for the inheritance of structure. Curr Opin Genet Dev 12: 178–187 [DOI] [PubMed] [Google Scholar]
- Grimes HL, Chan TO, Zweidler-McKay PA, Tong B, Tsichlis PN (1996) The Gfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG, and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol Cell Biol 16: 6263–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzano R, Montcouquiol M, Rashi-Elkeles S, Elkon R, Yücel R, Frankel WN, Rechavi G, Möröy T, Friedman TB, Kelley MW, Avraham KB (2004) Transcription profiling of inner ears from Pou4f3(ddl/ddl) identifies Gfi1 as a target of the Pou4f3 deafness gene. Hum Mol Genet 13: 2143–2153 [DOI] [PubMed] [Google Scholar]
- Hock H, Hamblen MJ, Rooke HM, Traver D, Bronson RT, Cameron S, Orkin SH (2003) Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity 18: 109–120 [DOI] [PubMed] [Google Scholar]
- Jegalian AG, Wu H (2002) Regulation of Socs gene expression by the proto-oncoprotein GFI-1B: two routes for STAT5 target gene induction by erythropoietin. J Biol Chem 277: 2345–2352 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Karsunky H, Zeng H, Schmidt T, Zevnik B, Kluge R, Schmid KW, Dührsen U, Möröy T (2002) Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat Genet 30: 295–300 [DOI] [PubMed] [Google Scholar]
- Lachner M, O'Sullivan RJ, Jenuwein T (2003) An epigenetic road map for histone lysine methylation. J Cell Sci 116 (Part 11): 2117–2124 [DOI] [PubMed] [Google Scholar]
- Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH (2003) Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13: 1192–1200 [DOI] [PubMed] [Google Scholar]
- Li Y, Kirschmann DA, Wallrath LL (2002) Does heterochromatin protein 1 always follow code? Proc Natl Acad Sci USA 99 (Suppl 4): 16462–16469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möröy T (2005) The transcription factor ‘Growth factor independence 1'—Gfi1 Internat. J Biochem Cell Biol 37: 541–546 [DOI] [PubMed] [Google Scholar]
- Osawa M, Yamaguchi T, Nakamura Y, Kaneko S, Onodera M, Sawada K, Jegalian A, Wu H, Nakauchi H, Iwama A (2002) Erythroid expansion mediated by the Gfi-1B zinc finger protein: role in normal hematopoiesis. Blood 100: 2769–2777 [DOI] [PubMed] [Google Scholar]
- Peters AH, Kubicek S, Mechtler K, O'Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JH, Jenuwein T (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12: 1577–1589 [DOI] [PubMed] [Google Scholar]
- Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107: 323–337 [DOI] [PubMed] [Google Scholar]
- Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD (2003) Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell 12: 1591–1598 [DOI] [PubMed] [Google Scholar]
- Rodriguez P, Bonte E, Krijgsveld J, Kolodziej KE, Guyot B, Heck AJ, Vyas P, de Boer E, Grosveld F, Strouboulis J (2005) GATA-1 forms distinct activating and repressive complexes in erythroid cells. EMBO J 24: 2354–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rödel B, Wagner T, Zörnig M, Niessing J, Möröy T (1998) The human homologue (GFI1B) of the chicken GFI gene maps to chromosome 9q34.13—a locus frequently altered in hematopoietic diseases. Genomics 54: 580–582 [DOI] [PubMed] [Google Scholar]
- Rothmann C, Cohen AM, malik Z (1997) Chromatin condensation in erythrpopoiesis resolved by multipixel spectral imagin: differentiation versus apoptosis. J Histochem Cytochem 45: 1097–1108 [DOI] [PubMed] [Google Scholar]
- Saleque S, Cameron S, Orkin SH (2002) The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev 16: 301–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T (2004) Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119: 603–614 [DOI] [PubMed] [Google Scholar]
- Scheijen B, Jonkers J, Acton D, Berns A (1997) Characterization of pal-1: a common proviral insertion site in murine leukemia virus-induced lymphomas of c-myc and Pim-1 transgenic mice. J Virol 71: 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T, Zörnig M, Beneke R, Möröy T (1996) MoMuLV proviral integrations identified by Sup-F selection in tumors from infected myc/pim bitransgenic mice correlate with activation of the gfi-1 gene. Nucleic Acids Res 24: 2528–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T (2004) A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev 18: 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev 16: 1779–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y (2005) Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev 19: 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong B, Grimes HL, Yang TY, Bear SE, Qin Z, Du K, El-Deiry WS, Tsichlis PN (1998) The Gfi-1B proto-oncoprotein represses p21WAF1 and inhibits myeloid cell differentiation. Mol Cell Biol 18: 2462–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassen L, Fiolka K, Mahlmann S, Möröy T (2005) Direct transcriptional repression of the genes encoding the zinc-finger proteins Gfi1b and Gfi1 by Gfi1b. Nucleic Acids Res 33: 987–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis D, Hamblen M, Zhou Y, Venken KJ, Schumacher A, Grimes HL, Zoghbi HY, Orkin SH, Bellen HJ (2003) The zinc finger transcription factor Gfi1, implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development 130: 221–232 [DOI] [PubMed] [Google Scholar]
- Yücel R, Karsunky H, Klein-Hitpass L, Möröy T (2003) The transcriptional repressor Gfi1 affects development of early, uncommitted c-Kit+ T cell progenitors and CD4/CD8 lineage decision in the thymus. J Exp Med 97: 831–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yücel R, Kosan C, Heyd F, Möröy T (2004) Gfi1:GFP knock-in mutant reveals differential expression and autoregulation of the gene growth factor independence 1 (Gfi1) during lymphocyte development. J Biol Chem 279: 40906–40917 [DOI] [PubMed] [Google Scholar]
- Zörnig M, Schmidt T, Karsunky H, Grzeschiczek A, Möröy T (1996) Zinc finger protein GFI-1 cooperates with myc and pim-1 in T-cell lymphomagenesis by reducing the requirements for IL-2. Oncogene 12: 1789–1801 [PubMed] [Google Scholar]
- Zweidler-Mckay PA, Grimes HL, Flubacher MM, Tsichlis PN (1996) Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol Cell Biol 16: 4024–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Text
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6