

Abstract
Cross-species infections are responsible for the majority of emerging and re-emerging viral diseases. However, little is known about the mechanisms that restrict viruses to a certain host species, and the factors viruses need to cross the species barrier and replicate in a different host. Cytomegaloviruses (CMVs) are representatives of the beta-herpesviruses that are highly species specific. They replicate only in cells of their own or a closely related species. In this study, the molecular mechanism underlying the cytomegalovirus species specificity was investigated. We show that infection of human cells with the murine cytomegalovirus (MCMV) triggers the intrinsic apoptosis pathway involving caspase-9 activation. MCMV can break the species barrier and replicate in human cells if apoptosis is blocked by Bcl-2 or a functionally analogous protein. A single gene of the human cytomegalovirus encoding a mitochondrial inhibitor of apoptosis is sufficient to allow MCMV replication in human cells. Moreover, the same principle facilitates replication of the rat cytomegalovirus in human cells. Thus, induction of apoptosis serves as an innate immune defense to inhibit cross-species infections of rodent CMVs.
Keywords: apoptosis, caspase, cytomegalovirus, species specificity, vMIA
Introduction
Viruses have gone through a coevolution with their hosts, during which they have adapted to them. As a result of this adaptation, many viruses have a limited host range. Occasionally, a virus acquires a mutation or a new gene that allows it to infect and replicate in a different host species. This may lead to severe disease in the newly infected species, to a local outbreak, or even a pandemic (Weiss, 2003) such as the Spanish flu of 1918/19 (Taubenberger et al, 2005). Another example of a cross-species infection is the AIDS pandemic. In this case, simian immunodeficiency viruses have acquired the ability to replicate and spread in humans (Hahn et al, 2000).
The molecular mechanisms underlying the species barrier have only begun to be understood. In some instances, the availability of an appropriate entry receptor on the cell surface limits the virus' host range (reviewed in Baranowski et al, 2003). In other cases, the virus is able to enter cells of other species, but fails to block intracellular defense mechanisms that inhibit virus replication. It has recently been shown for the human immunodeficiency virus that the ability of the viral protein Vif to bind cellular cytidine deaminases limits its host range (Mariani et al, 2003). If not blocked by Vif, these enzymes cause a lethal hypermutation of the viral genome (Lecossier et al, 2003). More recent work has demonstrated that the species specificity of myxoma virus, a poxvirus of rabbits, is a consequence of the virus' inability to inhibit the interferon response in cells of foreign species (Wang et al, 2004). Myxoma virus can break the species barrier and replicate in murine cells if activation of the interferon response is blocked by inhibitory drugs or with interferon neutralizing antibodies. Similarly, the host range of certain paramyxoviruses and an attenuated vaccinia virus also depends on this innate immune defense (Parisien et al, 2002; Hornemann et al, 2003).
For the cytomegaloviruses (CMVs), the molecular basis of their species specificity has not been determined. These viruses belong to the β subfamily of the herpesviruses. Representatives of this subfamily have been identified in numerous animal species, and these viruses elicit similar illnesses in their respective hosts (Mocarski and Courcelle, 2001). Human cytomegalovirus (HCMV) is an opportunistic pathogen that causes generally mild infections in healthy people, but can cause severe disease in immunocompromised individuals such as transplant recipients or AIDS patients. Since their first isolation in cell culture, the CMVs have been recognized as highly species specific (Weller, 1970). They replicate only in cells of their own or a closely related species. For instance, simian CMV can replicate in human fibroblasts (Lafemina and Hayward, 1988), and HCMV can replicate in simian fibroblasts (Perot et al, 1992). Similarly, murine cytomegalovirus (MCMV) was shown to be infectious for rat cells (Bruggeman et al, 1982; Smith et al, 1986), but the rat cytomegalovirus did not replicate in murine fibroblasts (Bruggeman et al, 1982). However, cells of other more distant species are usually nonpermissive. A number of studies have shown that CMVs can enter cells of other species and express a subset of viral genes (Kim and Carp, 1972; Fioretti et al, 1973; Lafemina and Hayward, 1988; Garcia-Ramirez et al, 2001). This has led to the conclusion that the restriction to CMV replication in nonpermissive cells is associated with a postpenetration block to viral gene expression and DNA replication, but not due to a failure to enter the cell (Mocarski and Courcelle, 2001). However, the molecular mechanism for this intracellular block to viral replication has remained elusive.
In this study, we show that MCMV infection of human cells activates programmed cell death (apoptosis). This prevents efficient viral DNA replication and expression of late genes. Inhibition of apoptosis by a Bcl-2-like protein of human CMV or a different virus is sufficient to allow replication of MCMV or the rat CMV in human cells.
Results
MCMV replicates in human 293 and 911 cells
Previous studies have shown that CMVs can enter cells of other species but do not replicate in them (Kim and Carp, 1972; Fioretti et al, 1973; Lafemina and Hayward, 1988). To analyze this phenomenon, we infected various human cell types with an MCMV expressing the green fluorescent protein (MCMV-GFP) at a low multiplicity of infection (MOI). MCMV-GFP infected human cells as indicated by GFP expression, but the infection did not spread from infected to uninfected neighboring cells, and the GFP expressing cells disappeared over time (not shown). Only in human embryonic kidney 293 and human embryonic retinoblast 911 cells, a cell-to-cell spread of infection was noticed: groups of green cells and occasionally even the formation of small plaques could be observed (Figure 1A and B). A growth kinetic experiment showed that human 293 and 911 cells can support MCMV replication, although virus release was delayed and reached lower titers as compared to permissive murine cells (Figure 1C). In primary human embryonic lung fibroblasts (MRC-5) and retinal pigment epithelial cells (RPE1) as well as in all other human cells analyzed, MCMV replication was not detected. RPE1 and MRC-5 cells were used for further analyses, because they are among the few cells that are permissive for HCMV. When infected at a low MOI, 293 and 911 cells did not release substantial amounts of virus into the supernatant (data not shown). Apparently, the cells infected at a low MOI produced enough virus to allow a limited spread to neighboring cells, but not enough to be detectable in the supernatant by plaque assay.
Figure 1.
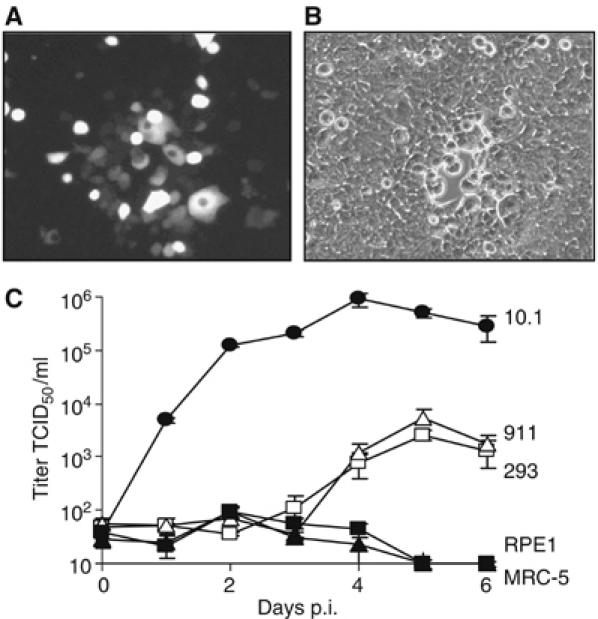
MCMV replication in human 293 and 911 cells. (A, B) Fluorescent and phase contrast images of 293 cells 6 days after infection with MCMV-GFP at a low MOI. (C) Growth kinetic of MCMV-GFP on murine 10.1 cells and human 293, 911, RPE1 and MRC-5 cells. Cells were infected at an MOI of 5 TCID50/cell. Virus titers were determined in the supernatant.
Impaired viral gene expression and apoptosis of nonpermissive human cells
To identify a block in the cascade of viral gene expression as a possible underlying mechanism of the species barrier, the expression of an immediate-early (IE1), an early (E1), an early-late (M44) and a late (gB) gene was analyzed in permissive and nonpermissive cells (Figure 2A). In 293 cells, all kinetic classes of genes were expressed, and viral DNA was replicated, again with delay as compared to the murine cells. In the nonpermissive RPE1 cells, by contrast, expression of gB was not detectable, and DNA replication occurred only transiently. Starting at day 2 postinfection, massive cell death was observed: cells disintegrated and detached from the culture dish. The extent of cell death can be seen in a time course experiment that measured cell viability up to 120 h postinfection (Figure 2B). An apoptosis-specific assay showed that nonpermissive human cells entered apoptosis upon MCMV infection (Figure 2C). The DNA degradation associated with apoptosis probably accounts for the loss of viral DNA seen in RPE1 cells (Figure 2A). Interestingly, Kim and Carp (1972) had already observed that none of the human WI-38 fibroblasts used in their study survived an MCMV infection, but apparently this observation was not further investigated.
Figure 2.
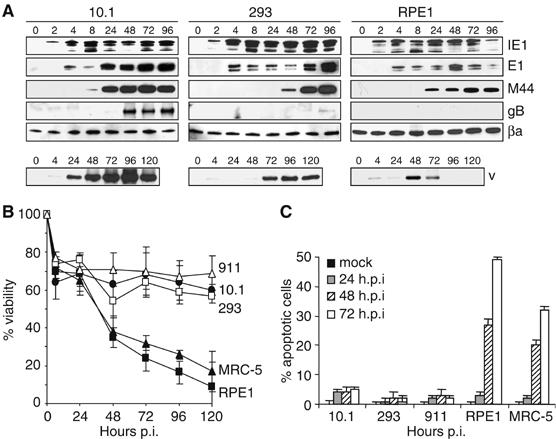
Impaired MCMV gene expression and apoptosis of nonpermissive human cells. (A) Permissive murine 10.1 cells, permissive human 293 cells, and nonpermissive human RPE1 cells were infected at an MOI of 5 TCID50/cell. Viral immediate-early (IE1), early (E1), early-late (M44) and late (gB) gene expression was analyzed by Western blot at the indicated hours postinfection. Viral DNA (vDNA) was detected by Southern blot hybridization. Both attached and floating cells were collected for Western and Southern blot analyses. (B) Viability of cells infected at the same MOI was analyzed by MTT assay. (C) Nuclear DNA fragmentation as a sign of apoptosis was analyzed by TUNEL assay on adherent cells on coverslips. Since apoptotic cells tend to detach from the coverslips, the true percentage of apoptotic cells is higher than the percentage determined by TUNEL assay.
A viral bcl-2 homolog is sufficient to facilitate MCMV replication in human cells
Based on these observations, we asked which properties of 293 and 911 cells made them resistant to MCMV-induced apoptosis and permissive for MCMV replication. These two cell lines differ from all other human cells we have analyzed in that they were transformed with adenovirus type 5 DNA and express adenoviral E1A and E1B genes (Graham et al, 1977; Fallaux et al, 1996). The 293 cell line also contains sequences from the adenovirus E4 region, but the 911 cell line does not (Fallaux et al, 1996). E1A is a strong transactivator of gene expression and is proapoptotic. The E1B genes are antiapoptotic either by binding and sequestering p53 (E1B-55k) or by functioning as a viral Bcl-2-like protein (E1B-19k) (Shenk, 2001). Therefore, we transduced human RPE1 cells with retroviral vectors encoding E1B-55k or E1B-19k, in order to determine if one of these genes could make these cells permissive for MCMV replication. Transgene expression was tested by Western blot (Figure 3A) and immunofluorescence (not shown). E1B-55k had no effect on the permissivity of RPE1 cells, whereas E1B-19k inhibited MCMV-induced apoptosis and rendered RPE1 cells fully permissive for MCMV replication (Figure 3B and C). Even after low MOI infection, the virus spread across the whole monolayer and reached remarkably high titers (Figure 3D). Similarly, overexpression of the cellular bcl-2 or bcl-XL gene conferred permissivity, as did the HCMV UL37x1 gene, which encodes a mitochondria-localized inhibitor of apoptosis (vMIA), a protein with a function similar to Bcl-2 (Goldmacher et al, 1999; Arnoult et al, 2004) (Figure 3B and C). Thus, a single gene of HCMV can facilitate MCMV replication in human cells.
Figure 3.
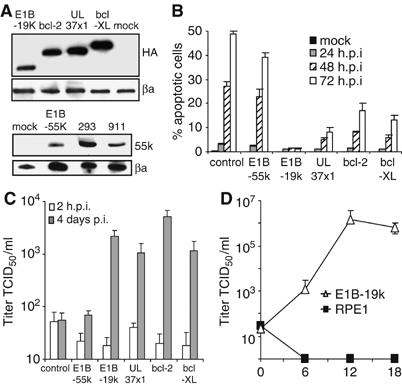
MCMV replication in RPE1 cells expressing E1B-19k. (A) Western blot of transduced RPE1 cells expressing HA-tagged E1B-19k, bcl-2, bcl-XL, UL37x1/vMIA, or untagged E1B-55k. Proteins were detected with an anti-HA or an anti-E1B-55k antibody, respectively. 293 and 911 cells served as positive controls for E1B-55k expression. βa, beta actin. (B) Apoptosis of transduced and control RPE1 cells after high-MOI infection with MCMV-GFP was determined by TUNEL assay as in Figure 2C. (C) Virus release after infection of RPE1 cells at an MOI of 5 TCID50/cell. The 2 h value represents the residual input virus after infection and washing. (D) Virus release after infection of RPE1 cells expressing E1B-19k at an MOI of 0.2 TCID50/cell.
If a bcl-2-like protein is sufficient to allow MCMV replication in cultured human cells, it should be possible to construct a recombinant MCMV that can replicate in human cells. To create space for the insertion of foreign genes, we deleted the region m02–m06 of the MCMV genome. This region contains well-characterized immune evasion genes that are necessary for MCMV virulence and pathogenicity in mice (Kleijnen et al, 1997; Reusch et al, 1999; Oliveira et al, 2002; Wagner et al, 2002). Therefore, the recombinant viruses should be nonvirulent in an immunocompetent host. A total of 12 different apoptosis-related genes and control genes, driven by a phosphoglycerate kinase (pgk) promoter, were inserted into the MCMV genome (Figure 4A and B). In addition to the adenoviral and bcl-2 family genes used in the previous experiments, a Bcl-2 homolog from Kaposi Sarcoma associated Herpesvirus (KSBcl-2) (Cheng et al, 1997) as well as the antiapoptotic genes crmA of cowpox virus (Miura et al, 1993; Tewari and Dixit, 1995), p35 of baculovirus (Xue and Horvitz, 1995), E3L of vaccinia virus (Garcia et al, 2002), and IE1 of HCMV (Zhu et al, 1995) were included. The recombinant viruses were tested for their ability to facilitate MCMV growth on human RPE1 and MRC-5 cells. Figure 4 shows that only MCMVs expressing a bcl-2-like gene were able to replicate in human cells, whereas MCMVs expressing viral genes that inhibit different checkpoints of apoptosis were not. Even after infection at a very low MOI, the recombinant viruses containing a bcl-2-like gene could form plaques on RPE1 cells (Figure 4E). The infection spread across the entire monolayer and could be passaged serially on RPE1 cells.
Figure 4.
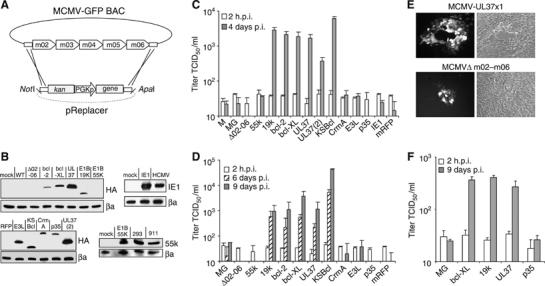
Growth of recombinant MCMVs in human cells. (A) Construction of recombinant MCMVs using the BAC technology. Inserted genes were driven by a murine pgk promoter (PGKp). (B) Expression of the inserted genes in cells infected with the recombinant viruses. HA-tagged proteins were detected with an anti-HA antibody. The HCMV IE1 protein and the E1B-55k protein were detected with specific antibodies. The wildtype MCMV-GFP (MG) and recombinant MCMVs carrying the monomeric red fluorescent protein (mRFP) gene or a deletion of m02–m06 only were used as controls. UL37(2) represents a recombinant virus, in which UL37x1/vMIA was inserted in place of the m152 gene. (C) Virus release after infection of RPE1 cells at an MOI of 5 TCID50/cell. The 2 h values represent the residual input virus after infection and washing. M, MCMV wild-type virus. (D) Virus release after infection of RPE1 cells at an MOI of 0.2 TCID50/cell. (E) Fluorescent and phase contrast image of RPE1 cells 8 days after a low-MOI infection with an MCMV expressing a bcl-2-like protein (MCMV-UL37x1) or a control virus (MCMVΔm02-m06). Efficient spread of the infection and plaque formation was only seen with MCMVs expressing a bcl-2-like protein. (F) Virus release after infection of MRC-5 cells at an MOI of 5 TCID50/cell.
To test if the site of insertion is important, the UL37x1/vMIA gene was also inserted at a different location. It replaced the immune evasion gene m152 (Ziegler et al, 1997) and was driven by the endogenous m152 promoter. This recombinant virus also replicated in RPE1 cells, albeit to slightly lower titers (Figure 4C). This may reflect the fact that the m152 promoter is an early promoter and as such does not provide the gene product immediately after infection.
An MCMV mutant expressing the adenovirus E1A gene could not be generated. Murine fibroblasts transfected with the recombinant genome showed morphological signs of apoptosis, probably because E1A is proapoptotic (Shenk, 2001). The infection did not spread to neighboring cells, and thus infectious virus could not be obtained.
Activation of caspase-9 by MCMV infection
Bcl-2 and its homologs block apoptosis by inhibiting mitochondrial cytochrome c release and subsequent activation of caspase-9 (Kuwana and Newmeyer, 2003). Our observation that bcl-2-like proteins inhibit MCMV-induced apoptosis of human cells suggested that MCMV infection causes activation of caspase-9 and cleavage of downstream effectors such as caspase-3 and poly(ADP-ribose) polymerase (PARP). Indeed, caspases-9 and -3 and PARP were cleaved in MCMV-infected RPE1 cells (Figure 5A). Similarly, these molecules were cleaved in cells infected with an HCMV (strain AD169) in which UL37x1/vMIA was deleted. This is consistent with a previous study, which has found PARP cleavage in human fibroblasts infected with a UL37x1 mutant of AD169 (Reboredo et al, 2004). Caspase-9, caspase-3, and PARP were not activated when viral DNA replication was inhibited by phosphonoacetic acid (PAA), indicating that viral DNA replication or events after DNA replication were responsible for the induction of apoptosis (Figure 5A). MCMV-induced death of RPE1 was significantly reduced when broad-spectrum caspase inhibitors were added after infection (Figure 5B and C), supporting the notion that caspase-mediated cell death inhibited viral replication. zVAD-fmk at 100 μM concentration was the most effective inhibitor (Figure 5B and C). Unfortunately, Boc-D-fmk could not be used at this high concentration, because it turned out to be toxic for cells (not shown).
Figure 5.
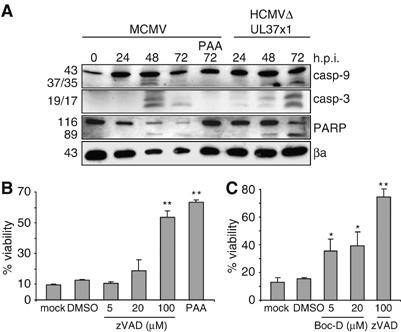
Caspase activation upon MCMV infection of human cells. (A) Human RPE1 cells were infected at an MOI of 5 TCID50/cell. Cleavage (activation) of caspases 9 and 3 and PARP was detected at 48 and 72 h postinfection, but not in the presence of PAA. Similarly, an HCMV UL37x1 deletion mutant induced activation of these molecules. (B, C) Cell death triggered by MCMV infection of RPE1 cells was inhibited by broad-spectrum caspase inhibitors zVAD-fmk and Boc-D-fmk or by an inhibitor of viral DNA replication (PAA, 250 μg/ml). Significance levels were calculated using ANOVA. *P<0.05; **P<0.001.
It has previously been shown that the cowpox virus CrmA (an inhibitor of caspase-1 and -8) and the baculovirus p35 protein (a substrate inhibitor of many caspases) can also inhibit caspase-9 in recombinant protein assays (reviewed in Ekert et al, 1999). However, later studies have shown that both, CrmA and p35, do not inhibit caspase-9-mediated cell death in living cells (Ryan et al, 2002). Nevertheless, we wanted to exclude the possibility that CrmA and p35 failed to facilitate MCMV replication (Figure 4), because the genes were inactivated or expressed at insufficient levels by the recombinant MCMVs. We also wondered, whether a recently discovered positional homolog of UL37x1 in MCMV, m38.5, might be expressed by MCMV at levels insufficient to inhibit MCMV-induced apoptosis of human cells. The m38.5 protein has a low-level sequence similarity to UL37x1/vMIA, localizes to mitochondria, and inhibits proteasome inhibitor-mediated, but not Fas-mediated apoptosis (McCormick et al, 2005). However, its activity against virus-induced apoptosis and its role for MCMV replication have not been studied yet. To address these questions, we transduced RPE1 cells with retroviral vectors encoding CrmA, p35, or m38.5, respectively. Expression of the proteins was verified by Western blot (not shown). The cells were then tested for resistance against proteasome inhibitor- and Fas-mediated cell death (as carried out by McCormick et al, 2005) and against MCMV-induced apoptosis. Figure 6 shows that CrmA was a potent inhibitor of Fas-meditated apoptosis, whereas m38.5 protected cells from apoptosis induced by the proteasome inhibitor MG-132. The baculovirus p35 had only a moderate activity against Fas- and proteasome inhibitor-induced cell death. However, none of these three proteins protected from MCMV-induced cell death. This confirms the results obtained with the recombinant MCMVs (Figure 4). The inability of CrmA and p35 to inhibit MCMV-induced apoptosis is consistent with previous studies, which have shown that CrmA and p35 expressed by recombinant Sindbis viruses conferred only very little protection against Sindbis virus-induced neuronal cell death in a mouse model of apoptosis (Nava et al, 1998; Ryan et al, 2002). By contrast, Bcl-2 and a dominant-negative caspase-9 protected mice efficiently in the same system (Levine et al, 1996; Ryan et al, 2002). Apparently, the inhibitory activity of p35 against effector caspases (Ekert et al, 1999) is insufficient for blocking Sindbis virus- or cytomegalovirus-induced apoptosis.
Figure 6.
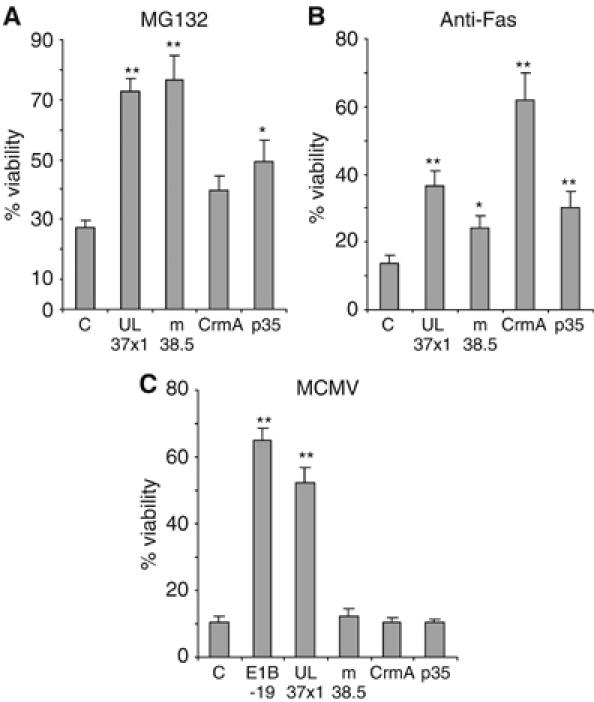
Transduced RPE1 cells stably expressing UL37x1/vMIA, m38.5, CrmA, or p35 were treated with (A) the proteasome-inhibitor MG-132 or (B) anti-Fas antibody plus cycloheximide, or (C) infected with MCMV to induce apoptosis. Cell viability was measured using an MTT assay. C, RPE1 control cells Significance levels were calculated using ANOVA. *P<0.05; **P<0.001.
Replication of RCMV in human cells
To analyze whether the requirement of a bcl-2-like gene is specific for MCMV or whether this principle is of more general importance, we infected RPE1 cells and RPE1 cells expressing E1B-19k with rat cytomegalovirus (RCMV, Maastricht strain). Like MCMV, RCMV also induced cell death in RPE1 cell, which could be suppressed by E1B-19k (Figure 7A). RCMV grew only in the presence of a bcl-2-like protein such as E1B-19k (Figure 7B). The amounts of virus released from RPE1-E1B-19k cells were relatively low, but the plaque formation seen in these cells after low-MOI infection clearly indicated virus replication and spread (Figure 7C). Taken together, the results suggest that the inability to inhibit apoptosis in human cells limits cross-species infections and represents an important determinant of MCMV's and RCMV's species specificity.
Figure 7.
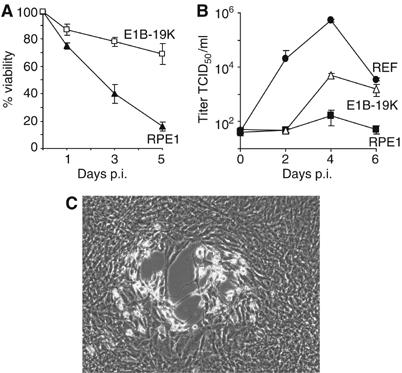
Growth of rat cytomegalovirus in human cells. (A) RPE1 cells died after infection with RCMV at an MOI of 5 TCID50/cell, but RPE1 cells expressing E1B-19k were mostly protected from RCMV-induced cell death. (B) RPE1 cells, RPE1-E1B-19k cells, and rat embryo fibroblasts (REF) were infected with RCMV at an MOI of 5 TCID50/cell, and titers in the supernatant were determined. (C) Phase contrast image of RPE1 cells expressing E1B-19k 7 days after low-MOI infection with RCMV. Plaque formation as an indication of virus replication and spread was only observed in RPE1-E1B-19k cells, but not in normal RPE1 cells.
Discussion
Previous analyses of the CMV species specificity have indicated that the host cell restriction to CMV replication observed in nonpermissive cells is the result of a postpenetration block to viral gene expression and not of a failure to enter cells (Kim and Carp, 1972; Fioretti et al, 1973; Lafemina and Hayward, 1988). However, the nature of this block has remained elusive.
Inside the cell, viruses need to overcome several innate immune defenses of the cell in order to replicate and spread efficiently. These include toll-like receptor signaling, triggering of the interferon response, activation of cellular stress responses, and induction of apoptosis. Here we show that MCMV infection of human cells leads to activation of caspase-9 and induction of apoptosis. Antiapoptotic Bcl-2 family proteins can inhibit mitochondrial release of cytochrome c and subsequent activation of caspase-9. This study demonstrates that expression of such a protein inhibits apoptosis induced by cytomegalovirus infection and allows MCMV replication in human cells. In this context, it should be noted that Bcl-2 and the HCMV vMIA protein can also inhibit caspase-independent cell death (Roumier et al, 2002). Therefore, we cannot exclude that this activity contributed to the inhibition of MCMV-induced apoptosis. However, the fact that two broad-spectrum caspase inhibitors, zVAD-fmk and Boc-D-fmk, were capable of inhibiting virus-induced cell death argues for an important role of caspases in this process. At high concentrations, zVAD-fmk can also inhibit calpains. These molecules are activated upon genotoxic stress and act upstream of caspases (Waterhouse et al, 1998). This suggests a possible role of calpains in MCMV-induced apoptosis and could explain why zVAD-fmk is a somewhat more potent inhibitor than Boc-D-fmk.
Our finding that the HCMV vMIA protein can facilitate MCMV replication in human cells is in agreement with previous results showing that this protein is required for efficient replication of the HCMV laboratory strain AD169varATCC (Brune et al, 2003; Yu et al, 2003; Reboredo et al, 2004) and the clinical strain VR1814/FIX (S Hölzer and W Brune, unpublished). However, it was recently reported that vMIA is not required for efficient replication of the HCMV strain Towne, although a UL37x1 deletion mutant caused increased apoptosis of infected fibroblasts (McCormick et al, 2005). Since Towne and FIX both encode a functional copy of the inhibitor of caspase-8 activation (vICA), a gene product of UL36, whereas AD169 does not, the requirement of vMIA appears to be strain-dependent, but not dependent on the function of vICA.
Insertion of the UL37x1 gene into the MCMV genome is sufficient to facilitate MCMV growth in human cells. This leaves two possible explanations: either MCMV does not encode an analogous protein and does not need it for replication in murine cells, or the virus does encode such a protein, but it functions in a species-specific manner (i.e. in murine but not in human cells). A recent reevaluation of the MCMV genome sequence has identified a previously unrecognized ORF, m38.5, which is a positional homolog of UL37x1/vMIA and shows a low-level sequence similarity to this HCMV protein (McCormick et al, 2003). The m38.5 protein localizes to mitochondria and inhibits cell death induced by a proteasome inhibitor, but does not inhibit Fas-induced apoptosis of human HeLa cells like vMIA does (McCormick et al, 2005). Our results confirm this activity and show that m38.5 cannot block MCMV-induced cell death in human cells. It remains to be determined whether the m38.5 protein is responsible for inhibiting virus-induced apoptosis in murine cells, which would suggest a species-specific function. Another recent study has detected increased levels of the cellular antiapoptotic bcl-2 family protein Bfl-1/A1, but also of the proapoptotic protein Bim in MCMV-infected dendritic cells (Andoniou et al, 2004). Although the study did not resolve whether the increased Bfl-1/A1 levels were responsible for the observed resistance of infected cells against apoptotic stimuli, it points out the possibility that MCMV could compensate for a lack of a Bcl-2-like protein by upregulating a cellular antiapoptotic gene.
The present study shows that inhibition of apoptosis enables not only the murine but also the rat cytomegalovirus to cross the species barrier and replicate in human cells. This indicates that the mechanism identified is not unique to MCMV. It also raises the question, whether—conversely—HCMV triggers apoptosis upon infection of rodent cells. Preliminary data from our laboratory indicate that infection of murine NIH-3T3 and 10.1 fibroblasts with HCMV strains AD169 or FIX does not induce significant levels of apoptosis, even when the cells were infected at a high MOI. A possible explanation for this can be found in a previous study, which has shown that HCMV does not reach the stage of viral DNA replication in mouse cells (Lafemina and Hayward, 1988). This is consistent with the observation that an inhibitor of viral DNA replication prevents the induction of apoptosis (Reboredo et al, 2004; and this study). It further suggests that the species specificity of HCMV depends on an additional intracellular mechanism, which prevents the onset of DNA replication. To date, it is not known whether viral DNA replication itself induces apoptosis, or whether later processes are responsible: viral DNA replication can activate the DNA damage response, which is known to be proapoptotic (Smith and Mocarski, 2005; Sinclair et al, 2006). Subsequently, DNA-filled capsids have to leave the nucleus, which is associated with dissolution of the nuclear lamina (Muranyi et al, 2002). This process is also likely to trigger apoptosis, if it does not occur at an appropriate time during the cell cycle. Viral glycoproteins traveling through the ER and Golgi could cause ‘ER stress'. This process, also known as ‘unfolded protein response', has recently been shown to be activated during HCMV replication (Isler et al, 2005).
Considering the large size and the protracted replication cycle of the CMVs, it seems likely that these viruses have found ways to subvert many if not all aspects of innate immunity. CMV proteins inhibiting apoptosis (Zhu et al, 1995; Goldmacher et al, 1999; Brune et al, 2001, 2003; Skaletskaya et al, 2001; Ménard et al, 2003) and the interferon response (reviewed in Hengel et al, 2005) have already been identified, and viral proteins blocking TLR signaling or the stress response are likely to follow. Analyses of viral inhibitors of the adaptive immune response encoded by HCMV and MCMV, for example, proteins downregulating MHC class I surface expression, have shown that some of the proteins do not function properly with target molecules of other species (Machold et al, 1997). Thus, it can be assumed that some of the viral inhibitors of the innate immune response will also operate in a species-specific manner. Inhibiting apoptosis of the infected cell is clearly a crucial task for the virus (Andoniou and Degli-Esposti, 2006) and can restrict the virus' cell tropism (Brune et al, 2001) and host range, as we show here. However, the fact that MCMV replicates slower and spreads less efficiently in human cells even in the presence of an antiapoptotic protein suggests that inhibition of apoptosis represents an important, but not the only limiting factor for efficient replication and spread.
We are only beginning to understand the molecular mechanisms underlying the species barrier of different viruses, and only few mechanisms have been identified to date. Failure to inhibit the interferon response was identified as a limiting factor for cross-species infections of certain paramyxo- and poxviruses (Parisien et al, 2002; Hornemann et al, 2003; Wang et al, 2004), which replicate in the cytoplasm. The present study shows that certain β-herpesviruses—large DNA viruses that replicate their genomes in the nucleus—induce apoptosis in cells of a foreign species, even though they can inhibit premature apoptosis in cells of their own species. A number of other viruses also depend on inhibition of apoptosis for normal replication, and consequently they encode potent cell death suppressors: Adenoviruses and γ-herpesviruses are two prominent examples (Pilder et al, 1984; White et al, 1984; Altmann and Hammerschmidt, 2005). It is possible that the species restriction of these viruses also depends (in part) on their ability to inhibit cell death in cells of a foreign species, even if in the end more than one mechanism should turn out to be involved, as it appears to be the case for human CMV.
Studies on viral species specificity teach us, how viruses counteract innate immune defenses, how these innate immune defenses operate, and how they differ from one species to another. These insights should lead to a better understanding of zoonotic infections in general.
Materials and methods
Cells
MRC-5 cells (ATCC CCL-171) are primary human embryonic lung fibroblasts (Jacobs et al, 1970). hTERT RPE1 cell (ATCC CRL-4000) are telomerase-immortalized retinal RPE1 (Bodnar et al, 1998). 293 cells (ATCC CRL-1573) are human embryonic kidney cells transformed with adenovirus 5 DNA (Graham et al, 1977). For the experiments in this study, the 293A subclone was used, which was selected for a flattened morphology (Invitrogen). 911 cells are human embryonic retinoblasts transformed with adenovirus 5 E1 genes (Fallaux et al, 1996). 10.1 and REF cells are spontaneously immortalized mouse and rat embryo fibroblasts, respectively (Burns et al, 1988; Harvey and Levine, 1991).
Plasmids and genes
The human bcl-2 and bcl-XL genes, adenovirus E1B-19k, cowpox virus crmA, baculovirus p35, and vaccinia virus E3L were cloned by PCR in pcDNA3 (Invitrogen), adding an HA tag to the 5′ end. Plasmids pcDNA-UL37x1HA and pcDNA-m38.5 contain the HCMV UL37x1 and the MCMV m38.5, respectively, tagged with an HA epitope at the 3′ end. pcDNA3-IE1 containing the HCMV IE1 gene was provided by Michael Nevels (University of Regensburg, Germany). Plasmid pRC-RSV-E1A containing the adenovirus E1A gene was provided by Thorsten Stiewe (University of Würzburg, Germany). The pSTK146 plasmid (Schiedner et al, 2000) containing the murine pgk promoter and the adenovirus 5 E1 region was provided by Stefan Kochanek (University of Ulm, Germany). The mRFP gene was excised from pRSET-mRFP1 (Campbell et al, 2002). The kanamycin resistance gene flanked by FRT sites was taken from pSLFRTkn (Atalay et al, 2002).
Retroviral transduction
The E1B-19k, E1B-55k, bcl-2, bcl-XL, and UL37x1 genes were inserted into the murine leukemia virus-based retroviral plasmid pLXSN (Clontech). Production of retroviral vectors using the Phoenix packaging cell line, and transduction of RPE1 cells was done as in previous studies (Brune et al, 2003). Transduced cells were selected with 700 μg/ml G418 and grown as bulk cultures without clonal selection.
Western blotting and immunofluorescence
For Western blot analysis, cells were lysed with lysis buffer containing 1% Triton X-100. Protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. For immunological detection, the following monoclonal antibodies were used: CROMA101 against MCMV IE1 and CROMA103 against E1 (both provided by Stipan Jonjic, University of Rijeka, Croatia), 2E8.21A against MCMV gB and 3B9.22A against M44 (both provided by Lambert Loh, University of Saskatchewan, Canada), 2A6 against E1B-55k and 1B12 against HCMV IE1 (both provided by Tom Shenk, Princeton University, USA). Antibodies against the HA epitope tag (16B12, Covance Research Products), β-actin (A5316, Sigma), caspases-3 and -9 (8G10 and 9502, Cell Signaling), and PARP (7D3–6, BD Biosciences) were purchased from suppliers as indicated. For immunofluorescence, cells were grown on coverslips, fixed with 3% paraformaldehyde, and permeabilized with 0.3% Triton X-100. Proteins were detected using the primary antibodies listed above and an AlexaFluor 594-coupled secondary antibody (Molecular Probes).
Viruses and growth kinetics
MCMV-GFP, a recombinant MCMV expressing the enhanced GFP, was constructed by Martin Messerle (Medical School Hannover, Germany) and has been used in previous studies (Brune et al, 2001, 2003). The Maastricht strain of RCMV (Bruggeman et al, 1982) was provided to us by Sebastian Voigt (Charité, Berlin, Germany). All recombinant viruses were constructed using bacterial artificial chromosome (BAC) technology (Brune et al, 2000) and are based on the MCMV-GFP BAC. Construction of these recombinant MCMVs has been approved by the Central Commision for Biological Safety (ZKBS) of the Federal Republic of Germany. To delete the immune evasion genes m02–m06 and insert foreign genes, a plasmid named pReplacer was constructed on the basis of pBluescriptII KS+ (Stratagene). It contains 50 nucleotide homologies to sequences upstream of m02 and downstream of m06, a kanamycin resistance gene, a pgk promoter, and a multiple cloning site as shown in Figure 4A. Foreign genes were inserted using the multiple cloning site. The mutagenesis cassette can be excised from the backbone of pReplacer with restriction enzymes NotI or SacII at the 5′ and ApaI or KpnI at the 3′ end. The linear recombination substrates were used for homologous recombination in Escherichia coli strain DY380 containing the MCMV-GFP BAC as previously described (Brune et al, 2003). Recombinant MCMV genomes were analyzed by restriction digest and Southern blot. Wild-type and recombinant MCMV were grown on murine 10.1 fibroblasts essentially as described (Brune et al, 1999), and RCMV was propagated of REFs. Titrations were performed on the same cells using the median tissue culture infectious dose (TCID50) method (Mahy and Kangro, 1996). For growth kinetics, cells were seeded in six-well plates and infected with MCMV at the indicated MOI. At 2 h after infection, cells were washed with PBS, and fresh medium was added. Medium was replaced at the indicated time points, and the content of virus in the supernatant was determined by titration. All growth kinetic experiments were performed in triplicate.
The HCMV mutant AD169ΔUL37x1 and the clinical HCMV strain VR1814 (FIX) have been described previously (Hahn et al, 2002; Brune et al, 2003).
Southern blot analysis
Adherent and floating (dead) cells were collected at the indicated time points, and DNA was extracted by standard procedures. Three micrograms of each DNA sample was digested with HindIII, separated on a 0.8% agarose gel, and transferred onto a nylon membrane. Hybridization with a digoxigenin-labeled probe directed against the GFP gene and chemiluminescent detection was performed using a DIG-High Prime DNA labeling and detection kit (Roche), according to the manufacturer's recommendations.
Apoptosis assays
Cell viability was determined by MTT assay, which measures mitochondrial activity, according to standard protocols. Briefly, cells were seeded in 96-well plates at 5 × 103 cells per well and treated with proapoptotic reagents or infected with MCMV. At appropriate time points after treatment, cells were incubated for 4 h with 100 μl medium containing 500 μg/ml 3-(4,5-dimethylthiazole-2-yl)-2,5- diphenyl tetrazolium bromide (MTT). The formazan crystals were solubilized with 100 μl of a 1:1 DMSO:ethanol mixture. The formazan concentration was measured at 570 nm using an ELISA plate reader. Every test was carried out with at least four replicates of each sample. Statistical analyses were carried out using the analysis of variance (ANOVA, F-test). To analyze nuclear DNA fragmentation as a late sign of apoptosis, cells were grown and infected on coverslips, fixed with 3% paraformaldehyde, and stained with a terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) assay kit (Roche). Nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI). The percentage of apoptotic cells was determined by counting 500 cells in about 20 random visual fields.
For induction of apoptosis, cells were treated with 10 μM of the proteasome inhibitor MG-132 (Calbiochem) or 0.2 μg/ml anti-Fas antibody (clone 7C11, Coulter) and 10 μg/ml cycloheximide (AppliChem). To analyze caspase and PARP cleavage, cells were infected at an MOI of 5 TCID50/ml, washed with PBS 2 h p.i., and harvested at the indicated time points. Lysates were prepared as described above, and proteins were detected by Western blot. The broad-spectrum caspase inhibitors zVAD-fmk and Boc-D-fmk were purchased from MBL International as 100 and 20 mM stock solutions in DMSO, respectively. RPE1 cells were infected at an MOI of 5. After 6 h, cells were washed and incubated with medium containing zVAD-fmk, Boc-D-fmk, DMSO, or PAA.
Acknowledgments
We thank S Erhard for technical assistance, Ed Mocarski, Ulrich Koszinowski, Tom Shenk, Sebastian Voigt, and Matthias Budt for a critical reading of the manuscript, and numerous colleagues for providing reagents. This study was supported by the Emmy Noether Program of the Deutsche Forschungsgemeinschaft (BR 1730/2) and SFB 479, TP B6.
References
- Altmann M, Hammerschmidt W (2005) Epstein–Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol 3: 2148–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoniou CE, Andrews DM, Manzur M, Ricciardi-Castagnoli P, Degli-Esposti MA (2004) A novel checkpoint in the Bcl-2-regulated apoptotic pathway revealed by murine cytomegalovirus infection of dendritic cells. J Cell Biol 166: 827–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoniou CE, Degli-Esposti MA (2006) Insights into the mechanisms of CMV-mediated interference with cellular apoptosis. Immunol Cell Biol 84: 99–106 [DOI] [PubMed] [Google Scholar]
- Arnoult D, Bartle LM, Skaletskaya A, Poncet D, Zamzami N, Park PU, Sharpe J, Youle RJ, Goldmacher VS (2004) Cytomegalovirus cell death suppressor vMIA blocks Bax—but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc Natl Acad Sci USA 101: 7988–7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atalay R, Zimmermann A, Wagner M, Borst E, Benz C, Messerle M, Hengel H (2002) Identification and expression of human cytomegalovirus transcription units coding for two distinct Fcgamma receptor homologs. J Virol 76: 8596–8608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranowski E, Ruiz-Jarabo CM, Pariente N, Verdaguer N, Domingo E (2003) Evolution of cell recognition by viruses: a source of biological novelty with medical implications. Adv Virus Res 62: 19–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279: 349–352 [DOI] [PubMed] [Google Scholar]
- Bruggeman CA, Meijer H, Dormans PH, Debie WM, Grauls GE, van Boven CP (1982) Isolation of a cytomegalovirus-like agent from wild rats. Arch Virol 73: 231–241 [DOI] [PubMed] [Google Scholar]
- Brune W, Hengel H, Koszinowski UH (1999) A mouse model for cytomegalovirus infection, unit 19.7. In Current Protocols in Immunology, Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W (eds), pp 19.7.1–19.7.13. New York: John Wiley & Sons [DOI] [PubMed] [Google Scholar]
- Brune W, Ménard C, Heesemann J, Koszinowski UH (2001) A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 291: 303–305 [DOI] [PubMed] [Google Scholar]
- Brune W, Messerle M, Koszinowski UH (2000) Forward with BACs: new tools for herpesvirus genomics. Trends Genet 16: 254–259 [DOI] [PubMed] [Google Scholar]
- Brune W, Nevels M, Shenk T (2003) Murine cytomegalovirus m41 open reading frame encodes a Golgi-localized antiapoptotic protein. J Virol 77: 11633–11643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns WH, Barbour GM, Sandford GR (1988) Molecular cloning and mapping of rat cytomegalovirus DNA. Virology 166: 140–148 [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY (2002) A monomeric red fluorescent protein. Proc Natl Acad Sci USA 99: 7877–7882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Nicholas J, Bellows DS, Hayward GS, Guo HG, Reitz MS, Hardwick JM (1997) A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc Natl Acad Sci USA 94: 690–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekert PG, Silke J, Vaux DL (1999) Caspase inhibitors. Cell Death Differ 6: 1081–1086 [DOI] [PubMed] [Google Scholar]
- Fallaux FJ, Kranenburg O, Cramer SJ, Houweling A, van Ormondt H, Hoeben RC, van der Eb AJ (1996) Characterization of 911: a new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum Gene Ther 7: 215–222 [DOI] [PubMed] [Google Scholar]
- Fioretti A, Furukawa T, Santoli D, Plotkin SA (1973) Nonproductive infection of guinea pig cells with human cytomegalovirus. J Virol 11: 998–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MA, Guerra S, Gil J, Jimenez V, Esteban M (2002) Anti-apoptotic and oncogenic properties of the dsRNA-binding protein of vaccinia virus, E3L. Oncogene 21: 8379–8387 [DOI] [PubMed] [Google Scholar]
- Garcia-Ramirez JJ, Ruchti F, Huang H, Simmen K, Angulo A, Ghazal P (2001) Dominance of virus over host factors in cross-species activation of human cytomegalovirus early gene expression. J Virol 75: 26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmacher VS, Bartle LM, Skaletskaya A, Dionne CA, Kedersha NL, Vater CA, Han J, Lutz RJ, Watanabe S, McFarland ED, Kieff ED, Mocarski ES, Chittenden T (1999) A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc Natl Acad Sci USA 96: 12536–12541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FL, Smiley J, Russell WC, Nairn R (1977) Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 36: 59–74 [DOI] [PubMed] [Google Scholar]
- Hahn G, Khan H, Baldanti F, Koszinowski UH, Revello MG, Gerna G (2002) The human cytomegalovirus ribonucleotide reductase homolog UL45 is dispensable for growth in endothelial cells, as determined by a BAC-cloned clinical isolate of human cytomegalovirus with preserved wild-type characteristics. J Virol 18: 9551–9555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn BH, Shaw GM, De Cock KM, Sharp PM (2000) AIDS as a zoonosis: scientific and public health implications. Science 287: 607–614 [DOI] [PubMed] [Google Scholar]
- Harvey DM, Levine AJ (1991) p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes Dev 5: 2375–2385 [DOI] [PubMed] [Google Scholar]
- Hengel H, Koszinowski UH, Conzelmann KK (2005) Viruses know it all: new insights into IFN networks. Trends Immunol 26: 396–401 [DOI] [PubMed] [Google Scholar]
- Hornemann S, Harlin O, Staib C, Kisling S, Erfle V, Kaspers B, Hacker G, Sutter G (2003) Replication of modified vaccinia virus Ankara in primary chicken embryo fibroblasts requires expression of the interferon resistance gene E3L. J Virol 77: 8394–8407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isler JA, Skalet AH, Alwine JC (2005) Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol 79: 6890–6899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JP, Jones CM, Baille JP (1970) Characteristics of a human diploid cell designated MRC-5. Nature 227: 168–170 [DOI] [PubMed] [Google Scholar]
- Kim KS, Carp RI (1972) Abortive infection of human diploid cells by murine cytomegalovirus. Infect Immun 6: 793–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleijnen MF, Huppa JB, Lucin P, Mukherjee S, Farrell H, Campbell AE, Koszinowski UH, Hill AB, Ploegh HL (1997) A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J 16: 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Newmeyer DD (2003) Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol 15: 691–699 [DOI] [PubMed] [Google Scholar]
- Lafemina RL, Hayward GS (1988) Differences in cell-type-specific blocks to immediate early gene expression and DNA replication of human, simian and murine cytomegalovirus. J Gen Virol 69: 355–374 [DOI] [PubMed] [Google Scholar]
- Lecossier D, Bouchonnet F, Clavel F, Hance AJ (2003) Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300: 1112. [DOI] [PubMed] [Google Scholar]
- Levine B, Goldman JE, Jiang HH, Griffin DE, Hardwick JM (1996) Bcl-2 protects mice against fatal alphavirus encephalitis. Proc Natl Acad Sci USA 93: 4810–4815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machold RP, Wiertz EJ, Jones TR, Ploegh HL (1997) The HCMV gene products US11 and US2 differ in their ability to attack allelic forms of murine major histocompatibility complex (MHC) class I heavy chains. J Exp Med 185: 363–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahy BWJ, Kangro HO (1996) Virology Methods Manual. San Diego, USA: Academic Press [Google Scholar]
- Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR (2003) Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114: 21–31 [DOI] [PubMed] [Google Scholar]
- McCormick AL, Meiering CD, Smith GB, Mocarski ES (2005) Mitochondrial cell death suppressors carried by human and murine cytomegalovirus confer resistance to proteasome inhibitor-induced apoptosis. J Virol 79: 12205–12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick AL, Skaletskaya A, Barry PA, Mocarski ES, Goldmacher VS (2003) Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology 316: 221–233 [DOI] [PubMed] [Google Scholar]
- Ménard C, Wagner M, Ruzsics Z, Holak K, Brune W, Campbell A, Koszinowski U (2003) Role of murine cytomegalovirus US22 gene family members for replication in macrophages. J Virol 77: 5557–5570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J (1993) Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell 75: 653–660 [DOI] [PubMed] [Google Scholar]
- Mocarski ES, Courcelle CT (2001) Cytomegaloviruses and their replication. In Fields Virology, Knipe DM, Howley PM (eds), 4th edn, pp 2629–2673. Philadelphia: Lippincott-Williams & Wilkins [Google Scholar]
- Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH (2002) Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297: 854–857 [DOI] [PubMed] [Google Scholar]
- Nava VE, Rosen A, Veliuona MA, Clem RJ, Levine B, Hardwick JM (1998) Sindbis virus induces apoptosis through a caspase-dependent, CrmA-sensitive pathway. J Virol 72: 452–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira SA, Park SH, Lee P, Bendelac A, Shenk TE (2002) Murine cytomegalovirus m02 gene family protects against natural killer cell-mediated immune surveillance. J Virol 76: 885–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien JP, Lau JF, Horvath CM (2002) STAT2 acts as a host range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J Virol 76: 6435–6441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perot K, Walker CM, Spaete RR (1992) Primary chimpanzee skin fibroblast cells are fully permissive for human cytomegalovirus replication. J Gen Virol 73: 3281–3284 [DOI] [PubMed] [Google Scholar]
- Pilder S, Logan J, Shenk T (1984) Deletion of the gene encoding the adenovirus 5 early region 1b 21,000-molecular-weight polypeptide leads to degradation of viral and host cell DNA. J Virol 52: 664–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboredo M, Greaves RF, Hahn G (2004) Human cytomegalovirus proteins encoded by UL37 exon 1 protect infected fibroblasts against virus-induced apoptosis and are required for efficient virus replication. J Gen Virol 85: 3555–3567 [DOI] [PubMed] [Google Scholar]
- Reusch U, Muranyi W, Lucin P, Burgert HG, Hengel H, Koszinowski UH (1999) A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J 18: 1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumier T, Vieira HL, Castedo M, Ferri KF, Boya P, Andreau K, Druillennec S, Joza N, Penninger JM, Roques B, Kroemer G (2002) The C-terminal moiety of HIV-1 Vpr induces cell death via a caspase-independent mitochondrial pathway. Cell Death Differ 9: 1212–1219 [DOI] [PubMed] [Google Scholar]
- Ryan CA, Stennicke HR, Nava VE, Burch JB, Hardwick JM, Salvesen GS (2002) Inhibitor specificity of recombinant and endogenous caspase-9. Biochem J 366: 595–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiedner G, Hertel S, Kochanek S (2000) Efficient transformation of primary human amniocytes by E1 functions of Ad5: generation of new cell lines for adenoviral vector production. Hum Gene Ther 11: 2105–2116 [DOI] [PubMed] [Google Scholar]
- Shenk T (2001) Adenoviridae: the viruses and their replication. In Fields Virology, Knipe DM, Howley PM (eds), 4th edn, pp 2265–2300. Philadelphia: Lippincott-Williams & Wilkins [Google Scholar]
- Sinclair A, Yarranton S, Schelcher C (2006) DNA-damage response pathways triggered by viral replication. Expert Rev Mol Med 8: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaletskaya A, Bartle LM, Chittenden T, McCormick AL, Mocarski ES, Goldmacher VS (2001) A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci USA 98: 7829–7834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GB, Mocarski ES (2005) Contribution of GADD45 family members to cell death suppression by cellular Bcl-xL and cytomegalovirus vMIA. J Virol 79: 14923–14932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CB, Wei LS, Griffiths M (1986) Mouse cytomegalovirus is infectious for rats and alters lymphocyte subsets and spleen cell proliferation. Arch Virol 90: 313–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG (2005) Characterization of the 1918 influenza virus polymerase genes. Nature 437: 889–893 [DOI] [PubMed] [Google Scholar]
- Tewari M, Dixit VM (1995) Fas- and tumor necrosis factor-induced apoptosis is inhibited by the poxvirus crmA gene product. J Biol Chem 270: 3255–3260 [DOI] [PubMed] [Google Scholar]
- Wagner M, Gutermann A, Podlech J, Reddehase MJ, Koszinowski UH (2002) Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J Exp Med 196: 805–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Ma Y, Barrett JW, Gao X, Loh J, Barton E, Virgin HW, McFadden G (2004) Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol 5: 1266–1274 [DOI] [PubMed] [Google Scholar]
- Waterhouse NJ, Finucane DM, Green DR, Elce JS, Kumar S, Alnemri ES, Litwack G, Khanna K, Lavin MF, Watters DJ (1998) Calpain activation is upstream of caspases in radiation-induced apoptosis. Cell Death Differ 12: 1051–1061 [DOI] [PubMed] [Google Scholar]
- Weiss RA (2003) Cross-species infections. Curr Top Microbiol Immunol 278: 47–71 [DOI] [PubMed] [Google Scholar]
- Weller TH (1970) Cytomegaloviruses: the difficult years. J Infect Dis 122: 532–539 [DOI] [PubMed] [Google Scholar]
- White E, Grodzicker T, Stillman BW (1984) Mutations in the gene encoding the adenovirus early region 1B 19,000-molecular-weight tumor antigen cause the degradation of chromosomal DNA. J Virol 52: 410–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue D, Horvitz HR (1995) Inhibition of the Caenorhabditis elegans cell-death protease CED-3 by a CED-3 cleavage site in baculovirus p35 protein. Nature 377: 248–251 [DOI] [PubMed] [Google Scholar]
- Yu D, Silva MC, Shenk T (2003) Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci USA 100: 12396–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Shen Y, Shenk T (1995) Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol 69: 7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler H, Thale R, Lucin P, Muranyi W, Flohr T, Hengel H, Farrell H, Rawlinson W, Koszinowski UH (1997) A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 6: 57–66 [DOI] [PubMed] [Google Scholar]