

Abstract
Mouse p53 is phosphorylated at Ser18 and Ser23 after DNA damage. To determine whether these two phosphorylation events have synergistic functions in activating p53 responses, we simultaneously introduced Ser18/23 to Ala mutations into the endogenous p53 locus in mice. While partial defects in apoptosis are observed in p53S18A and p53S23A thymocytes exposed to IR, p53-dependent apoptosis is essentially abolished in p53S18/23A thymocytes, indicating that these two events have critical and synergistic roles in activating p53-dependent apoptosis. In addition, p53S18/23A, but not p53S18A, could completely rescue embryonic lethality of Xrcc4−/− mice that is caused by massive p53-dependent neuronal apoptosis. However, certain p53-dependent functions, including G1/S checkpoint and cellular senescence, are partially retained in p53S18/23A cells. While p53S18A mice are not cancer prone, p53S18/23A mice developed a spectrum of malignancies distinct from p53S23A and p53−/− mice. Interestingly, Xrcc4−/−p53S18/23A mice fail to develop tumors like the pro-B cell lymphomas uniformly developed in Xrcc4−/− p53−/− animals, but exhibit developmental defects typical of accelerated ageing. Therefore, Ser18 and Ser23 phosphorylation is important for p53-dependent suppression of tumorigenesis in certain physiological context.
Keywords: apoptosis, gene expression, phosphorylation, tumor suppression
Introduction
The importance of p53 in tumor suppression is highlighted by studies demonstrating that p53 is the most commonly mutated tumor suppressor gene in human cancers (Hollstein et al, 1991). This essential role of p53 is evolutionarily conserved, as mice lacking p53 uniformly develop and die of cancers (Donehower et al, 1992; Jacks et al, 1994). While p53 is inert and relatively unstable in cells, the stability and activity of p53 are significantly induced after various cellular and genotoxic stresses, leading to cell cycle arrest, apoptosis and DNA repair (Ko and Prives, 1996). In this context, the interaction between p53 and Mdm2, Pirh2, or COPI, all E3 ubiquitin ligases and p53 transcriptional targets, leads to p53 ubiquitination and degradation (Haupt et al, 1997; Kubbutat et al, 1997; Leng et al, 2003; Dornan et al, 2004). In response to various stresses, p53 undergoes post-translational modifications, including phosphorylation and acetylation, leading to the disruption of its interaction with negative regulators and enhanced stability and activity (Martinez et al, 1997; Leng et al, 2003; Dornan et al, 2004).
Multiple phosphorylation sites at the N-terminus of p53, including Ser3, 6, 9, 15, 20, 33, 37 and Thr18 could be phosphorylated in response to DNA damage (Saito et al, 2002). Ser15 and Ser20 are highly conserved evolutionarily and correspond to Ser18 and Ser23 of mouse p53. Biochemical and cell line transfection studies suggest that these phosphorylation sites are involved in p53 stabilization and activation (Xu, 2003). The physiological roles of these phosphorylation sites have also been addressed in the knockin mice with either Ser18 or Ser23 to Ala mutations (p53S18A and p53S23A, respectively). These studies indicate an important role for Ser18 phosphorylation in activating p53-dependent apoptosis and cell cycle arrest after DNA damage (Chao et al, 2003; Sluss and Jones, 2003; Borges et al, 2004). However, p53S18A mice are not predisposed to spontaneous tumorigenesis, indicating that p53 Ser18 phosphorylation is not required for p53-dependent tumor suppression in ageing animals (Chao et al, 2003; Sluss and Jones, 2003). Studies of p53 responses in p53S23A embryonic stem (ES) cells and mouse embryonic fibroblasts (MEFs) indicate that Ser23 phosphorylation is dispensable for p53 stabilization and activation in response to DNA damage (Wu et al, 2002). Consistent with this finding, p53 responses to DNA damage in MEFs derived from germline p53S23A mice are also normal (MacPherson et al, 2004). However, in this study, p53 stabilization is modestly impaired in thymocytes after IR but significantly impaired in neurons of p53S23A mice after DNA damage. Therefore, Ser23 phosphorylation appears to stabilize p53 in a cell-type-dependent manner.
Since phosphorylation at Ser15 and Ser20 occur simultaneously after various types of DNA damage, and might be mediated by ATM-dependent signaling pathways after DNA double-strand breaks, the two phosphorylation events might have synergistic roles in activating p53-dependent tumor suppression (Ye et al, 2001; Saito et al, 2002; Kurz et al, 2004). To address this issue in a physiological context, we introduced S18/23A mutations into the endogenous p53 gene in mice. Our findings indicate that the two phosphorylation events are critical to activate p53-dependent apoptosis. In addition, they are important for p53-dependent suppression of spontaneous tumorigenesis, but dispensable for p53-dependent tumor suppression induced by excessive DNA damage as a result of DNA repair deficiency.
Results
Generation of p53S18/23A germline mice
The knockin strategy to introduce S18/23A mutations into the p53 gene is described in Figure 1. Heterozygous mutant ES cells were used to generate chimeric mice that transmitted the p53S18/23A allele into the germline. The F1 mutant heterozygous mice were bred with CMV-Cre transgenic mice to excise the LoxP-flanked PGK-NeoR gene from the targeted allele. The PGK-NeoR gene deleted heterozygous mice were intercrossed to generate homozygous mutant mice. The entire p53 cDNA derived from homozygous mutant MEFs was sequenced to verify that only the S18/23A mutations but not other mutations were present in the p53 gene of p53S18/23A mice. p53S18/23A mice were born at an expected Mendelian ratio (Figure 1F) and displayed no apparent developmental abnormalities.
Figure 1.
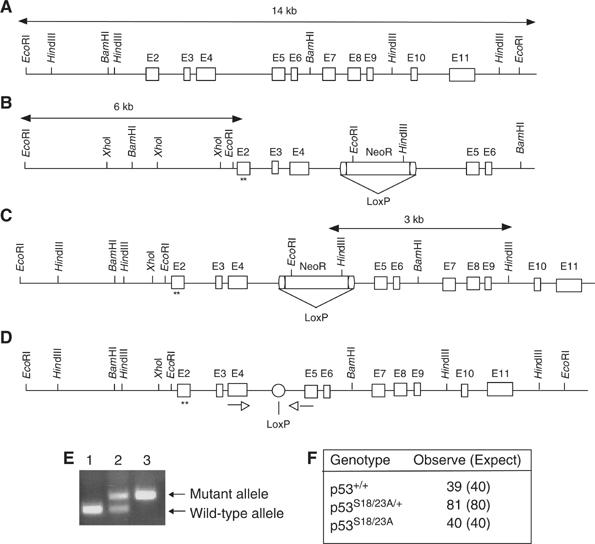
Generation of p53S18/23A knockin mice. (A) The endogenous mouse p53 locus. (B) The targeting construct. The PGK-NeoR cassette flanked by two loxP sites was inserted into intron 4. S18/23A mutations are indicated by asterisks. (C) The targeted p53 locus following homologous recombination between the endogenous p53 allele and targeting vector. (D) Knockin p53 allele following LoxP/Cre-mediated deletion of the PGK-NeoR gene. (E) PCR analysis of the genomic DNA isolated from the tails of WT (lane 1), p53S18/23A/+ (lane 2) and p53S18/23A mice (lane 3). The PCR products depicting the WT and knockin mutant allele are indicated to the right. The primers are indicated by arrowheads. (F) The number of offsprings in various genotypes derived from the breeding of p53S18/23A/+ mice. The expected number based on the Mendelian ratio is also shown.
p53 responses to DNA damage in p53S18/23A MEFs
Neither p53 Ser18 nor Ser23 phosphorylation affects p53 stability in MEFs after DNA damage (Wu et al, 2002; Chao et al, 2003; Sluss and Jones, 2003; MacPherson et al, 2004). To test whether these two phosphorylation events have synergistic roles in p53 stabilization in MEFs after DNA damage, protein levels of p53 in p53S18/23A MEFs at various time points after DNA damage were examined. Similarly to p53S18A MEFs (Chao et al, 2003), p53 protein levels were slightly higher in the p53S18/23A MEFs than in wild-type (WT) control MEFs in response to DNA damage induced by doxorubicin or UVC light (Figure 2A and B). To test whether Ser18 and Ser23 phosphorylation have synergistic roles in activating p53 transcription activities in MEFs, the mRNA levels of a number of p53 target genes in p53S18/23A MEFs after DNA damage were determined by quantitative real-time PCR (Figure 2C). When compared with those in WT MEFs, the mRNA levels of p21, Noxa, Bax and K/DR5 were similarly reduced in p53S18/23A MEFs and p53S18A MEFs after doxorubicin treatment (Figure 2C) as well as after IR (data not shown). In addition, similarly to p53S18A MEFs (Chao et al, 2003), p53-dependent cell cycle G1/S checkpoint is partially impaired in p53S18/23A MEFs after IR (Figure 2D, Chao et al, 2003). Therefore, Ser18 and Ser23 phosphorylation have no apparent functional synergy in p53 stabilization and activation in MEFs after DNA damage.
Figure 2.
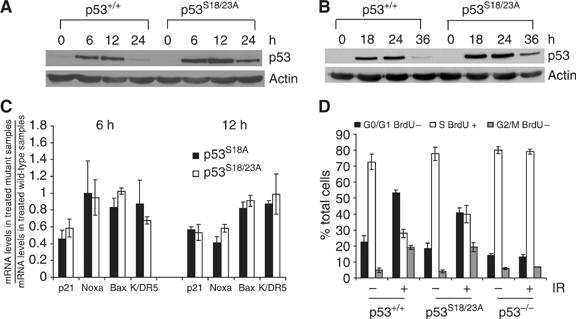
p53 stabilization and activity in p53S18/23A MEFs after DNA damage. Protein levels of p53 in the WT and p53S18/23A MEFs at various time points after exposure to 0.25 μM doxorubicin (A) or 60 J/M2 UVC (B). Time points and genotypes are indicated on the top. p53 and actin are indicated on the right. (C) Real-time PCR analysis of the p53-dependent transcription of p21, Noxa, Bax and K/DR5 at 6 and 12 h after doxorubicin treatment. The ratio of the mRNA levels in the untreated p53S18A or p53S18/23A MEFs versus those in treated WT MEFs is shown. Mean values from three independent experiments are shown with standard deviation. (D) Cell cycle G1/S arrest in WT, p53S18/23A and p53−/− MEFs after 20 Gy of IR. The genotypes are indicated at the bottom.
Cellular proliferation and genetic stability of p53S18/23A cells
To test whether p53S18/23A mutation affects lymphocyte proliferation, the proliferation of thymocytes derived from WT, p53S18/23A and p53−/− mice was analyzed at various time points after stimulation with PMA and Ionomycin as described (Chao et al, 2000; Kang et al, 2005). p53S18/23A thymocytes proliferated similarly to WT thymocytes but more slowly than p53−/− thymocytes at the later time point (Figure 3A). p53 is required for the replicative senescence of MEFs. Therefore, to test whether p53S18/23A mutation impacts on p53-dependent cellular senescence, we examined the proliferation and senescence of WT, p53S18/23A and p53−/− MEFs following a standard 3T3 proliferation protocol. As expected, p53−/− MEFS completely escaped senescence in culture (Figure 3B). p53S18/23A MEFs proliferated modestly faster than WT MEFs but significantly slower than p53−/− MEFs (Figure 3B). In addition, p53S18/23A MEFs reached senescence similarly to WT MEFs, but underwent spontaneous immortalization more easily than WT MEFs (Figure 3B).
Figure 3.
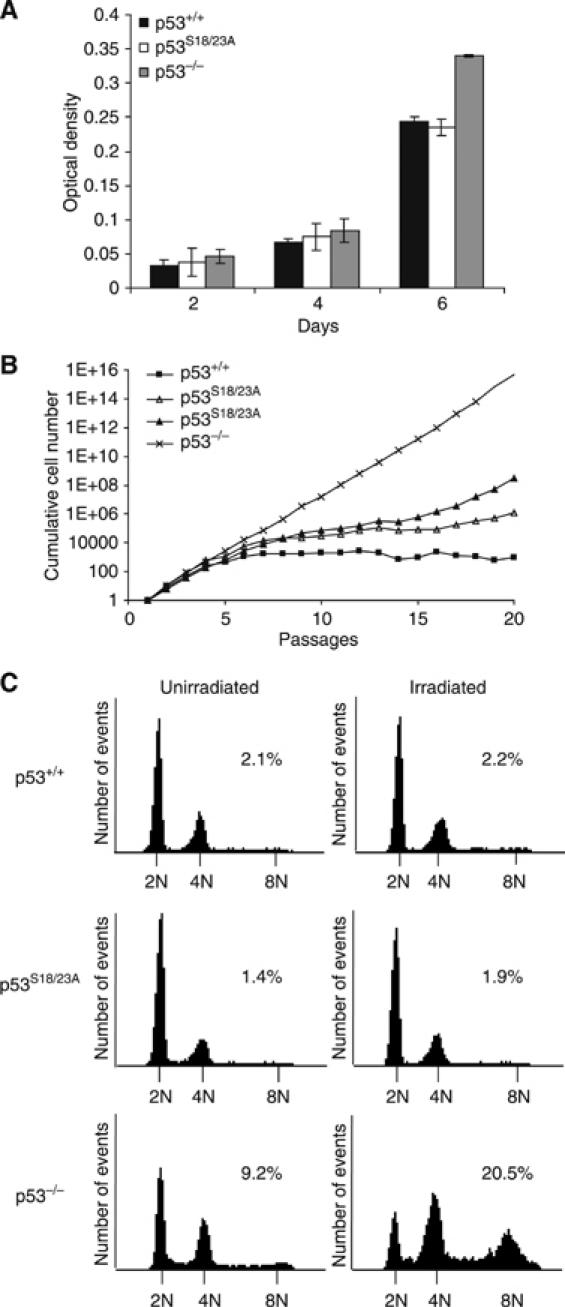
Cellular proliferation and polyploidy of p53S18/23A cells. (A) Proliferation of thymocytes derived from WT, p53S18/23A and p53−/− mice. Thymocytes were activated with 5 ng/ml PMA, 500 ng/ml ionomycin, and proliferation was measured after 2, 4 or 6 days of stimulation. Mean values from triplicate wells are shown with error bars. (B) 3T3 proliferation assay of WT, p53S18/23A and p53−/− MEFs. MEFs were serially passaged with a plating density of 3 × 105 cells per 6 cm plate and counted once every 3 days up to 20 passages. Cumulative cell numbers, based on the average of duplicate plates for each passage, are shown. Data from two independent p53S18/23A MEFs are presented. (C) Polyploidy in WT, p53S18/23A and p53−/− MEFs before (left panels) and 24 h after IR (right panels). Genotypes are indicated to the left. Histograms show DNA content on X-axis versus cell number on Y-axis. The peaks representing 2N, 4N and 8N cells are indicated. The percentage of total cells with over 8N DNA content is shown.
Polyploidy is the type of genetic instability consistently observed in p53−/− cells (Bischoff et al, 1990; Bouffler et al, 1995; Fukasawa et al, 1996) Therefore, we examined the polyploidy in the WT, p53S18/23A and p53−/− MEFs before and after DNA damage. In contrast to p53−/− MEFs that had high levels of polyploidy especially after IR, p53S18/23A MEFs exhibited normal diploid DNA content both before and after IR (Figure 3C). Therefore, p53-dependent suppression of polyploidy is retained in p53S18/23A cells.
p53-dependent apoptosis in p53S18/23A thymocytes
To test whether Ser18 and Ser23 play synergistic roles in p53 apoptotic activities, p53-dependent apoptosis in WT, p53S18A and p53S18/23A thymocytes was compared as previously described (Chao et al, 2003). Consistent with previous findings, p53-dependent apoptosis was partially impaired in p53S18A thymocytes after IR (Chao et al, 2003) (Figure 4A). In contrast, similarly to p53−/− thymocytes, p53S18/23A thymocytes were essentially resistant to p53-dependent apoptosis after IR (Figure 4A). Since p53-dependent apoptosis is only modestly reduced in p53S23A thymocytes after IR (MacPherson et al, 2004), these findings indicate that Ser18 and Ser23 phosphorylation play critical and synergistic roles in activating p53-dependent apoptosis after DNA damage. Consistent with this finding, when compared with that in WT thymocytes, p53-dependent induction of target genes was much more dramatically impaired in p53S18/23A thymocytes than in p53S18A thymocytes after IR (Figure 4B). In addition, with the exception of Puma, the extent of reduction of other analyzed p53 target genes was similar between p53S18/23A and p53−/− thymocytes after IR, further underscoring the importance of Ser18 and Ser23 phosphorylation in activating p53 activities in thymocytes after IR.
Figure 4.
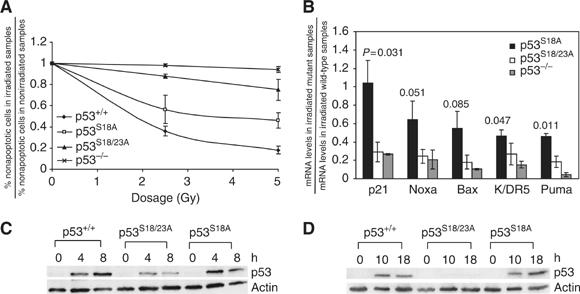
p53 stabilization and activity in p53S18/23A thymocytes after IR. (A) p53-dependent apotosis of WT, p53−/−, p53S18A and p53S18/23A thymocytes 24 h after exposure to 2.5 Gy and 5 Gy IR. Mean values from three independent experiments are shown with standard deviation. (B) p53-dependent transcription of its target genes in p53S18A, p53S18/23A and p53−/− thymocytes as compared to that in WT thymocytes 18 h after 5 Gy IR. Mean values from two independent experiments are shown with error bars. P-values between the levels of reduction in p53S18/23A thymocytes and p53S18A thymocytes are given. Protein levels of p53 in WT, p53S18/23A and p53S18A thymocytes at earlier (C) or later time points (D) after 5 Gy IR. Genotypes and time points are indicated on the top. p53 and actin are indicated on the right.
To understand this functional synergy, p53 protein levels in p53S18/23A thymocytes after IR were compared to those in WT and p53S18A thymocytes. As expected, Ser18Ala mutation has no apparent impact on p53 stabilization in thymocytes after IR (Chao et al, 2003). When compared to those in WT and p53S18A thymocytes, p53 protein levels were modestly reduced in p53S18/23A thymocytes at earlier time points after IR, but much more dramatically reduced at later time points after IR (Figure 4C and D). Therefore, the combination of the reduced p53 protein levels and its activities contribute to the loss of p53-dependent apoptosis in p53S18/23A thymocytes after IR.
p53S18/23A completely rescued embryonic lethality of Xrcc4−/− mice
Xrcc4 is a critical member of the mammalian nonhomologous end joining pathway (Li et al, 1995). Xrcc4-deficiency results in extensive DNA damage, accompanied by massive neuronal apoptosis in the embryo, leading to embryonic lethality (Gao et al, 2000). Both the neuronal apoptosis and embryonic lethality phenotypes can be completely rescued by p53 deficiency, indicating that neuronal apoptosis in Xrcc4−/− mice is p53-dependent (Gao et al, 2000). To test whether p53S18/23A mutation impairs p53 apoptotic activities in developing neurons in the presence of unrepaired DNA damage, p53S18/23A mutation was introduced into Xrcc4−/− mice. Similarly to Xrcc4−/−p53−/− mice, Xrcc4−/−p53S18/23A mice were born at the expected Mendelian ratio, indicating that p53S18/23A mutation can suppress the p53-dependent neuronal apoptosis in Xrcc4−/− embryos (Figure 5A). In support of this notion, the levels of apoptosis in the brain of Xrcc4−/−p53S18/23A embryo were similar to those in WT embryo, but much lower than those in Xrcc4−/−p53+/+ embryonic brain (Figure 5C). Therefore, p53S18/23A mutation essentially abolishes the p53-dependent apoptosis in the developing neurons in the presence of DNA damage. Consistent with the findings that Ser18A mutation only partially impairs p53 apoptotic activities (Chao et al, 2003), S18A mutation could rescue the embryonic lethality of Xrcc4−/− mice only at a very low frequency (Figure 5B).
Figure 5.
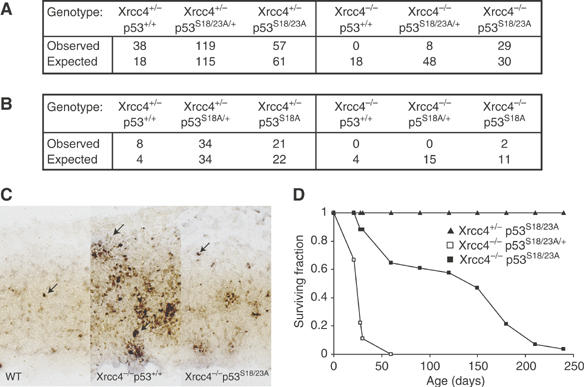
p53S18/23A mutation completely rescues the embryonic lethality of Xrcc4−/− mice. (A) The number of offspring in various genotypes derived from the following breeding: Xrcc4+/−p53S18/23A/+ × Xrcc4+/−p53S18/23A/+ or Xrcc4+/−p53S18/23A × Xrcc4+/−p53S18/23A. The expected number based on the Mendelian ratio is also shown. (B) The number of offsprings in various genotypes derived from either Xrcc4+/−p53S18A/+ × Xrcc4+/−p53S18A/+ or Xrcc4+/−p53S18A × Xrcc4+/−p53S18A breeding. (C) p53-dependent apoptosis in the embryonic cerebral cortex as shown by ISEL+. Saggital sections from E12.5 embryos were shown. The apoptotic cells are indicated by arrowheads. (D) The surviving percentage of Xrcc4+/−p53S18/23A (n=16), Xrcc4−/−p53S18/23A/+ (n=10) and Xrcc4−/−p53S18/23A (n=34) mice at various times after birth. N represents the number of mice monitored. P-value is 0.0004 for the comparison between Xrcc4−/−p53S18/23A/+ and Xrcc4+/−p53S18/23A mice and 0.001 for the comparison between Xrcc4−/−p53S18/23A and Xrcc4+/−p53S18/23A mice.
p53S18/23A mice are cancer prone
Since p53 apoptotic activities are greatly compromised in p53S18/23A mice, we monitored the spontaneous tumorigenesis in p53S18/23A and control WT mice. p53−/− animals primarily develop thymic lymphomas within 6 months of age (Donehower et al, 1992; Jacks et al, 1994). p53S18/23A mice were also cancer prone but developed tumors in a wide spectrum of tissues with a significantly delayed onset (Figure 6A). In this context, lymphomas of the spleen and lymph node, but not thymus are common in p53S18/23A mice. In addition, tumors rarely detected in p53−/− mice, including leukemias, fibrosarcomas, adenomas and granuloma, had also been observed in p53S18/23A mice (Figure 6B, Supplementary Figure 1). Therefore, Ser18 and Ser23 phosphorylation are important for p53-dependent suppression of spontaneous tumorigenesis in aging animals.
Figure 6.
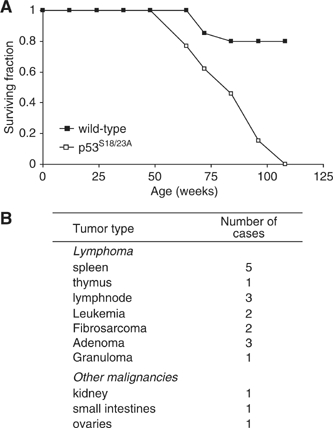
Aging p53S18/23A mice are prone to spontaneous cancer. (A) Survival curve of 20 pairs of WT and p53S18/23A mice. P-value is 0.04 for the comparison of the survival rate of p53S18/23A mice and WT controls. Animals were monitored for spontaneous tumorigenesis over the course of 2 years. (B) The spectrum of tumors developed in p53S18/23A mice.
p53S18/23A was able to suppress tumorigenesis in Xrcc4−/− mice
Xrcc4−/−p53−/− animals uniformly develop and die of B cell lymphomas by 8 weeks of age (Gao et al, 2000). The accelerated tumorigenesis is caused by widespread genetic instability since p53-deficiency allows the survival of cells with extensive DNA damage caused by DNA repair deficiency. While p53-dependent apoptosis is greatly reduced in p53S18/23A mice, leading to the complete rescue of embryonic lethality of Xrcc4-deficient mice, Xrcc4−/−p53S18/23A mice were not highly prone to tumors, since only two mice out of a cohort of 34 animals developed thymic tumors at 93 and 128 days of age. Despite the increased longevity of Xrcc4−/−p53S18/23A mice compared to Xrcc4−/−p53−/− animals, the median life span of Xrcc4−/−p53S18/23A mice was still significantly reduced compared to the Xrcc4+/− p53S18/23A controls (Figure 5D). In this context, Xrcc4−/− p53S18/23A mice were runted and displayed several aging-related phenotypes, including an acute spinal curvature, reduced thickness of the skin and testicular atrophy (Figure 7). In addition, Xrcc4−/− mice are immunodeficient due to the lack of mature lymphocytes (Gao et al, 2000). These findings suggest that Xrcc4−/−p53S18/23A mice die prematurely due to aging associated phenotypes.
Figure 7.
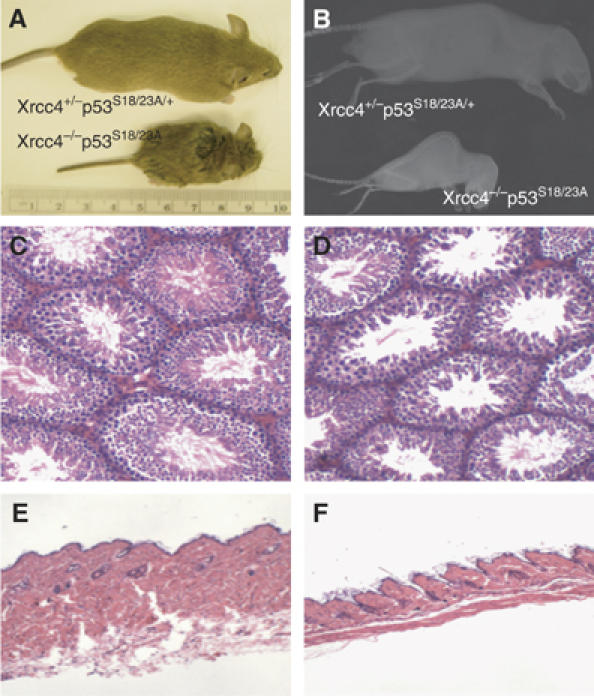
Xrcc4−/−p53S18/23A mice show accelerated aging phenotype. (A) Picture of 8-week-old male Xrcc4+/−p53S18/23A/+ and Xrcc4−/−p53S18/23A littermates. Histological analysis of testes (C, D) and skin (E, F) of the 8-week-old Xrcc4+/−p53S18/23A/+ (C, E) and Xrcc4−/−p53S18/23A (D, F) littermates. (B) X-ray image showing the spine curvature of 17-week-old Xrcc4+/−p53S18/23A/+ and Xrcc4−/−p53S18/23A littermates.
Discussion
Phosphorylation of human p53 at Ser15 and Ser20 occurs simultaneously after DNA damage (Saito et al, 2002). To address the physiological roles of these phosphorylation events, we introduced Ser18/23 (corresponding to Ser15 and Ser20 of human p53) to Ala mutations into the endogenous p53 gene in mice. p53-dependent apoptotic activities are greatly reduced in p53S18/23A thymocytes after IR. In addition, p53S18/23A mutation completely rescues the embryonic lethality of Xrcc4-deficient mice, which die of massive p53-dependent apoptosis in the embryonic neurons (Gao et al, 2000). Therefore, these two phosphorylation events are critical to activate p53-dependent apoptotic activities after DNA damage. The extent of the defective p53-dependent apoptosis after DNA damage appears to be much more severe in p53S18/23A mice than in p53S18A or p53S23A mice, indicating that Ser18 and Ser23 play synergistic roles in activating p53-mediated apoptotic activities after DNA damage (Chao et al, 2003; MacPherson et al, 2004). In this context, Ser18 phosphorylation has been shown to be important for activating p53-dependent transcription by recruiting co-activators to p53 target promoters (Lambert et al, 1998; Dumaz and Meek, 1999; Chao et al, 2003), and Ser23 phosphorylation is important for p53 stabilization in a cell-type-dependent manner (MacPherson et al, 2004). However, p53 stability and activity are similar between p53S18/23A and p53S18A MEFs after DNA damage, indicating that the functional synergy between these two phosphorylation events is cell-type specific.
Despite significant impairment of p53 responses to DNA damage in p53S18A mice, these mice are resistant to spontaneous tumorigenesis (Chao et al, 2003). In contrast, aging p53S18/23A mice spontaneously develop tumors at a high frequency. While the loss of p53-dependent apoptosis in p53S18/23A mice is similar to that in p53−/− mice, the onset of tumorigenesis in p53S18/23A mice is much slower than that in p53−/− mice, likely due to the partial retention of other p53-dependent functions. In this context, p53-dependent cell cycle G1/S checkpoint and cellular senescence are partially retained in p53S18/23A cells. In addition, p53S18/23A is sufficient to suppress polyploidy typically associated with p53-deficiency. The tumor spectrum in p53S18/23A mice is also different from that in p53−/− mice, which predominantly develop thymic lymphomas (Donehower et al, 1992; Jacks et al, 1994). The spectrum of tumors detected in p53S18/23A mice is much more close to the published tumor spectrum in p53+/− mice (Jacks et al, 1994). While p53S23A mice are also cancer prone and predominantly develop B cell lymphomas (MacPherson et al, 2004), p53S18/23A mice develop lymphomas as well as tumors in a number of other cell types. Therefore, the impact of Ser23Ala mutation on p53 tumor suppression activities might be more cell type-restricted than Ser18/23Ala mutations. Alternatively, the difference in tumor spectrum between the p53S18/23A and p53S23A mice could be partially due to the different genetic background of the two mutant strains of mice.
Nonhomologous end joining machinery is critical to maintain genetic stability in lymphocytes (Xu, 2006). In the absence of NHEJ and a functional p53, widespread genetic instability, particularly chromosomal translocations, leads to facilitated tumorigenesis in B lineage cells (Frank et al, 2000; Gao et al, 2000). In this context, p53 deficiency completely rescues the embryonic lethality of Xrcc4−/− mice, and Xrcc4−/−p53−/− mice uniformly die of B cell tumors by 3 months of age (Gao et al, 2000). p53S18/23A can also completely rescue the embryonic lethality of Xrcc4−/− mice. Interestingly, the Xrcc4−/−p53S18/23A do not develop tumors, but succumb to aging associated phenotypes. These findings indicate that despite the increased levels of DNA damage as a result of DNA repair deficiency, p53-dependent tumor suppression activity is retained in p53S18/23A mice. Since p53-dependent apoptosis upon damage is greatly reduced in p53S18/23A mice, these findings support the recent conclusion that p53-dependent apoptosis is not essential for p53-dependent tumor suppression (Liu et al, 2004). Considering that p53S18/23A animals are susceptible to spontaneous tumorigenesis, these findings also suggest that the type and/or level of stimuli to activate p53 tumor suppression in aging animals are different from those in DNA repair deficient mice. In conclusion, our studies demonstrate that Ser18 and Ser23 phosphorylation function synergistically in activating p53 apoptotic activities, and are required for p53-dependent tumor suppression in certain contexts.
Materials and methods
Generation of p53S18/23A knockin ES cells
Ser18 and Ser23 are both encoded by exon 2 of mouse p53. A mouse genomic fragment including p53 exon 2 was isolated, and nucleotides encoding Ser18 and Ser23 were simultaneously mutated to those encoding Ala residues by site-directed mutagenesis. The PGK-neomycing resistance gene (PGK-NeoR) flanked by two LoxP sites was inserted into the intron 4. The targeting construct was linearized with XbaI and electroporated in mouse J-1 ES cells, which were derived from 129 strain of inbred mice. Homologous recombination was confirmed by Southern blotting.
Introduction of p53 mutant alleles into Xrcc4-deficient mice
Xrcc4+/− mice were bred with either p53S18A or p53S18/23A homozygous mutant mice to generate Xrcc4+/−p53S18/+ or Xrcc4+/−p53S18/23A/+ mice, which were subsequently intercrossed to generate the double mutant mice.
Western blot analysis
Whole-cell lysates from 4 × 105 MEFs or 5 × 106 mouse thymocytes were separated by a 10% SDS–PAGE gel and transferred onto a nitrocellulose membrane. The membranes were blocked in 5% nonfat milk and probed with a polyclonal antibody against p53 (CM-5; Vector Laboratories Inc.) overnight at 4°C. The filter was then incubated with a horseradish peroxidase-conjugated secondary antibody, and developed with ECL Plus (Amersham Biosciences). To ensure that the protein amounts in all lanes were similar, the membrane was stripped and probed with a goat polyclonal antibody directed against β-actin (I-19; Santa Cruz Biotechnology).
Proliferation and cell cycle assays
The cell cycle G1/S checkpoint was analyzed as previously described (Xu et al, 1998). Briefly, 1.5 × 106 MEFs were synchronized at G0 by culturing in Dulbecco's modified Eagle's medium (DMEM) supplemented with 0.5% FCS for 4 days. Cells were either untreated or irradiated with 20 Gy IR and simultaneously released in normal growth media (10% FCS in DMEM) in the presence of 10 μM BrdU. After 24 h, MEFs were harvested and fixed in 70% ethanol, stained for BrdU and DNA content and analyzed by flow cytometry.
Proliferation of MEFs was analyzed following a standard 3T3 proliferation protocol. Briefly 3 × 105 MEFs were serially passaged in 6 cm dishes and counted every 3 days up to 20 passages. Two plates of each genotype were counted at each passage.
Thymocyte proliferation was performed as previously described (Chao et al, 2000). Briefly, single-cell suspension of thymocytes was cultured in growth media alone (RPMI 1640 media supplemented with 10% FCS, and 100 μg/ml penicillin/streptomycin) or in the presence of 5 ng/ml PMA and 500 ng/ml Ionomycin at a density of 106 cells/ml. Cell numbers were analyzed after 2, 4 or 6 days in culture using the CellTiter 96 Cell Proliferation kit (Promega).
p53-dependent apoptosis in mouse thymocytes
p53-dependent apopotosis was analyzed as previously described (Chao et al, 2003). Single-cell suspension of thymocytes was cultured in DMEM supplemented with 5% FCS and 10 mM HEPES (pH 7.0) at a density of 106 cells/ml. The cells were subsequently exposed to 0, 2.5 and 5 Gy of IR. The percentage of apoptotic cells was analyzed 24 h following IR by staining with flourescein isothiocyanate-labeled Annexin V (Pharmingen) and analyzed by flow cytometry.
Quantitative real-time PCR analysis
Total RNA from MEFs or thymocytes were isolated using Trizol Reagent (invitrogen) followed by Rneasy RNA cleanup (Qiagen) and on-column Dnase digestion (Qiagen).
One microgram of RNA was reverse transcribed using First Strand Superscript Synthesis System (Invitrogen). Real-time PCR was performed on an ABI Prism 7000 Sequence Detection System with SYBR Green PCR Master Mix (Applied Biosystems). The following PCR conditions were used: 10 min hot start at 95°C, followed by 40 cycle of 30 s at 95°C and 1 min at 60°C. Samples were run in triplicate and the mean threshold cycle (Ct) for each target gene analyzed was normalized to the Ct for the housekeeping gene Gapdh. Primer sequences for p21, Noxa, K/DR5, Bas, Puma and Gapdh were described previously (Chao et al, 2003).
Histology analysis
Tissues or tumor samples were harvested from animals upon death and fixed in 10% buffered formalin, embedded in paraffin and sliced in 6 μM sections. All sections were stained with hematoxylin and eosin for histological assessment. X-ray images of euthanized animals were taken using a Faxitron X-ray system.
Detection of neuronal apoptosis
In situ end-labeling plus (ISEL+) was performed essentially as described previously (Blaschke et al, 1996; Blaschke and Chun, 1998) Briefly, 20 μm-thick sections were obtained from freshly frozen embryos and collected on Superfrost plus slides (Fisher), fixed in 4% paraformaldehyde, acetylated, dehydrated through an ethanol series, and either used fresh or stored at −80°C. DNA was end-labeled with digoxygenin-11-dUTP (Roche) by incubation with terminal deoxynucleotidyl transferase (Invitrogen) for 1 h at 37°C. dUTP incorporation was detected by binding with an alkaline phosphatase-conjugated sheep antidigoxygenin antibody (1:2000)(Roche) and visualized by reacting with 5-bromo-4-chloro-3-indoxyl phosphate/tetranitroblue tetrazolium (Chemicon). Images were captured under direct illumination using a Zeiss Axio Imager.
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank Dr Nissi Varki for help with mouse pathology. This work was supported by grants from NIH (CA 94254) and Wadsworth foundation to YX and MH51699 and MH01723 to JC.
References
- Bischoff F, Yim S, Pathak S, Grant G, Siciliano M, Giovanella B, Strong L, Tainsky M (1990) Spontaneous abnormalities in normal fibroblasts from patients with Li-Fraumeni cancer syndrome: aneuploidy and immortalization. Cancer Res 50: 7979–7984 [PubMed] [Google Scholar]
- Blaschke JAW, Chun J (1998) Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J Comp Neurol 396: 39–50 [DOI] [PubMed] [Google Scholar]
- Blaschke A, Staley K, Chun J (1996) Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 122: 1165–1174 [DOI] [PubMed] [Google Scholar]
- Borges HL, Chao C, Xu Y, Linden R, Wang JY (2004) Radiation-induced apoptosis in developing mouse retina exhibits dose-dependent requirement for ATM phosphorylation of p53. Cell Death Differ 11: 494–502 [DOI] [PubMed] [Google Scholar]
- Bouffler S, Kemp C, Balmain A, Cox R (1995) Spontaneous and ionizing radiation-induced chromosomal abnormalities in p53-deficient mice. Cancer Res 55: 3883–3889 [PubMed] [Google Scholar]
- Chao C, Hergenhahn M, Kaeser MD, Wu Z, Saito S, Iggo R, Hollstein M, Appella E, Xu Y (2003) Cell type- and promoter-specific roles of Ser18 phosphorylation in regulating p53 responses. J Biol Chem 278: 41028–41033 [DOI] [PubMed] [Google Scholar]
- Chao C, Yang EM, Xu Y (2000) Rescue of defective T cell development and function in Atm−/− mice by a function. J Immunol 164: 345–349 [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS, Allan B (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215–221 [DOI] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O'Rourke K, Koeppen H, Dixit VM (2004) The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429: 86–92 [DOI] [PubMed] [Google Scholar]
- Dumaz N, Meek DW (1999) Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J 18: 7002–7010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW (2000) DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell 5: 993–1002 [DOI] [PubMed] [Google Scholar]
- Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF (1996) Abnormal centrosome amplification in the absence of p53. Science 271: 1744–1747 [DOI] [PubMed] [Google Scholar]
- Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, Chaudhuri J, Horner J, DePinho RA, Alt FW (2000) Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 404: 897–900 [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387: 296–299 [DOI] [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC (1991) p53 mutations in human cancers. Science 253: 49–53 [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA (1994) Tumor spectrum analysis in p53-mutant mice. Curr Biol 4: 1–7 [DOI] [PubMed] [Google Scholar]
- Kang J, Ferguson D, Song H, Bassing C, Eckersdorff M, Alt FW, Xu Y (2005) Functional interaction of H2AX, NBS1, and p53 in ATM-dependent DNA damage responses and tumor suppression. Mol Cell Biol 25: 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko LJ, Prives C (1996) p53: puzzle and paradigm. Genes Dev 10: 1054–1072 [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299–303 [DOI] [PubMed] [Google Scholar]
- Kurz EU, Douglas P, Lees-Miller SP (2004) Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J Biol Chem 279: 53272–53281 [DOI] [PubMed] [Google Scholar]
- Lambert PF, Kashanchi F, Radonovich MF, Shiekhattar R, Brady JN (1998) Phosphorylation of p53 Serine 15 increases interaction with CBP. J Biol Chem 273: 33048–33053 [DOI] [PubMed] [Google Scholar]
- Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S (2003) Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112: 779–791 [DOI] [PubMed] [Google Scholar]
- Li Z, Otevrel T, Cheng HL, Seed B, Stamato TD, Taccioli GE, Alt FW (1995) The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell 83: 1079–1089 [DOI] [PubMed] [Google Scholar]
- Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S, Lozano G (2004) Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 36: 63–68 [DOI] [PubMed] [Google Scholar]
- MacPherson D, Kim J, Kim T, Rhee BK, Van Oostrom CT, DiTullio RA, Venere M, Halazonetis TD, Bronson R, De Vries A, Fleming M, Jacks T (2004) Defective apoptosis and B-cell lymphomas in mice with p53 point mutation at Ser 23. EMBO J 23: 3689–3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez JD, Craven MT, Joseloff E, Milczarek G, Bowden GT (1997) Regulation of DNA binding and transactivation in p53 by nuclear localization and phosphorylation. Oncogene 14: 2511–2520 [DOI] [PubMed] [Google Scholar]
- Saito SI, Goodarzi AA, Higashimoto Y, Noda Y, Lees-Miller SP, Appella E, Anderson CW (2002) ATM mediates phosphorylation at multiple p53 sites, including Ser46, in response to ionizing radiation. J Biol Chem 277: 12491–12494 [DOI] [PubMed] [Google Scholar]
- Sluss HK, Jones SN (2003) Analysing p53 tumour suppressor functions in mice. Expert Opin Ther Targets 7: 89–99 [DOI] [PubMed] [Google Scholar]
- Wu Z, Earle J, Saito S, Anderson CW, Appella E, Xu Y (2002) Mutation of mouse p53 Ser23 and the response to DNA damage. Mol Cell Biol 22: 2441–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y (2003) Regulation of p53 responses by post-translational modifications. Cell Death Differ 10: 400–403 [DOI] [PubMed] [Google Scholar]
- Xu Y (2006) DNA damage: a trigger of innate immunity but a requirement for adaptive immune homeostasis. Nat Rev Immunol 6: 261–270 [DOI] [PubMed] [Google Scholar]
- Xu Y, Yang EM, Brugarolas J, Jacks T, Baltimore D (1998) Involvement of p53 and p21 in cellular defects and tumorigenesis in Atm−/− mice. Mol Cell Biol 18: 4385–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye R, Bodero A, Zhou BB, Khanna KK, Lavin MF, Lees-Miller SP (2001) The plant isoflavenoid genistein activates p53 and Chk2 in an ATM-dependent manner. J Biol Chem 276: 4828–4833 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1