

Abstract
An essential component of the ATR (ataxia telangiectasia-mutated and Rad3-related)-activating structure is single-stranded DNA. It has been suggested that nucleotide excision repair (NER) can lead to activation of ATR by generating such a signal, and in yeast, DNA damage processing through the NER pathway is necessary for checkpoint activation during G1. We show here that ultraviolet (UV) radiation-induced ATR signaling is compromised in XPA-deficient human cells during S phase, as shown by defects in ATRIP (ATR-interacting protein) translocation to sites of UV damage, UV-induced phosphorylation of Chk1 and UV-induced replication protein A phosphorylation and chromatin binding. However, ATR signaling was not compromised in XPC-, CSB-, XPF- and XPG-deficient cells. These results indicate that damage processing is not necessary for ATR-mediated S-phase checkpoint activation and that the lesion recognition function of XPA may be sufficient. In contrast, XP-V cells deficient in the UV bypass polymerase η exhibited enhanced ATR signaling. Taken together, these results suggest that lesion bypass and not lesion repair may raise the level of UV damage that can be tolerated before checkpoint activation, and that XPA plays a critical role in this activation.
Keywords: ATR, checkpoint, nucleotide excision repair, Pol eta, XPA
Introduction
Eukaryotic cells have evolved highly conserved DNA damage response pathways called checkpoints that are responsible for coordinating various stress responses such as cell cycle arrest, lesion repair, adaptation and apoptosis (Zhou and Elledge, 2000; Melo and Toczyski, 2002). In mammalian cells, these pathways are primarily controlled by two phosphatidylinositol kinase-related protein kinases, ATM (ataxia telangiectasia-mutated) and ATR (ATM and Rad-3 related) (Abraham, 2001; Bakkenist and Kastan, 2004). The ATM kinase has been shown to be primarily activated by double-stranded DNA breaks caused by ionizing radiation (IR). In contrast, ATR is responsible for signaling in response to multiple types of DNA damage, including those induced by ultraviolet (UV) radiation, methyl methanesulfonate, cis-platinum, IR and the DNA replication inhibitors aphidicolin and hydroxyurea.
ATR checkpoint signaling is known to involve the independent translocation of multicomponent protein complexes to sites of damage (Kondo et al, 2001; Melo et al, 2001; Zou et al, 2002; Lee et al, 2003), which leads to the phosphorylation of ATR's main effector kinase Chk1 (Zhou and Elledge, 2000; Melo and Toczyski, 2002). Although the exact checkpoint signal is not yet known, it is thought to involve a replication protein A (RPA)-coated, single-stranded DNA (ssDNA) region adjacent to a primer (reviewed by Cortez, 2005; O'Connell and Cimprich, 2005). One process that has been shown to generate an ATR-activating signal is the inhibition of DNA replication (Cliby et al, 1998; Kumagai et al, 1998; Hekmat-Nejad et al, 2000; Liu et al, 2000; Brown and Baltimore, 2003). Experiments in the Xenopus extract system have shown that both DNA-damaging agents and replication inhibitors stall the replicative polymerases, resulting in continued unwinding of DNA by the MCM helicase (Pacek and Walter, 2004; Byun et al, 2005). This uncoupling of the polymerase activity from the helicase activity leads to an accumulation of ssDNA, increased binding of RPA and subsequent phosphorylation of Chk1 by ATR. ATR is thought to be recruited to RPA through its interacting protein, ATRIP, which is also required for ATR phosphorylation of Chk1 (Cortez et al, 2001; Zou and Elledge, 2003).
In addition to replication, nucleotide excision repair (NER) has also been implicated in ATR checkpoint activation (O'Driscoll et al, 2003; Giannattasio et al, 2004a; reviewed in Rouse and Jackson, 2002). In mammalian cells, defects in various NER proteins have been linked to a number of related DNA damage sensitivity disorders including xeroderma pigmentosum (XP), Cockayne syndrome (CS) and trichothiodystrophy (Ford and Hanawalt, 1997; Lehmann, 2003). Seven complementation groups have been identified for XP syndrome (XPA-XPG) and two for CS (CSA and CSB). Proteins involved in NER can be further classified by their roles in either transcription-coupled repair (TCR), a more rapid form of repair for the transcribed strand of expressed genes, or global genomic repair (GGR), which is needed for the rest of the genome. Regardless of the form of repair through which a DNA lesion is recognized (TCR or GGR), lesion processing proceeds through a common mechanism of damaged DNA base excision resulting in an ssDNA gap intermediate (de Laat et al, 1999; Hanawalt, 2002; Costa et al, 2003).
Evidence of a role for NER in contributing to checkpoint activation has come mainly from studies in yeast (Neecke et al, 1999; Giannattasio et al, 2004a). Mec1, the Saccharomyces cerevisiae ATR homolog, has been shown to have reduced activity in Rad14 (XPA) and Rad2 (XPG) mutants, which are defective in NER (Giannattasio et al, 2004b). Physical interactions between the checkpoint protein Ddc1 (human Rad9) and the NER protein Rad14 (XPA) have also been observed (Giannattasio et al, 2004a). Importantly, these studies were conducted in yeast arrested in G1, providing evidence that NER is needed to generate a checkpoint-inducing signal in the absence of replication. It has been speculated that this signal is an RPA-coated, ssDNA-gapped structure (O'Driscoll et al, 2003; Giannattasio et al, 2004a). Such a structure could be generated upon excision of the UV lesion and is similar to the checkpoint-activating structure generated at a stalled replication fork.
Whether or not NER directly contributes to activation of the ATR-dependent checkpoint in higher eukaryotes is unclear. A previous study found defects in UV-induced H2AX phosphorylation in non-replicating XPA-deficient cells but not Cockayne syndrome A (CSA)-deficient cells (O'Driscoll et al, 2003). This defect was similar to that seen in Seckel syndrome cells, which have reduced ATR expression. In addition, accumulation of p53 is reduced in XPA-deficient cells that are unable to progress through S phase (Nelson and Kastan, 1994). Another study found that cycling cells deficient in XPC, which is needed for the recruitment of XPA during GGR, did not have a defect in ATR phosphorylation of Chk1 in response to UV (Ward et al, 2004). Chromatin immunoprecipitation has also revealed that upstream checkpoint proteins are recruited to UV-damaged DNA in XPC- and XPA-deficient cells, and in this study at least some phosphorylation of Chk1 was observed during G1 and S phase in these cells (Jiang and Sancar, 2006). Importantly, none of these prior studies assessed ATR function in NER-deficient cells in comparison to isogenic, complemented control cell lines, nor did they study the role of lesion excision in UV-induced checkpoint activation in mammalian cells.
Similar to NER, it is also not known what role UV trans-lesion synthesis plays in checkpoint activation. One trans-lesion synthesis polymerase, polymerase η (Polη), is mutated in variant forms of xeroderma pigmentosum (XP-V) that are proficient in the repair of UV DNA damage (Masutani et al, 1999; Lehmann, 2005). Polη is an error-prone polymerase on undamaged DNA templates but can specifically bypass UV-damaged bases during replication in an error-free manner by inserting adenines across from a cyclobutane pyrimidine dimer (CPD) (Kannouche and Stary, 2003; Lehmann, 2005). Cells deficient in Polη exhibit a prolonged UV-induced S-phase arrest and have greatly enhanced H2AX phosphorylation following UV treatment, indicative of double-strand break formation (Limoli et al, 2002). These breaks may arise from damage-induced replication fork collapse, which is also observed upon loss of ATR function (Brown and Baltimore, 2003).
In this study, we sought to determine the contribution of NER and Polη-mediated UV lesion bypass to ATR checkpoint activation. To do so, we assessed translocation of the ATR-associated protein, ATRIP, to sites of UV damage in various NER-deficient cell lines and in complemented, isogenic controls. We also assessed the activation of ATR following UV treatment in these cells by analyzing the ATR-dependent phosphorylation of Chk1 at S345. We found that cells deficient in XPA have a defect in ATRIP translocation and Chk1 phosphorylation following UV treatment. XPA-depleted Xenopus egg extracts also exhibited a defect in Chk1 phosphorylation. Importantly, these defects were observed during S phase. Moreover, we found that XPA-deficient cells have decreased UV-induced RPA chromatin binding and phosphorylation. Chk1 phosphorylation was unaffected in XP-C, CS-B, XP-G and XP-F cell lines. We also found that XP-V cells, which are deficient in Polη, had increased Chk1 phosphorylation relative to their complemented controls following UV treatment. Consistent with these results, we found that damage-induced RPA phosphorylation was also modestly increased in XP-V cells. Overall, our findings suggest that XPA and Polη may have opposing effects on the S-phase activation of ATR following UV-induced DNA damage. They also suggest that although XPA plays a role in Chk1 phosphorylation, excision of the UV-induced lesion is not necessary for S-phase checkpoint activation in mammalian cells.
Results
ATRIP colocalization with UV-induced DNA damage is reduced in XPA-deficient cells
Both ATR and its associated protein ATRIP have been shown to translocate to punctate nuclear foci following treatment of cells with IR and UV (Tibbetts et al, 1999; Cortez et al, 2001; Barr et al, 2003). This translocation event is thought to be one of the earliest indicators of the ATR-mediated DNA damage response. By locally UV irradiating portions of the nucleus through a 5 μm porous filter (Volker et al, 2001), it has been shown that ATR staining is enhanced in regions containing UV photoproducts (Ward et al, 2004). Using this method, we assessed the translocation of ATRIP to sites of UV damage. As shown in Figure 1A (and data not shown), ATRIP, like ATR, colocalizes with regions of the nucleus also recognized by an antibody to CPDs. We did not observe ATRIP translocation to regions of the nucleus that did not stain positive for CPDs; however, in many cells containing regions that stained positive for CPDs, ATRIP colocalization was not observed. Importantly, when cells were synchronized by a double-thymidine block and released into S phase, the percentage of cells in which ATRIP translocated to sites of UV damage increased significantly, moving from 38% in asynchronous cells to 77% in S-phase synchronized cells (Supplementary Figure 1A). This increase correlates with the increase in the percentage of S-phase cells observed. These observations indicate that ATRIP can translocate to UV lesions during S phase and are consistent with the idea that ATR activation is dramatically enhanced in S-phase cells (Lupardus et al, 2002; Ward et al, 2004).
Figure 1.
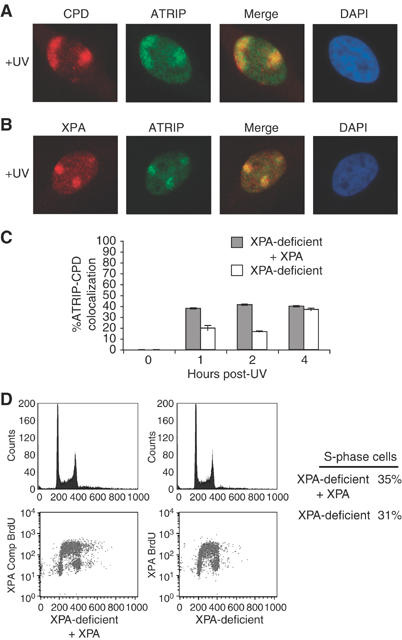
XPA facilitates ATRIP recruitment to sites of UV damage. HeLa cells grown on coverslips were exposed to 50 J/m2 UV through a porous membrane and allowed to recover for 1 h. Cells were fixed and stained with an anti-ATRIP rabbit polyclonal antibody and an anti-CPD mouse monoclonal antibody (A) or an anti-XPA mouse monoclonal antibody (B), followed by staining with a fluorescent goat anti-rabbit antibody Alexa 488 (green) and goat anti-mouse antibody Alexa 595 (red). Nuclei were counterstained with DAPI. Colocalization of labeled proteins is shown as yellow in merge. (C) XPA-complemented cells (gray bar) and XPA-deficient cells (white bar) were UV treated and stained as in panel (A) at the times shown. The recruitment of ATRIP to CPD lesions was determined for a total of 200 nuclei containing CPD foci. Shown graphically is the percentage of nuclei with ATRIP foci that colocalized with CPD foci. Error bars indicate standard error of three samples from a representative experiment (n=3) performed in triplicate. (D) Percentage of S-phase cells was determined by flow cytometry using propidium iodide staining (top panels) and by measuring BrdU incorporation (bottom panels).
As seen in Figure 1B, ATRIP also colocalizes with the NER protein XPA after UV treatment through these filters. Thus, we asked if XPA was required for UV-induced ATRIP translocation. Utilizing the protocol described in Figure 1A, we assessed the colocalization of ATRIP with CPDs at different time points over 4 h in an XPA-deficient cell line and an isogenic line complemented with wild-type XPA. As shown in Figure 1C, in the XPA-deficient cell line, there was a marked decrease in the percentage of cells exhibiting colocalization of ATRIP with sites of UV damage 1 and 2 h following UV treatment. At 4 h following UV treatment, however, the localization of ATRIP to sites of UV damage was similar in both XPA-deficient and XPA-complemented cells. The defect in ATRIP translocation at 1 and 2 h does not appear to be due to a difference in the cell cycle distributions of these two cell lines as propidium iodide staining and BrdU incorporation indicate that both have similar cell cycle profiles (Figure 1D).
UV-induced Chk1 phosphorylation is decreased in XPA-deficient cells
The defect in translocation of ATRIP to sites of UV damage suggests that ATR signaling may be reduced in XPA-deficient cells. Chk1 is a downstream effector of ATR that is phosphorylated on serine 345 in an ATR-dependent manner following several types of DNA damage (Guo et al, 2000; Hekmat-Nejad et al, 2000; Liu et al, 2000), and the phosphorylation of Chk1 is thought to be necessary for Chk1 activation (Zhao and Piwnica-Worms, 2001). Using an antibody that recognizes Chk1 phosphoserine 345 (S345), we tested whether Chk1 phosphorylation was decreased in XPA-deficient cells. Asynchronous XPA-complemented and XPA-deficient cells were treated with varying doses of UV (Figure 2A) and harvested 1 h post-treatment. In the XPA-complemented cells, UV induced the phosphorylation of Chk1 S345. However, the phosphorylation of Chkl was greatly reduced in the XPA-deficient cells at all of the doses tested. We then assessed the effect of XPA loss on Chk1 phosphorylation at several times following UV treatment. Consistent with the results shown in Figure 1C for ATRIP localization to CPDs, the effect of XPA loss was most evident at early times after UV treatment (Figure 2B).
Figure 2.
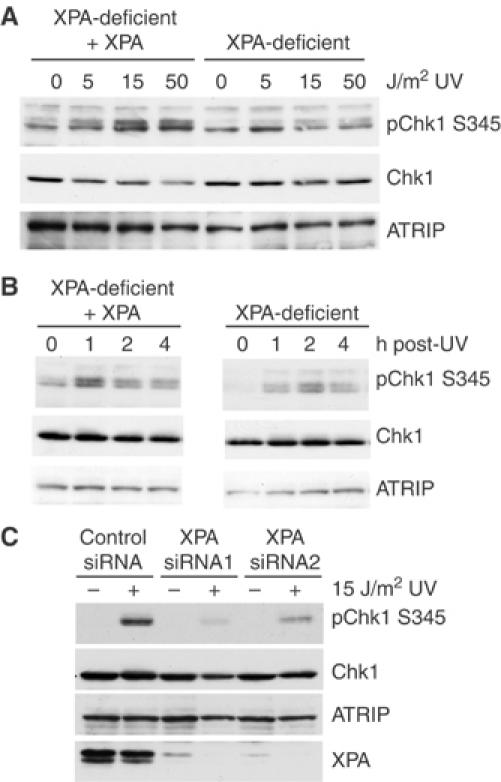
XPA-deficient cells show reduced Chk1 phosphorylation following UV treatment. (A) Isogenic XPA-deficient and XPA-complemented cells were either mock treated or exposed to 0, 5, 15 or 50 J/m2 UV. Cells were harvested after 1 h and lysates were resolved by SDS–PAGE and analyzed by Western blot using phospho-serine 345 Chk1, Chk1 and ATRIP antibodies. (B) XPA-complemented and XPA-deficient cells were mock treated or exposed to 15 J/m2 UV and then harvested at the times shown. Lysates were analyzed as described in panel A. (C) HeLa cells were transfected with control siRNA or one of two siRNAs to XPA and treated with 15 J/m2 UV 48 h later. At 1 h after UV treatment, lysates were prepared and resolved by SDS–PAGE and analyzed by Western blot using phospho-serine 345 Chk1, Chk1, ATRIP and XPA antibodies.
To further demonstrate a role for XPA in UV-induced ATR activation, we examined the effect of XPA loss on UV-induced Chk1 phosphorylation in HeLa cells. For this, we used small interfering (si) RNA-mediated XPA depletion. Depletion of XPA using either of two different XPA-specific siRNAs resulted in decreased phosphorylation of Chk1 following 15 J/m2 of UV treatment (Figure 2C). In addition, XPA-deficient mouse embryo fibroblasts (Ichikawa et al, 2000) also exhibited poor Chk1 phosphorylation relative to control 3T3 cells (Supplementary Figure 2).
To determine if the role of XPA in the phosphorylation of Chk1 is specific to UV-like DNA-damaging agents, we also analyzed the phosphorylation of Chk1 in XPA-deficient and XPA-complemented cells following several other types of DNA damage. As shown in Figure 3A, Chk1 phosphorylation was significantly decreased in XPA-deficient cells following treatment with the UV mimetic, 4-nitroquinoline oxide (4-NQO). However, there was no defect in Chk1 S345 phosphorylation following aphidicolin treatment (Figure 3B). Likewise, XPA-deficient and XPA-complemented cells had similar levels of IR-induced Chk1 phosphorylation (Figure 3C). Taken together, these results suggest that XPA is necessary for the timely and efficient activation of Chk1 following damage induced by UV and UV-like agents.
Figure 3.
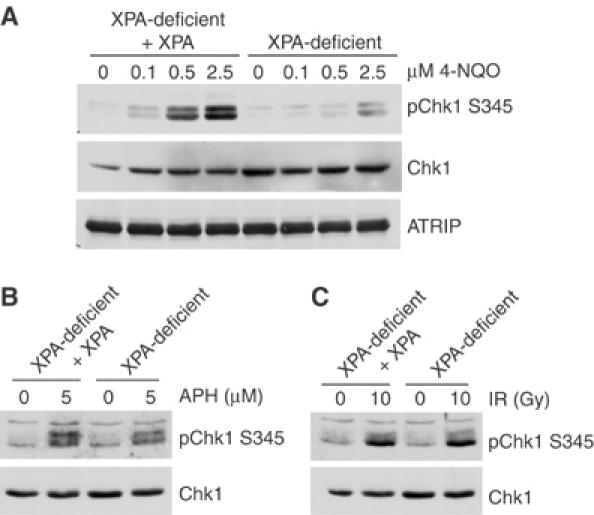
XPA is required for full Chk1 phosphorylation induced by 4-NQO, but not aphidicolin or IR. Isogenic XPA-complemented and XPA-deficient cells were exposed to increasing concentrations of the UV-mimetic drug 4-NQO for 1 h (A), 5 μM aphidicolin for 6 h (B) or 10 Gy IR followed by a 2 h recovery (C). Lysates were prepared and analyzed as described in Figure 2.
UV-induced Chk1 phosphorylation is decreased in Xenopus egg extracts lacking XPA
The UV-induced phosphorylation of Chk1 has been reported to be restricted to S-phase cells (Ward et al, 2004). Consistent with this finding, we have also found that Chk1 phosphorylation is enhanced in S-phase synchronized cells (Supplementary Figure 1B). Because ATRIP translocation to sites of UV damage and Chk1 phosphorylation both appear to occur predominantly in S phase, our results suggest that XPA may be required for the S-phase checkpoint. As another way of assessing the role of XPA in this checkpoint, we used the Xenopus model system to test whether depletion of XPA from egg extracts also leads to a defect in Chk1 phosphorylation. In Xenopus egg extracts, UV-induced phosphorylation of Chk1 requires initiation of replication (Lupardus et al, 2002). Using an antibody raised against the full-length, recombinant Xenopus XPA (xXPA) protein, we found that xXPA was recruited to chromatin after UV damage but not after aphidicolin treatment (Figure 4A). Notably, when this antibody was used to immunodeplete xXPA from extracts, we observed a significant decrease in the UV-induced phosphorylation of XChk1 on S344, the site analogous to S345 in Chk1 (Figure 4B). These results are consistent with our studies in XPA-deficient mammalian cells and support the idea that the requirement for XPA in ATR checkpoint signaling following UV is primarily in S phase.
Figure 4.
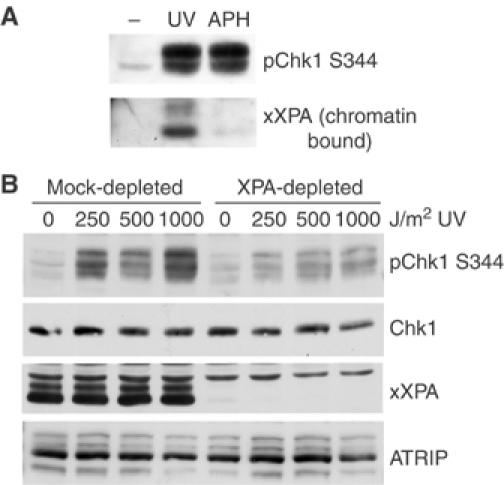
Depletion of XPA from Xenopus egg extracts inhibits UV-induced Chk1 phosphorylation. (A) Xenopus egg extracts were incubated with either UV-damaged sperm chromatin (1000 J/m2) or treated with 150 μM aphidicolin. After 90 min, total extract samples were taken and an aliquot was removed for analysis of Chk1 S344 phosphorylation. From the remaining sample, chromatin was isolated through a sucrose cushion, sheared and boiled in sample buffer. Samples were resolved by SDS–PAGE and analyzed by Western blot using the indicated antibodies. (B) Xenopus extract was either mock-depleted or XPA-depleted using rabbit IgG (mock) or an xXPA antibody coupled to protein A beads. Undamaged or UV-treated sperm chromatin was added to the extracts and incubated for 90 min before addition of sample buffer. Equal amounts of extract were resolved by SDS–PAGE and analyzed by Western blot using the indicated antibodies.
XPC- and CSB-deficient cells show no defect in Chk1 phosphorylation
Because cells deficient in XPA, a protein central to both TCR and GGR pathways (de Laat et al, 1999; Hanawalt, 2002; Costa et al, 2003), exhibit a defect in the ATR response, we asked whether one of these NER repair pathways was specifically involved. To do this, we utilized XPC- and CSB-deficient cells, which are defective in GGR and TCR, respectively, to perform the ATRIP-CPD colocalization assay described in Figure 1A. As seen in Figure 5A, there is a slight decrease in colocalization of ATRIP with CPD lesions in XPC-deficient cells compared to an isogenic, XPC-complemented cell line. Importantly, however, this decrease was not accompanied by a significant decrease in Chk1 S345 phosphorylation (Figure 5B). These results are in agreement with a previous report that XPC-deficient cells do not have a significant defect in the ATR response (Ward et al, 2004). We also found that there was no difference in ATRIP translocation or Chk1 S345 phosphorylation following UV treatment between CSB-deficient and isogenic, complemented cell lines (Figures 5C and D). Furthermore, the percentage of S-phase cells was similar in both XPC- and CSB-deficient and complemented cell lines. These observations indicate that the defect seen in the XPA-deficient cells is not likely attributed to a specific arm of the NER pathway.
Figure 5.
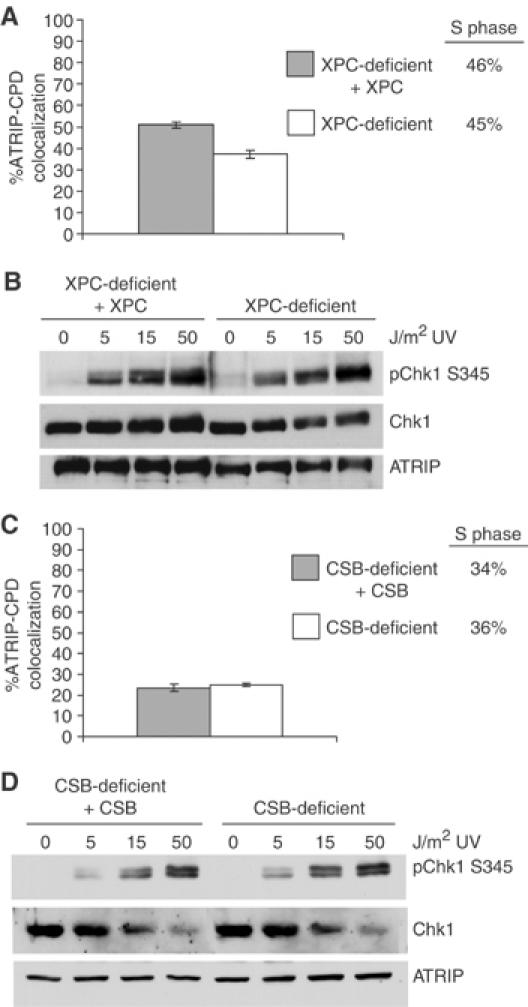
ATRIP translocation and Chk1 phosphorylation are unaffected by loss of XPC or CSB. Isogenic XPC-deficient (white bar) and XPC-complemented (gray bar) cells (A) or CSB-deficient (white bar) and CSB-complemented (gray bar) cells (C) were UV treated and stained as in Figure 1A. Colocalization of ATRIP and CPD was determined as in Figure 1C. Percentage of S-phase cells was determined by flow cytometry as shown in Figure 1D. XPC-deficient and XPC-complemented cells (B) or CSB-deficient and CSB-complemented cells (D) were treated with increasing amounts of UV and allowed to recover for 1 h. Equal amounts of cell lysate were resolved by SDS–PAGE and analyzed by Western blot using the indicated antibodies.
XPF- and XPG-deficient cells show no defect in Chk1 phosphorylation
A late step in NER is excision of the DNA surrounding the UV-damaged bases by the endonucleases XPF-ERCC1 and XPG (de Laat et al, 1999; Hanawalt, 2002; Costa et al, 2003). In S. cerevisiae, the XPG homolog Rad2 has been shown to be required for UV-dependent activation of the ATR homolog, Mec1, in G1 cells (Giannattasio et al, 2004a). Using cells deficient in either XPF or XPG and their complemented, isogenic lines, we assessed Chk1 S345 phosphorylation 1 h after treatment with UV. As shown in Figure 6A, there was no decrease in Chk1 S345 phosphorylation in XPF-deficient cells. There was also no significant decrease in Chk1 S345 phosphorylation in the XPG-deficient cells relative to control cells (Figure 6B). Together, these results suggest that excision of the UV lesion is not required for Chk1 S345 phosphorylation in asynchronous cells.
Figure 6.
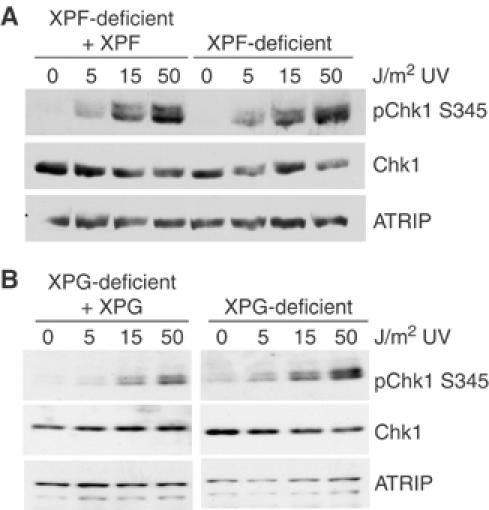
XPF and XPG are not required for UV-induced Chk1 phosphorylation. Isogenic XPF-deficient and XPF-complemented cells (A) or XPG-deficient and XPG-complemented cells (B) were either mock treated or exposed to UV radiation and allowed to recover for 1 h. Equal amounts of cell lysate were resolved by SDS–PAGE and analyzed by Western blot using the indicated antibodies.
XPA deficiency results in decreased RPA phosphorylation and chromatin binding
RPA is an ssDNA binding protein that is thought to be a component of the ATR checkpoint signal (reviewed in O'Connell et al, 2000; Cortez, 2005). RPA accumulates on chromatin following UV treatment in both mammalian cells and Xenopus extracts (Lupardus et al, 2002; Zou and Elledge, 2003). In vitro studies using purified proteins have shown that RPA is sufficient to recruit ATRIP to ssDNA (Zou and Elledge, 2003), although RPA-independent modes of DNA binding and checkpoint signaling also exist (Bomgarden et al, 2004; Ball et al, 2005; Kim et al, 2005). RPA is also phosphorylated during normal cell cycle progression as well as hyperphosphorylated in response to DNA damage, and a subset of these phosphorylation events are ATR-dependent (Barr et al, 2003; Binz et al, 2004). As Chk1 phosphorylation is decreased in XPA-deficient cells and XPA-depleted Xenopus egg extracts, we asked whether RPA chromatin binding and/or phosphorylation was altered in XPA-deficient cells. To do so, cells were treated with UV damage and chromatin-bound RPA was isolated by subcellular fractionation. ORC2 binding was also analyzed to normalize for loading of equal amounts of chromatin. As shown in Figure 7, RPA chromatin binding and phosphorylation are significantly reduced in XPA-deficient cells compared to the complemented cells. These observations suggest that XPA may be involved in establishing an ATR-activating structure following UV treatment in S phase.
Figure 7.
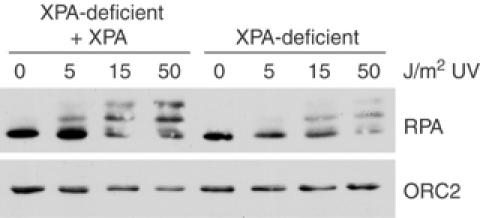
RPA chromatin binding and phosphorylation is decreased in XPA-deficient cells. XPA-deficient and XPA-complemented cells were treated with increasing amounts of UV radiation and fractionated. Proteins of the insoluble chromatin fraction were resolved by SDS–PAGE and analyzed by Western blot using the indicated antibodies.
Chk1 and RPA phosphorylation are enhanced in UV-treated XP-V cells
Activation of ATR by UV during S phase is thought to be due, at least in part, to the accumulation of ssDNA, which results from uncoupling of helicase and polymerase activities after stalling of the replicative polymerase at the lesion (Byun et al, 2005). In the light of this model, one might expect that the loss of NER would lead to increased ATR signaling as the additional unrepaired lesions present in repair-deficient cells could stall an increased number of replicative polymerases. Thus, it is somewhat surprising that loss of XPA leads to a defect in the S-phase checkpoint and that loss of other NER factors has no effect on ATR activation. It is possible, however, that loss of repair does not increase ATR signaling during S phase because other processes are able to compensate for the increased number of lesions.
Lesion bypass by trans-lesion synthesis DNA polymerases is one mechanism through which cells can tolerate UV DNA damage (Prakash and Prakash, 2002), and this process might be expected to reduce checkpoint activation by allowing bypass of UV lesions and preventing sustained blockage of replication forks. To test whether lesion bypass affects UV-induced checkpoint activation, the phosphorylation of Chk1 was assessed in XP-V cells, which lack activity of the trans-lesion Polη. As shown in Figure 8A, Chk1 S345 phosphorylation was increased in a sustained manner in Polη-deficient cells following treatment with UV relative to the same cell line complemented with the full-length wild-type Polη cDNA. In addition, Polη-deficient cells and Polη-complemented cells exhibited no difference in S345 phosphorylation following aphidicolin treatment (Figure 8B). These data suggest that loss of Polη leads to enhanced ATR signaling following UV damage.
Figure 8.
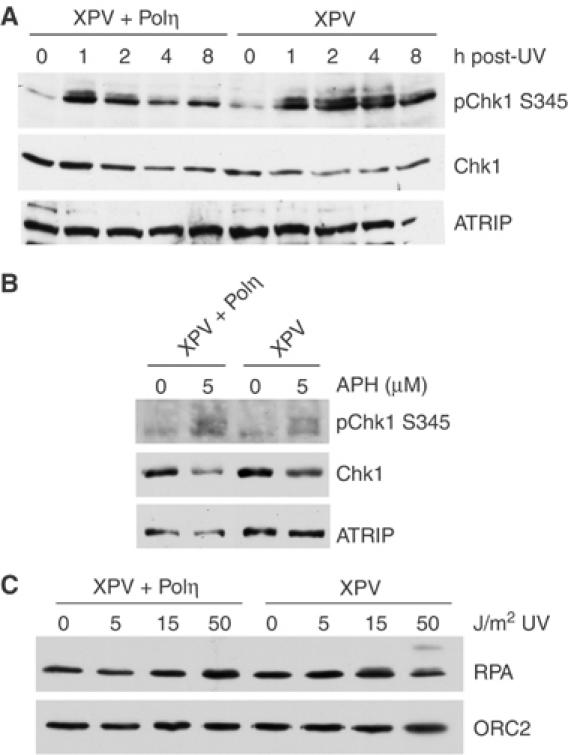
Chk1 and RPA phosphorylation are increased in XPV cells. (A) Polη-deficient (XP-V) and Polη-complemented (XP-V+Polη) cells were either mock treated or exposed to 15 J/m2 UV and allowed to recover for the times indicated. Equal amounts of cell lysate were resolved by SDS–PAGE and analyzed by Western blot using the indicated antibodies. (B) Polη-deficient and Polη-complemented cells were treated with 5 μM aphidicolin for 6 h and then analyzed as described in panel A. (C) Polη-deficient and Polη-complemented cells were treated with increasing amounts of UV and fractionated. Proteins of the insoluble chromatin fraction were resolved by SDS–PAGE and analyzed by Western blot using the indicated antibodies.
To further test the effects of Polη loss on ATR signaling, RPA chromatin binding and phosphorylation were assessed following increasing amounts of UV damage. As shown in Figure 8C, RPA chromatin binding was only modestly increased in UV-treated Polη-deficient cells compared to control cells, and the effect was most noticeable at lower UV doses. However, in Polη-deficient cells, RPA phosphorylation was increased compared to complemented controls, particularly after treatment with 50 J/m2 of UV (Figure 8C). This observation is consistent with the observed increase in Chk1 phosphorylation seen in XP-V cells (Figure 8A). Taken together, these results suggest that a lack of UV trans-lesion synthesis causes increased activation of the ATR pathway.
Discussion
Previous studies in mammalian cells and Xenopus egg extracts have shown that activation of the ATR-dependent checkpoint after UV irradiation is dependent upon DNA replication (Lupardus et al, 2002; Ward et al, 2004). This is thought to be due to stalling of the replicative polymerases by UV DNA damage, resulting in the accumulation of ssDNA and recruitment of ATR-ATRIP to sites of damage (Lupardus et al, 2002; Zou and Elledge, 2003; Byun et al, 2005). By such a model, one might predict that any process that alters the amount of UV-damaged DNA in the nucleus might significantly contribute to checkpoint activation caused by polymerase stalling. Here, we investigated the role of two processes, NER and UV lesion bypass, on ATR checkpoint activation. Interestingly, we found opposing roles for two proteins involved in these processes during S-phase checkpoint activation. After UV damage, cells deficient in the repair protein XPA were found to have a defect in ATR signaling, whereas cells deficient in the trans-lesion synthesis polymerase Polη exhibited enhanced ATR signaling. Our results may have also uncovered a role for XPA in checkpoint signaling that is distinct from its function in NER.
To address the role of NER in ATR activation, we analyzed a panel of cell lines that are defective in various steps of this repair pathway. Our results indicate that there is a defect in ATR activation when XPA is lost. This is supported by a decrease in ATRIP and ATR localization to sites of UV damage in XPA-deficient cells, as well as a decrease in UV-induced S345 Chk1 phosphorylation in cells from XPA patients and XPA−/− mice, in HeLa cells upon siRNA-mediated knockdown of XPA and in Xenopus egg extracts upon depletion of XPA. In addition, we found no substantial defect in ATR activation in XP-C, CS-B, XP-G and XP-F cell lines. Because XPC and CSB function in GGR and TCR, respectively (de Laat et al, 1999; Hanawalt, 2002; Costa et al, 2003), these results suggest that the defect in ATR activation is not due to a specific branch of NER. However, XPA is required for both types of repair and functions downstream of XPC and CSB (Costa et al, 2003). Thus, it is possible that both GGR and TCR must be compromised in mammalian cells in order to observe the reduced ATR activation seen in XP-A cells. Indeed, this is the case in yeast where loss of both GGR and TCR, but neither alone, leads to loss of Rad53 phosphorylation (Giannattasio et al, 2004a). In this context, it is worth noting that there was a slight reduction in ATRIP accumulation at sites of UV damage in XPC-deficient cells, suggesting that XPC may have some effect on the ATR response that does not translate to a defect in Chk1 phosphorylation.
The lack of a defect in XPF- and XPG-deficient cells is somewhat surprising. In contrast to XPA, which is involved in damage recognition, XPF and XPG are involved in damage processing, acting as endonucleases to remove the lesion and surrounding nucleotides, and they are needed for both TCR and GGR (de Laat et al, 1999; Hanawalt, 2002; Costa et al, 2003). Our results differ from a previous study in yeast, which suggests that the NER-dependent activation of ATR's homolog, Mec1, might result from damage processing and generation of a ssDNA gap by XPF and XPG. Importantly, this conclusion is based largely on experiments with G1 synchronized yeast where a defect in Rad53 phosphorylation is observed in mutants of Rad14 and Rad2, the yeast homologs of human XPA and XPG, respectively (Giannattasio et al, 2004a). In contrast, the phosphorylation of Chk1 and ATRIP relocalization that we observe following UV treatment occurs predominantly in S-phase cells. This indicates that the activation we are studying is largely S-phase specific and is consistent with our finding that XPA depletion from Xenopus egg extracts also reduces Chk1 phosphorylation. It should also be noted that ATR activation could be occurring at significantly lower levels outside of S phase and that our methods are not sensitive enough to detect this activation. Thus, we cannot exclude the possibility that activation of ATR outside of S phase may still require NER. Indeed, this possibility is consistent with experiments in non-replicating cells showing a requirement for XPA in UV-induced H2AX phosphorylation (O'Driscoll et al, 2003).
Altogether, our results provide the first evidence that XPA, but not NER-mediated damage processing, is necessary for ATR checkpoint activation in replicating human cells. The requirement for XPA could indicate that damage recognition by XPA may be sufficient for UV-induced S-phase checkpoint activation, although other unidentified functions of XPA are also possible. Regardless of the mechanism, this role for XPA appears to be specific to bulky lesions, like those produced by UV radiation or 4-NQO because no defect in the aphidicolin-induced phosphorylation of Chk1 was observed upon loss of XPA. We previously proposed a model in which UV lesions and replication inhibitors activate ATR in S phase through a common mechanism that involves slowing down of replicative polymerases and increased unwinding of DNA by the MCM helicases (Pacek and Walter, 2004; Byun et al, 2005). This leads to accumulation of RPA-coated ssDNA, which is thought to be involved in recruiting and/or stabilizing the interaction of ATR and ATRIP with DNA (Lupardus et al, 2002; Zou and Elledge, 2003). In our experiments, we observed less RPA chromatin binding and phosphorylation in UV-treated XPA-deficient cells. Thus, one possibility is that the specific binding of XPA at sites of UV or UV-like damage may enhance stalling of the replicative DNA polymerases at these lesions, resulting in increased activation of the ATR pathway. Although the naked lesion itself is sufficient to slow replicative polymerases in vitro (Fleck and Schar, 2004), the presence of XPA at lesions during DNA replication could facilitate checkpoint signaling by prolonging arrest at these sites, possibly by competing with bypass polymerases. Alternatively, as XPA directly binds to RPA during lesion repair (Matsuda et al, 1995; Stigger et al, 1998; Costa et al, 2003), XPA may help to stabilize the RPA-coated ssDNA intermediate formed when polymerases stall at UV lesions.
It is important to note that complete loss of Chk1 phosphorylation was not observed in cells from XPA patients or upon depletion of XPA. Moreover, at later times after UV treatment, the effect of XPA loss was less pronounced than at earlier times. Similarly, the effect of XPA loss on ATRIP localization to sites of UV damage was most noticeable at earlier times after UV treatment. There are at least two possible explanations for the residual Chk1 phosphorylation observed. As described above, the phosphorylation that does occur could reflect less efficient formation of the ATR-activating structure, ssDNA, through helicase and polymerase uncoupling. However, it is also possible that the Chk1 phosphorylation that is observed is induced in a different manner. Previous studies have linked loss of the ATR–Chk1 pathway to double-strand break formation and replication fork collapse (Osborn et al, 2002; Brown and Baltimore, 2003; Zachos et al, 2003, 2005). Because XPA is needed for activation of the ATR–Chk1 pathway, the Chk1 phosphorylation observed could result from collapse of replication forks in the absence of XPA, leading to generation of ssDNA from the collapsed fork and activation of ATR by the break and/or ensuing ssDNA. Additional work will be required to discriminate between these and other possibilities.
NER is the major mechanism by which UV DNA damage is repaired in mammalian cells. Because loss of NER activity leads to an increase in UV damage but does not cause an increase in ATR-mediated checkpoint signaling, it seems likely that another process can compensate for the loss of NER at the doses used here in a manner that affects the ability of UV lesions to stall polymerases in S phase and activate ATR. Supporting this hypothesis, we found that cells deficient in trans-lesion synthesis polymerase Polη exhibit increased phosphorylation of Chk1 in response to UV. Because these cells are repair-proficient and the level of damage does not change, these observations suggest that the ability to bypass lesions helps to suppress checkpoint activation. We also observed increased phosphorylation of RPA in XP-V cells as well as a modest increase in RPA chromatin binding. The accumulation of RPA suggests that additional ssDNA is present owing to an inability to efficiently bypass the lesion and is consistent with recent EM studies in yeast showing that cells lacking trans-lesion synthesis accumulate small ssDNA gaps along the replicated strands (Lopes et al, 2006). Recently, it was reported that cells deficient in Polκ, which is needed to bypass non-UV-mediated bulky adducts, also had increased Chk1 phosphorylation in response to benzo(α)pyrene-dihydrodiol epoxide (Bi et al, 2005). Taken together, these findings suggest that trans-lesion synthesis polymerases raise the threshold level of damage that can be tolerated before the checkpoint is activated. They are also consistent with a requirement for polymerase stalling in checkpoint activation and suggest that Polη may raise this threshold by reducing replication fork arrest.
Cells that are deficient in XPA and Polη (XPV) are derived from patients suffering from XP. Although defects in these genes result in increased sensitivity to UV, patients lacking XPA have much more severe clinical phenotypes than patients with XP-V (Masutani et al, 1999). This difference in severity is generally attributed to the increased accumulation of UV DNA damage in repair-deficient XP-A patients relative to repair-proficient XP-V patients (Lehmann, 2003). Our finding that XPA-deficient cells have decreased ATR checkpoint signaling may suggest an additional reason for the more severe UV-sensitive phenotype of these patients. Likewise, the enhanced ATR signaling we observed in Polη-deficient cells may also result in a less severe UV sensitivity owing to a more active checkpoint and upregulation of p53-mediated response genes such as XPC and DDB2 (Adimoolam and Ford, 2003). Ultimately, further investigation will be required to understand the contribution of ATR checkpoint activation to these UV-sensitive syndromes.
Materials and methods
Cell lines and cell culture
HeLa cells purchased from the National Cell Culture Center were maintained in DMEM (Invitrogen) supplemented with 10% FBS, penicillin, streptomycin and glutamine. XPA-complemented (GM15876A) and XPA-deficient (XP12R0) lines purchased from the Coriell Cell Repository were maintained in DMEM. XPA−/− MEFs (Ichikawa et al, 2000) and NIH 3T3 cells (ATCC) were maintained in DMEM. CSB-complemented and CSB-deficient (CS1AN.S3.G2) cell lines were maintained in MEM plus 400 μg/ml G418 (Tuo et al, 2002). XPF-complemented and XPF-deficient (GM08437) cell lines were maintained in MEM plus 300 μg/ml G418 (Chen et al, 2004). XPG-complemented lines were made by stable transfection of XPG-deficient cell lines (94RD270/XP3BR-SV) (Klein et al, 1990) with the XPG cDNA and selection in G418 (800 μg/ml). Stable colonies were screened for expression of XPG by Western blot and correction of UV sensitivity was assessed using an MTT assay (data not shown). XPG-complement cells were maintained in McCoy's media with 10% FBS and 200 μg/ml G418. Complemented and deficient Polη (GM2359) cell lines were maintained in DMEM plus 200 μg/ml G418 (Bassett et al, 2004). The following siRNA duplexes were purchased from Dharmacon (Lafayette, CO): GCUACUGGAGGCAUGGCU (XPA siRNA-1) and GGAGACGAUUGUUCAUCAA (XPA siRNA-2), the latter of which was previously reported (Lin et al, 2006). SiCONTROL (Dharmacon) was used as the non-targeting siRNA. All siRNA transfections were performed using 50 nM of oligonucleotide and Lipofectamine 2000 (Invitrogen).
Cells were washed once with PBS, then exposed to UV-C using a Stratalinker (Stratagene) except when local irradiation was used (see below). The media were replaced and cells were allowed to recover for 1 h in the incubator. 4-NQO (Sigma) treatment was performed by adding drug to cells in serum free media for 1 h. IR treatment was performed using a 137Cs irradiator followed by a 2 h recovery. Aphidicolin (Sigma) treatment was for 6 h at 5 μM in normal media.
Antibodies
The Chk1 S345 phospho-specific antibody (Cell Signaling), Chk1 antibody (Santa Cruz Biotech), human ORC2 antibody (BD Bioscience), XPA 12F5 antibody (Lab Vision), BrdU antibody (Roche), XPG 8H7 antibody (Santa Cruz) and human RPA AB3 antibody (Oncogene) are commercially available. The anti-CPD mouse monoclonal antibody (TDM2) was a gift of Toshio Mori, Nara Medical University, Nara, Japan (Mori et al, 1991). The human ATRIP antibody (Bomgarden et al, 2004) and the Xenopus ATRIP antibody (Byun et al, 2005) were described previously. The xXPA rabbit polyclonal antibody used for immunodepletion was generated at a commercial facility (Josman, Napa, CA) from full-length HIS-xXPA expressed using the Bac-To-Bac system (Invitrogen) in Hi5 insect cells.
Immunofluorescence staining
For the ATRIP-CPD filter-based assay, 2 × 105 cells were plated onto coverslips and incubated 12 h before staining. The published protocol (Fitch et al, 2003) was modified as follows. Cells were rinsed once with PBS before treatment with 50 J/m2 UV-C through a 5 μm filter membrane (Whatman) followed by a 1 h recovery. Cells were washed twice with PBS and fixed with 3% paraformaldehyde in PBS plus 0.1% Triton X-100 (PBST) for 20 min. Cells were then washed once with PBS, permeabilized for 15 min with 0.1% PBST and treated with 2 N HCl for 5 min. Acid was neutralized by washing five times with PBS. Cells were then stained with affinity-purified anti-ATRIP (1:600) and anti-CPD (1:150) primary antibodies diluted in 0.1% BSA/PBST for 1 h in a humidified chamber at 37°C. After washing three times with 0.1% BSA/PBST, cells were stained with secondary Alexa rabbit 488 and mouse 595 antibodies (Molecular Probes) diluted 1:400 in 0.1% BSA/PBST. Cells were washed five times with 0.1% BSA/PBST before staining with 1:10 000 DAPI (Sigma) and fixing with Prolong anti-fade (Molecular Probes). All slides were visualized with a Zeiss Axioskope microscope. Images were prepared using Adobe Photoshop.
Extract preparation and depletions
To prepare whole-cell extracts, cells were harvested by trypsinization, washed in PBS, resuspended in modified TGN buffer (50 mM Tris pH 7.5, 150 mM NaCl, 50 mM β-glycerophosphate, 10% glycerol, 1% Tween 20, 0.2% Nonidet P-40, 1 mM NaF, 1 mM Na3VO4, 1 mM DTT and 10 μg/ml leupeptin, pepstatin, aprotinin) for 20 min on ice and cleared by centrifugation (10 min, 16 000 g).
Xenopus extracts were prepared as described (Murray, 1991; Lupardus et al, 2002). Aphidicolin was used at 150 μM, and sperm chromatin was UV damaged in a Stratalinker (Stratagene). Chromatin binding from interphase extracts has been previously described (Lupardus et al, 2002). Immunodepletion (three rounds, 30 min each) of XPA from interphase extract was carried out at 4°C using Affiprep Protein-A beads (Bio-Rad). XPA antiserum was bound to beads at a 1:2 ratio of beads to serum. Phospho-S344 Chk1 blots for Xenopus Chk1 were performed with a phospho-Ser345 human Chk1 antibody raised by Zymed Inc.
Chromatin binding
Small-scale nuclear extracts were prepared using 1 × 106 cells harvested with trypsin, washed with PBS and resuspended in 200 μl buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 10 mM NaF, 1 mM Na3VO4 and protease inhibitor cocktail) plus 5% Triton X-100 for 5 min on ice. Nuclei were pelleted by centrifugation at 1300 g in swinging bucket Eppendorf 5417C centrifuge for 4 min. The nuclei were washed with 200 μl buffer A without detergent and respun. The pellet was then resuspended with 200 μl buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT), incubated on ice for 10 min and centrifuged at 1700 g for 4 min. Chromatin-bound proteins were washed with buffer B, respun and resuspended in 200 μl sample buffer before shearing DNA using a 28-gauge syringe needle.
Cell synchronization
Cells were synchronized in S phase by addition of 2 mM thymidine to cell media for 16 h, replacement with fresh media for 8 h and addition of 2 mM thymidine again for 16 h before release with fresh media for S-phase time course. BrdU incorporation was performed as described previously (Barr et al, 2003). Cell cycle analysis was performed using a Beckton Dickenson FACS Caliber.
Supplementary Material
Supplementary Legends
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
We thank Sharon Barr, Elbert Chang, Maureen Fitch and Narasimham Jammi for helpful preliminary studies. We are also thankful to Dr V Bohr, Dr P Cooper, Dr G Wang and Dr W Kaufmann for the generous gift of various cell lines and reagents. This work was supported by a grant awarded to KAC from the National Institutes of Health (GM62193). KAC is a Leukemia and Lymphoma Society Scholar.
References
- Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 [DOI] [PubMed] [Google Scholar]
- Adimoolam S, Ford JM (2003) p53 and regulation of DNA damage recognition during nucleotide excision repair. DNA Repair (Amst) 2: 947–954 [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2004) Initiating cellular stress responses. Cell 118: 9–17 [DOI] [PubMed] [Google Scholar]
- Ball HL, Myers JS, Cortez D (2005) ATRIP binding to RPA-ssDNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol Biol Cell 16: 2372–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr SM, Leung CG, Chang EE, Cimprich KA (2003) ATR kinase activity regulates the intranuclear translocation of ATR and RPA following ionizing radiation. Curr Biol 13: 1047–1051 [DOI] [PubMed] [Google Scholar]
- Bassett E, King NM, Bryant MF, Hector S, Pendyala L, Chaney SG, Cordeiro-Stone M (2004) The role of DNA polymerase eta in translesion synthesis past platinum-DNA adducts in human fibroblasts. Cancer Res 64: 6469–6475 [DOI] [PubMed] [Google Scholar]
- Bi X, Slater DM, Ohmori H, Vaziri C (2005) DNA polymerase {kappa} is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J Biol Chem 280: 22343–22355 [DOI] [PubMed] [Google Scholar]
- Binz SK, Sheehan AM, Wold MS (2004) Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amst) 3: 1015–1024 [DOI] [PubMed] [Google Scholar]
- Bomgarden RD, Yean D, Yee MC, Cimprich KA (2004) A novel protein activity mediates DNA binding of an ATR–ATRIP complex. J Biol Chem 279: 13346–13353 [DOI] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D (2003) Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 17: 615–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA (2005) Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 19: 1040–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Xu XS, Harrison J, Wang G (2004) Defining the function of xeroderma pigmentosum group F protein in psoralen interstrand cross-link-mediated DNA repair and mutagenesis. Biochem J 379: 71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH (1998) Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J 17: 159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D (2005) Unwind and slow down: checkpoint activation by helicase and polymerase uncoupling. Genes Dev 19: 1007–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ (2001) ATR and ATRIP: partners in checkpoint signaling. Science 294: 1713–1716 [DOI] [PubMed] [Google Scholar]
- Costa RM, Chigancas V, Galhardo Rda S, Carvalho H, Menck CF (2003) The eukaryotic nucleotide excision repair pathway. Biochimie 85: 1083–1099 [DOI] [PubMed] [Google Scholar]
- de Laat WL, Jaspers NG, Hoeijmakers JH (1999) Molecular mechanism of nucleotide excision repair. Genes Dev 13: 768–785 [DOI] [PubMed] [Google Scholar]
- Fitch ME, Cross IV, Ford JM (2003) p53 responsive nucleotide excision repair gene products p48 and XPC, but not p53, localize to sites of UV-irradiation-induced DNA damage, in vivo. Carcinogenesis 24: 843–850 [DOI] [PubMed] [Google Scholar]
- Fleck O, Schar P (2004) Translesion DNA synthesis: little fingers teach tolerance. Curr Biol 14: R389–R391 [DOI] [PubMed] [Google Scholar]
- Ford JM, Hanawalt PC (1997) Role of DNA excision repair gene defects in the etiology of cancer. Curr Top Microbiol Immunol 221: 47–70 [DOI] [PubMed] [Google Scholar]
- Giannattasio M, Lazzaro F, Longhese MP, Plevani P, Muzi-Falconi M (2004a) Physical and functional interactions between nucleotide excision repair and DNA damage checkpoint. EMBO J 23: 429–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Lazzaro F, Siede W, Nunes E, Plevani P, Muzi-Falconi M (2004b) DNA decay and limited Rad53 activation after liquid holding of UV-treated nucleotide excision repair deficient S. cerevisiae cells. DNA Repair (Amst) 3: 1591–1599 [DOI] [PubMed] [Google Scholar]
- Guo Z, Kumagai A, Wang SX, Dunphy WG (2000) Requirement for ATR in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev 14: 2745–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC (2002) Subpathways of nucleotide excision repair and their regulation. Oncogene 21: 8949–8956 [DOI] [PubMed] [Google Scholar]
- Hekmat-Nejad M, You Z, Yee M-C, Newport J, Cimprich KA (2000) Xenopus ATR is a replication-dependent chromatin binding protein required for the DNA replication checkpoint. Curr Biol 10: 1565–1573 [DOI] [PubMed] [Google Scholar]
- Ichikawa M, Nakane H, Marra G, Corti C, Jiricny J, Fitch M, Ford JM, Ikejima M, Shimada T, Yoshino M, Takeuchi S, Nakatsu Y, Tanaka K (2000) Decreased UV sensitivity, mismatch repair activity and abnormal cell cycle checkpoints in skin cancer cell lines derived from UVB-irradiated XPA-deficient mice. Mutat Res 459: 285–298 [DOI] [PubMed] [Google Scholar]
- Jiang G, Sancar A (2006) Recruitment of DNA damage checkpoint proteins to damage in transcribed and nontranscribed sequences. Mol Cell Biol 26: 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannouche P, Stary A (2003) Xeroderma pigmentosum variant and error-prone DNA polymerases. Biochimie 85: 1123–1132 [DOI] [PubMed] [Google Scholar]
- Kim SM, Kumagai A, Lee J, Dunphy WG (2005) Phosphorylation of Chk1 by ATM- and Rad3-related (ATR) in Xenopus egg extracts requires binding of ATRIP to ATR but not the stable DNA-binding or coiled-coil domains of ATRIP. J Biol Chem 280: 38355–38364 [DOI] [PubMed] [Google Scholar]
- Klein B, Pastink A, Odijk H, Westerveld A, van der Eb AJ (1990) Transformation and immortalization of diploid xeroderma pigmentosum fibroblasts. Exp Cell Res 191: 256–262 [DOI] [PubMed] [Google Scholar]
- Kondo T, Wakayama T, Naiki T, Matsumoto K, Sugimoto K (2001) Recruitment of Mec1 and Ddc1 checkpoint proteins to double-strand breaks through distinct mechanisms. Science 294: 867–870 [DOI] [PubMed] [Google Scholar]
- Kumagai A, Guo ZJ, Emami KH, Wang SX, Dunphy WG (1998) The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J Cell Biol 142: 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG (2003) Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol Cell 11: 329–340 [DOI] [PubMed] [Google Scholar]
- Lehmann AR (2003) DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 85: 1101–1111 [DOI] [PubMed] [Google Scholar]
- Lehmann AR (2005) Replication of damaged DNA by translesion synthesis in human cells. FEBS Lett 579: 873–876 [DOI] [PubMed] [Google Scholar]
- Limoli CL, Giedzinski E, Bonner WM, Cleaver JE (2002) UV-induced replication arrest in the xeroderma pigmentosum variant leads to DNA double-strand breaks, gamma-H2AX formation, and Mre11 relocalization. Proc Natl Acad Sci USA 99: 233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Dion V, Wilson JH (2006) Transcription promotes contraction of CAG repeat tracts in human cells. Nat Struct Mol Biol 13: 179–180 [DOI] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ (2000) Chk1 is an essential kinase that is regulated by ATR and required for the G(2)/M DNA damage checkpoint. Genes Dev 14: 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Foiani M, Sogo JM (2006) Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21: 15–27 [DOI] [PubMed] [Google Scholar]
- Lupardus PJ, Byun T, Yee MC, Hekmat-Nejad M, Cimprich KA (2002) A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev 16: 2327–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F (1999) The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 399: 700–704 [DOI] [PubMed] [Google Scholar]
- Matsuda T, Saijo M, Kuraoka I, Kobayashi T, Nakatsu Y, Nagai A, Enjoji T, Masutani C, Sugasawa K, Hanaoka F, Yasui A, Tanaka K (1995) DNA repair protein XPA binds replication protein A (RPA). J Biol Chem 270: 4152–4157 [DOI] [PubMed] [Google Scholar]
- Melo J, Toczyski D (2002) A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol 14: 237–245 [DOI] [PubMed] [Google Scholar]
- Melo JA, Cohen J, Toczyski DP (2001) Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev 15: 2809–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Nakane M, Hattori T, Matsunaga T, Ihara M, Nikaido O (1991) Simultaneous establishment of monoclonal antibodies specific for either cyclobutane pyrimidine dimer or (6–4)photoproduct from the same mouse immunized with ultraviolet-irradiated DNA. Photochem Photobiol 54: 225–232 [DOI] [PubMed] [Google Scholar]
- Murray AW (1991) Cell-cycle extracts. Methods Cell Biol 36: 581–605 [PubMed] [Google Scholar]
- Neecke H, Lucchini G, Longhese MP (1999) Cell cycle progression in the presence of irreparable DNA damage is controlled by a Mec1- and Rad53-dependent checkpoint in budding yeast. EMBO J 18: 4485–4497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WG, Kastan MB (1994) DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol Cell Biol 14: 1815–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell MJ, Cimprich KA (2005) G2 damage checkpoints: what is the turn-on? J Cell Sci 118: 1–6 [DOI] [PubMed] [Google Scholar]
- O'Connell MJ, Walworth NC, Carr AM (2000) The G2-phase DNA-damage checkpoint. Trends Cell Biol 10: 296–303 [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Osborn AJ, Elledge SJ, Zou L (2002) Checking on the fork: the DNA-replication stress-response pathway. Trends Cell Biol 12: 509–516 [DOI] [PubMed] [Google Scholar]
- Pacek M, Walter JC (2004) A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J 23: 3667–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S, Prakash L (2002) Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev 16: 1872–1883 [DOI] [PubMed] [Google Scholar]
- Rouse J, Jackson SP (2002) Interfaces between the detection, signaling, and repair of DNA damage. Science 297: 547–551 [DOI] [PubMed] [Google Scholar]
- Stigger E, Drissi R, Lee SH (1998) Functional analysis of human replication protein A in nucleotide excision repair. J Biol Chem 273: 9337–9343 [DOI] [PubMed] [Google Scholar]
- Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 13: 152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo J, Jaruga P, Rodriguez H, Dizdaroglu M, Bohr VA (2002) The cockayne syndrome group B gene product is involved in cellular repair of 8-hydroxyadenine in DNA. J Biol Chem 277: 30832–30837 [DOI] [PubMed] [Google Scholar]
- Volker M, Mone MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JH, van Driel R, van Zeeland AA, Mullenders LH (2001) Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell 8: 213–224 [DOI] [PubMed] [Google Scholar]
- Ward IM, Minn K, Chen J (2004) UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J Biol Chem 279: 9677–9680 [DOI] [PubMed] [Google Scholar]
- Zachos G, Rainey MD, Gillespie DA (2003) Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J 22: 713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos G, Rainey MD, Gillespie DA (2005) Chk1-dependent S–M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol Cell Biol 25: 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Piwnica-Worms H (2001) ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21: 4129–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439 [DOI] [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
- Zou L, Cortez D, Elledge SJ (2002) Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev 16: 198–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Legends
Supplementary Figure 1
Supplementary Figure 2