

Abstract
The Rad23/Rad4 nucleotide excision repair (NER) protein complex functions at an early stage of the NER reaction, possibly promoting the recognition of damaged DNA. Here we show that Rad4 protein is ubiquitinated and degraded in response to ultraviolet (UV) radiation, and identify a novel cullin-based E3 ubiquitin ligase required for this process. We also show that this novel ubiquitin ligase is required for optimal NER. Our results demonstrate that optimal NER correlates with the ubiquitination of Rad4 following UV radiation, but not its subsequent degradation. Furthermore, we show that the ubiquitin–proteasome pathway (UPP) regulates NER via two distinct mechanisms. The first occurs independently of de novo protein synthesis, and requires Rad23 and a nonproteolytic function of the 19S regulatory complex of the 26S proteasome. The second requires de novo protein synthesis, and relies on the activity of the newly identified E3 ubiquitin ligase. These studies reveal that, following UV radiation, NER is mediated by nonproteolytic activities of the UPP, via the ubiquitin-like domain of Rad23 and UV radiation-induced ubiquitination of Rad4.
Keywords: DNA repair, NER, proteasome, ubiquitin, yeast
Introduction
The ubiquitin–proteasome pathway (UPP) is the major pathway for degradation of cellular proteins (Pickart, 2004). This occurs through two discrete steps: covalent attachment of multiple ubiquitin molecules (conjugation) to target proteins, and subsequent degradation of the tagged proteins by the 26S proteasome (comprised of the catalytic 20S core and the 19S regulator) (Glickman and Ciechanover, 2002). This canonical role of the UPP is associated with housekeeping functions, regulation of protein turnover, and antigenic-peptide generation (Ciechanover and Ben-Saadon, 2004). More recently, it has become evident that both components of this pathway have noncanonical (nonproteolytic) functions that include the regulation of transcription (Ferdous et al, 2001, 2002; Salghetti et al, 2001; Gonzalez et al, 2002; Gillette et al, 2004), endocytosis (Strous and Govers, 1999), and DNA repair (Russell et al, 1999; Gillette et al, 2001; Dupre et al, 2004).
Nucleotide excision repair (NER) removes lesions in DNA caused by ultraviolet (UV) light. This process requires the coordinated activity of over 30 proteins, the majority of which are conserved from yeast to man. Defective NER has been documented in the hereditary cancer-prone disease Xeroderma pigmentosum (XP) and is the primary cellular phenotype of the disease (Hanawalt, 2001). The frequent association of the XP homozygous state with various types of skin cancers established the importance of NER as a fundamental mechanism for protecting the functional integrity of the human genome (Friedberg, 2001).
In the yeast Saccharomyces cerevisiae, the ubiquitin-like (Ubl) domain of the NER protein Rad23 is required for survival after exposure to UV light (Watkins et al, 1993; Russell et al, 1999). One of the first links between the UPP and NER was the observation that the Ubl domain of Rad23 protein interacted directly with the 26S proteasome (Schauber et al, 1998). We previously demonstrated that the 19S regulatory complex (19S RC) of the 26S proteasome has a nonproteolytic function in NER that is mediated through the Ubl domain of Rad23 protein (Russell et al, 1999). In vivo, the 19S RC negatively regulates the rate of lesion removal during NER. Rad23-deleted cells show reduced rates of excision of UV-radiation-induced DNA damage, and reduced UV survival. However, this phenotype is significantly rescued by introducing mutations in specific 19S RC subunits (Gillette et al, 2001). Importantly, mutations in 20S core proteasome subunits, which promote a severe defect in proteolysis, fail to rescue the NER deficiency associated with rad23-deleted cells (Gillette et al, 2001). These findings and more recent observations indicate that defective proteolysis does not correlate with the ability to rescue the NER deficiency associated with deletion of the RAD23 gene (Ortolan et al, 2004; Xie et al, 2004).
Here we demonstrate that Rad4 is targeted for degradation by the 26S proteasome following UV radiation. We show that a novel cullin-based E3 ubiquitin ligase ubiquitinates Rad4 following UV, and demonstrate that cellular survival after UV correlates with the ubiquitination of Rad4, but not its subsequent degradation. Finally, we show that two pathways that are distinct in their requirements for de novo protein synthesis regulate the NER response to UV radiation. Pathway I operates independently of de novo protein synthesis, while pathway II relies on de novo protein synthesis. We show that pathway I is regulated by the previously reported nonproteolytic activity of the 19S RC and the Ubl domain of Rad23, while pathway II requires the activity of the newly described E3 ligase. These studies demonstrate that nonproteolytic activities of the ubiquitin/proteasome system operate via two distinct pathways during NER, and reveal novel insights about the regulation of NER in response to UV radiation.
Results
Rad4 protein is degraded by the UPP in response to UV
It has been suggested that, following exposure of cells to UV irradiation, the proteolytic degradation of Rad4 is attenuated, resulting in accumulation of this repair factor and enhanced NER (Lommel et al, 2002; Ng et al, 2003). A similar model was originally proposed for the Rad4 homologue, XPC. More recent evidence using an antibody to the endogenous protein suggests that XPC is stable, with a half-life of over 6 h (Okuda et al, 2004). Since both of these earlier reports employed overexpressed, epitope-tagged versions of Rad4/XPC, we wanted to examine the stability of native Rad4 protein following exposure of cells to UV light, using an antibody specific to endogenously expressed Rad4 protein.
In the absence of UV light, the steady-state level of Rad4 protein does not alter significantly over a 3-h period following incubation of cells with the protein synthesis inhibitor cycloheximide, indicating that, like XPC, Rad4 protein is stable during this time period, and is not rapidly turned over by the proteasome (Figure 1A, top). Figure 1A (bottom) shows that, under the same conditions, an unrelated protein with a shorter half-life, cyclophilin A, shows a decrease in steady-state levels over the 3-h period, demonstrating the activity of the cycloheximide in blocking protein synthesis during the experiment.
Figure 1.
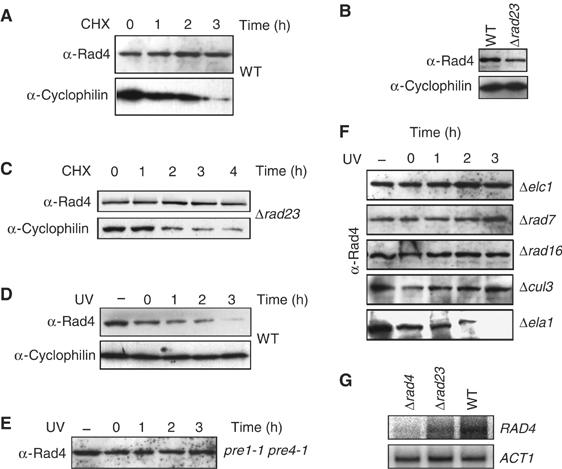
Post-UV degradation of Rad4 is dependent on proteolysis. (A) Anti-Rad4 Western blot of WGC4a (WT) extracts from a strain grown in the protein synthesis inhibitor cycloheximide for the time indicated (h), along with an anti-cyclophilin control of the same blot. (B) Anti-Rad4 Western blot showing the relative steady-state levels of Rad4 in a WT and Δrad23 mutant strain. The bottom panel shows anti-cyclophilin loading control. (C) Anti-Rad4 Western blot of Δrad23 extracts from a strain grown in the protein synthesis inhibitor cycloheximide for the time indicated (h), along with an anti-cyclophilin control of the same blot. (D) Anti-Rad4 Western blot of WGC4a (WT) extracts from a strain either unirradiated (−) or UV irradiated (40 J) and allowed to recover for the times indicated (h), along with an anti-cyclophilin control of the same blot. (E) Anti-Rad4 Western blot of pre 1-1, pre 4-1 extracts from cells either unirradiated (−) or UV irradiated (40 J) and allowed to recover for the times indicated (h). (F) Deletion of genes encoding the ECS ligase components results in stabilization of Rad4 post-UV. Anti-Rad4 Western blots of extracts from Research Genetics mutant strains listed either unirradiated (−) or UV irradiated and allowed to repair for the times indicated (h). (G) Northern blot analysis of RAD4 transcript levels in the indicated strains. ACT1 transcript levels are shown as a loading control.
It has been suggested that, in the absence of Rad23, Rad4 protein is rapidly degraded (Lommel et al, 2002). This was based on the observation that steady-state levels of epitope-tagged Rad4 are significantly reduced in rad23-deleted cells. We observed that the steady-state levels of native Rad4 (in the absence of cycloheximide) are also reduced in a Δrad23 strain (Figure 1B). We used the protein synthesis inhibitor cycloheximide to examine whether the lower steady-state levels of native Rad4 were due to increased turnover of Rad4 in the absence of its interacting partner Rad23. Surprisingly, the steady-state level of Rad4 protein in this strain did not alter significantly over a 4-h period following incubation of cells with the protein synthesis inhibitor cycloheximide (Figure 1C). We confirmed these observations in the absence of cycloheximide by performing pulse-chase experiments following the metabolic radiolabelling of cellular proteins and found the half-life of Rad4 to be between 3 and 4 h both in wild-type (WT) and rad23-deleted cells (see Supplementary data). These results show that the reduced levels of Rad4 observed in rad23-deleted cells are not due to a reduction in the half-life of Rad4. To determine the cause of reduced Rad4 levels in rad23-deleted cells, we investigated the transcriptional levels of Rad4 in WT and rad23-deleted cells. Northern blot analysis revealed that Rad4 transcript levels are significantly reduced in rad23-deleted cells compared with WT cells (Figure 1G). We conclude that the reduced levels of Rad4 protein in rad23-deleted cells are due to reduced Rad4 transcript levels, and not due to the increased proteolytic degradation of Rad4 in the absence of its interacting partner Rad23.
In contrast to earlier studies which used ectopically expressed Rad4, we found that endogenously expressed Rad4 protein is not stabilized after UV treatment; rather, it is rapidly degraded following exposure to UV light. Figure 1D (top) shows the decrease in steady-state levels of Rad4 protein over a 3-h period following exposure of WT cells to UV radiation. Rad4 steady-state levels returned to the pre-UV state within 5–6 h following UV irradiation (data not shown). The steady-state level of cyclophilin remains unchanged following exposure of cells to UV light Figure 1D (bottom). We do not detect a ladder of polyubiquitinated Rad4 following UV radiation, but longer running of samples on 5–15% gradient SDS–PAGE gels revealed the presence of a higher molecular weight form of Rad4, which is consistent with the conjugation of a single ubiquitin (Supplementary data S4). Using an isogenic strain carrying conditional mutations in two subunits of the 20S proteasome (pre1-1, pre4-1), we confirmed that the UV-dependent degradation of Rad4 protein is mediated by the proteolytic activity of the 26S proteasome. This temperature-sensitive strain shows a marked decrease in the ability to degrade proteins that have been targeted by the ubiquitin-dependent proteolytic pathway even at temperatures permissive for growth (Russell and Johnston, 2001). Figure 1E shows that in the pre1-1, pre4-1 strain, Rad4 protein steady-state levels do not alter following exposure to UV light. We also examined the stability of Rad4 protein post-UV radiation using a chemical approach. The proteolytic function of the 26S proteasome can be inhibited in yeast strains mutated in the ise1 gene by growing cells in the presence of the aldehyde peptide inhibitor (MG132) (Lee and Goldberg, 1996). Inhibition of proteolysis using this method also resulted in a stabilization of Rad4 protein following UV radiation (data not shown). These data show that endogenous Rad4 protein is targeted for degradation by the UPP in a UV-dependent fashion. This observation may have been overlooked in earlier studies due to the inherent instability of the epitope-tagged Rad4 protein used (Lommel et al, 2002; Ortolan et al, 2004).
UV-dependent degradation of Rad4 requires a novel cullin-based E3 ubiquitin ligase
Genetic and biochemical evidence suggests that, in addition to its ATPase activity (which is fundamental to the role of Rad16 in an early stage of the global genome repair (GGR) mechanism), the Rad7/Rad16 complex in concert with Elc1 may also have an additional, separate function as an E3 ubiquitin ligase (Ramsey et al, 2004). Previous studies have shown that a specific component of the cullin-based E3 ubiquitin ligases functions as a substrate-specific adaptor, the protein target usually interacting with the adaptor through a protein–protein interacting domain such as an SH2 domain or a leucine-rich repeat (LRR) motif. Previous protein–protein interaction studies predicted that Rad4 might be a primary target for this putative Rad7-containing E3 ligase, since it was shown that the LRR domain of Rad7 is required for a two-hybrid interaction between Rad7 and Rad4 (Wang et al, 1997; Pintard et al, 2004). To test whether this putative E3 ligase is required for the degradation of Rad4 protein in response to UV radiation in vivo, we deleted or mutated individual components of the Rad7 E3 ligase.
Deletion of any of the components of the Rad7-containing E3 ubiquitin ligase resulted in stabilization of Rad4 protein following exposure of these cells to UV light. Figure 1F shows that steady-state levels of Rad4 protein are stable following UV in the Δelc1 strain, showing that Elc1 protein is required for the degradation of Rad4 protein after UV. Steady-state levels of Rad4 protein were also stable in mutants defective in each of the other predicted components of the Elongin–Cullin–Socs-box (ECS) ligase complex, including Rad7 and Rad16 proteins (Figure 1F). Rad7 is an SOCS box-containing protein (Ho et al, 2002), suggesting that the Rad7 E3 ligase may be a novel member of the cullin-based ECS-type E3 ubiquitin ligases, a subclass of the Skp1–Cullin–F-box (SCF) type family of ubiquitin ligases. Therefore, we examined a number of cullin mutants, including cul3- and cul8-deleted strains and crl2, 4, and 7 temperature-sensitive mutants. We showed that CUL3 was uniquely required for the degradation of Rad4 protein following UV irradiation (Figure 1F, second to bottom panel). Deletion of an unrelated SOCS box gene, ELA1, did not result in the stabilization of Rad4 protein after UV (Figure 1F, bottom panel). We also examined the effect of other mutants on Rad4 stability post-UV, including the E2 encoding genes UBC4, UBC5, RAD6, and the cullin CUL8. Deletion of these genes did not result in the stabilization of Rad4 protein after UV (data not shown). This demonstrates that the inhibition of Rad4 protein degradation post-UV is specifically related to deletion of the genes that encode components of the cullin-based Rad7-containing E3 ligase complex.
Rad7 is a component of a cullin-based E3 ubiquitin ligase
The physical interaction of Rad7, Rad16, Elc1, and Cul3 proteins was confirmed by co-immunoprecipitation. A Rad7 polyclonal antibody was used to immunoprecipitate Rad7 protein from a WT extract. Figure 2A shows that, following immunoprecipitation and Western blotting, Rad16 protein co-precipitates with Rad7 protein in the WT extract. We found that, in addition to Elc1 protein, Cul3 protein also co-precipitated with Rad7 protein. A significant amount of both Elc1 and Cul3 proteins do not co-precipitate with Rad7 protein. It is likely that they associate with other protein complexes, possibly functioning in different ubiquitin ligases. None of these proteins immunoprecipitated in the control experiment using an extract from Δrad7 cells (Figure 2A, right panel). The specificity of the antibodies used was determined by comparing the Western blots of either WT or mutant extracts prepared from strains deleted of the corresponding genes (Supplementary data S1). The detection of Cul3, Elc1, and Rad16 proteins co-precipitating with Rad7 protein further suggests that this complex is a member of the cullin-based family of E3 ubiquitin ligases.
Figure 2.
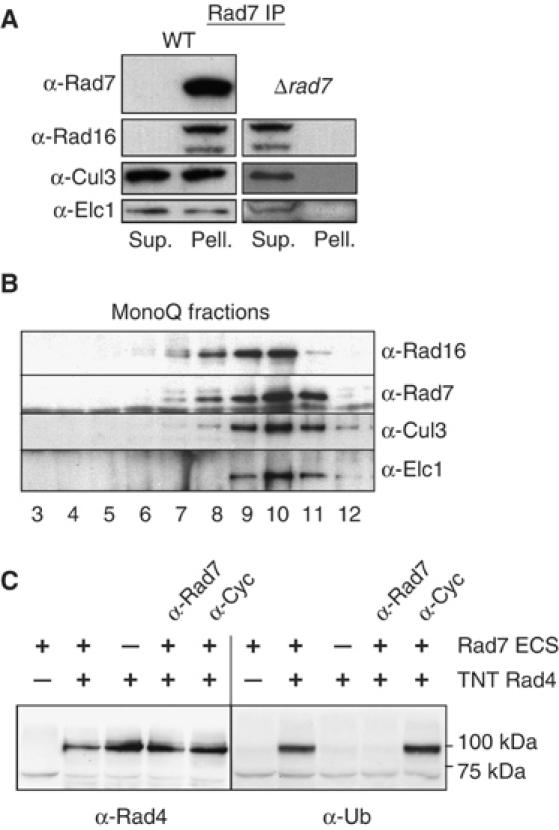
Characterization and activity of the Rad7 ECS E3 ligase. (A) Co-immunoprecipitation of ECS ligase components with Rad7. Western blot of Rad7 immunoprecipitation probed with the antibodies listed (Sup., supernatant; Pell., pellet). WT extract is on the left, control extract (Δrad7) lacking Rad7 protein is on the right. (B) Purification of the Rad7 ECS ligase. Western blot of MonoQ fractions probed with the antibodies listed (see Materials and methods for details). (C) In vitro ubiquitination of Rad4 by purified Rad7 ligase. Left panel, anti-Rad4 Western blot of ubiquitination reaction with components indicated (TNT—TNT reaction with control vector). Anti-rad7 antibody or the anti-cyclophilin antibody were added in lanes 4 and 5, respectively. Right panel, the left-hand blot was reprobed with ubiquitin antibody. Approximate MW markers indicated on the right.
The Rad7-containing E3 ligase complex ubiquitinates Rad4 in vitro
Collectively, the above observations suggest the existence of a complex of proteins that constitute a novel E3 (ECS-type) ubiquitin ligase. Previous studies describing the architecture of cullin-based E3 ligases and our genetic evidence suggest that Rad4 is a primary target of this E3 ligase (Wang et al, 1997; Pintard et al, 2004). We performed biochemical experiments to examine this. Previously, we described the purification of epitope-tagged Rad7 protein (Reed et al, 1999). Cul3 and Elc1 proteins co-purify with Rad7 and Rad16 proteins through a number of chromatographic steps that include Ni-NTA, p-11, DEAE Sephacel, and Mono-Q columns. This results in a nearly 15 000-fold enrichment of Rad7/Rad16. Western blotting showed that fractions eluted from the mono-Q column of the Rad7 protein purification (Reed et al, 1998; Figure 2B) contain Rad7, Rad16, Elc1, and Cul3 proteins. We expressed Rad4 protein using a coupled transcription/translation system. The peak mono-Q fractions (Figure 2B, lanes 9 and 10) were added to in vitro expressed Rad4 protein in an ubiquitination assay. Figure 2C shows that these fractions were capable of supporting the ubiquitination of Rad4 protein in vitro. The addition of an antibody specific to Rad7 (or a Rad16-specific antibody, data not shown) inhibited this activity (Figure 2C, second to last lane), confirming that the ubiquitin ligase function was dependent specifically on the E3 ligase complex and was not an unrelated contaminant. A non-specific antibody (Figure 2C, last lane) or preimmune serum (data not shown) had no effect.
Post-UV Rad4 degradation does not correlate with UV survival
To determine the influence of Rad4 protein stability on cellular survival after UV, we examined UV survival in deletion strains for each of the components of the ECS ligase. Rad4 steady-state levels are stable post-UV in each of the deletion strains (Figure 1F), but the effect on UV survival varies (Figure 3A). In the case of Δrad7 (Figure 3A) and Δrad16 (data not shown) strains, UV survival is significantly reduced following exposure of these mutants to UV light, as expected (Verhage et al, 1996). However, Δelc1- and Δcul3-deleted cells are not significantly UV sensitive (Figure 3A). This shows that, under the conditions tested, loss of the E3 ligase activity does not significantly affect UV survival. We speculated that site-directed mutations made to the SOCS box domain of the Rad7 protein might specifically affect its E3 ligase activity, leaving intact its fundamental role in GGR (Figure 3B). Rad4 protein stability and UV survival of a Δrad7 strain carrying a centromeric plasmid coding for WT Rad7 (pRad7) or the SOCS box-mutated Rad7 (psocs) proteins are shown in Figure 3C and D, respectively. The psocs strain exhibits stabilization of Rad4 protein post-UV (Figure 3C). However, the psocs strain, like those carrying deletions in Elc1 and Cul3, is not sensitive to UV (Figure 3A and D). These results demonstrate that altering the stability of Rad4 protein in response to UV does not affect UV survival in yeast.
Figure 3.
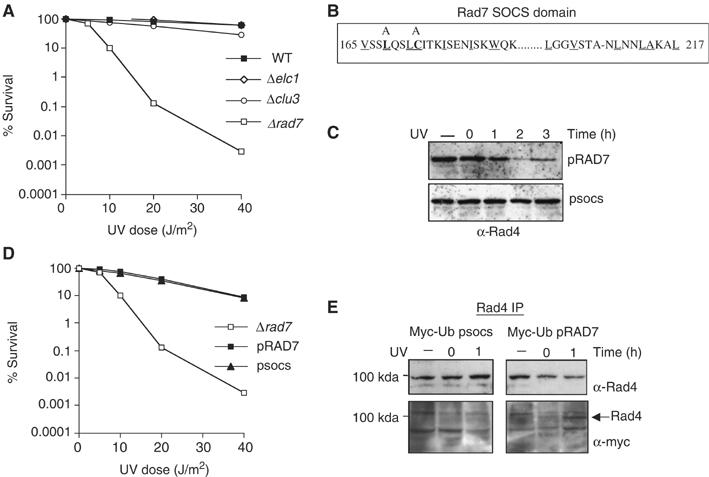
Rad7 SOCS box mutation stabilizes Rad4 without affecting UV survival. (A) UV survival for the mutant strains listed. (B) The amino-acid sequence of the SOCS box domain of Rad7. Conserved residues are underlined. Alanine mutations made in the pSOCS are noted on the top, with the mutated residues in bold. (C) Post-UV steady-state levels of Rad4 shown by anti-Rad4 Western blot. The cells were either unirradiated (−) or UV irradiated and allowed to repair for the times indicated (h). Top panel, extracts from an Sc507-derived rad7-deleted yeast strain transformed with a centromeric vector expressing WT Rad7 (top panel pRad7) or SOCS box domain mutant (bottom panel pSOCS) from the endogenous Rad7 promoter. (D) UV survival for the mutant strains listed. (E) The ubiquitination status of Rad4 at 0 and 1 h post-UV in the WT (pRad7), and SOCS box-mutated (psocs) strains expressing Myc-tagged ubiquitin. These strains were either unirradiated (−) or UV irradiated and allowed to repair for 0 or 1 h. Lower panel, Rad4 was immunoprecipitated from the extracts and then blotted with anti-Myc antibody. The upper panel shows the same blot probed with Rad4 antibody.
SOCS-mutated rad7 cells fail to ubiquitinate Rad4 after UV radiation
To determine whether the Rad7 E3 ubiquitin ligase is required for the ubiquitination of Rad4 protein in response to UV radiation in vivo, we immunoprecipitated Rad4 protein in a strain expressing Myc epitope-tagged ubiquitin protein (Hochstrasser et al, 1991) and either WT Rad7 or SOCS box-mutated Rad7 (psocs) proteins. Immunoprecipitation of Rad4 protein was followed by Western blotting with anti-Myc antibody to detect the presence of Myc-tagged ubiquitin. Figure 3E shows that Rad4 protein is ubiquitinated in response to UV in the pRAD7 strain (Figure 3E, lower right panel), but not in the psocs strain (Figure 3E, lower left panel). Reprobing the same blot with anti-Rad4 antibody confirmed the position of Rad4 protein (Figure 3E, upper left and right panels). This result was confirmed in an independent Rad4 co-immunoprecipitation experiment using an antibody raised against ubiquitin by genetic immunization (Chambers and Johnston, 2003; Supplementary data S2). This result demonstrates that the Rad7 E3 ligase activity is required for the ubiquitination of Rad4 protein in response to UV radiation in vivo.
The Rad7-containing E3 ubiquitin ligase regulates NER in the absence of Rad23
Collectively, our results show that the Rad7-containing E3 ubiquitin ligase targets Rad4 protein for ubiquitination in response to UV radiation, but neither the ubiquitination nor the UV-dependent degradation of Rad4 correlate with cellular survival following UV radiation. Therefore, what is the significance of the Rad7 E3 ubiquitin ligase on NER and UV survival? Since Rad4 and Rad23 are interacting partners and Rad23 has a Ubl domain that is required for normal rates of NER, we tested whether ubiquitination of Rad4 plays a role in UV survival in the absence of Rad23. Figure 4A shows that a Δrad23 strain carrying an additional mutation in the SOCS box domain of Rad7 protein is more sensitive to UV than a strain deleted only in the RAD23 gene. Figure 4B confirms that, following UV, Rad4 protein is degraded in the Δrad23 mutant (Δrad23/pRAD7), but in the Δrad23/psocs strain Rad4 protein is stable. It is interesting to note that, while the steady-state levels of Rad4 are lower in the Δrad23 mutant, the increased steady-state levels of Rad4 in the psocs mutant do not result in an increase in UV survival. This is not surprising, since we and others have previously shown that inhibiting the proteolytic activity of the 26S proteasome does not significantly influence NER in a Δrad23 strain (Gillette et al, 2001; Lommel et al, 2002). We conclude that the effect of the Rad7-containing E3 ligase on UV survival correlates with its ability to ubiquitinate Rad4 protein. Figure 4C shows that a Δrad23 mutant additionally mutated in the Elongin C component of the ECS ligase, elc1, also demonstrates an increased UV sensitivity. This suggests that the increase in UV sensitivity in the psocs mutant is due specifically to the loss of Rad7-containing E3 ligase activity, and is not an unrelated activity of the Rad7 protein. Furthermore, this effect is specific to genes encoding components of the Rad7-containing E3 ligase, since Δrad23 strains additionally mutated in an unrelated SOCS box gene, Ela1, do not exhibit this phenotype (Figure 4C). These results show that the effect of the E3 ubiquitin ligase on UV survival includes the ubiquitination of Rad4 protein in response to UV. They also demonstrate that the Rad7-containing E3 ligase and Rad23 proteins have overlapping functions in UV survival (Bertolaet et al, 2001a; Gillette et al, 2001).
Figure 4.
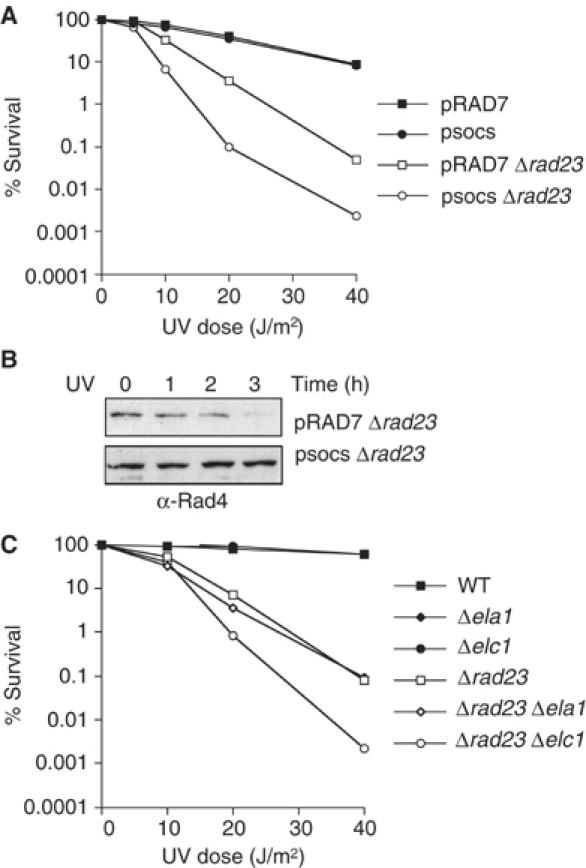
Effect of ECS ligase activity on UV survival in a Δrad23 mutant. (A) UV survival of the Sc507-derived strains listed. (B) Anti-Rad4 Western blot of protein extracts prepared from pRad7, Δrad23 (top), or psocs, Δrad23 (bottom). Strains were either unirradiated (−) or UV irradiated and allowed to repair for the times indicated (h). (C) UV survival of the Research Genetics strains listed.
The Rad23 protein has two functional domains that significantly influence NER: the Ubl domain and the C-terminal Rad4 protein interacting domain (Bertolaet et al, 2001a, 2001b; Gillette et al, 2001). The UBA domain of Rad23 does not appear to have a significant influence on NER (Bertolaet et al, 2001b), although a more recent study suggests that there might be some role for this domain in UV survival (Heessen et al, 2005). We next examined whether the overlapping function of Rad23 and the Rad7 E3 ligase activity was associated with the Ubl domain of Rad23. Figure 5A shows that an E3 ligase defective strain (psocs) expressing a Ubl-deleted version of Rad23 (Δubl) protein also exhibits increased UV sensitivity compared to a strain expressing Ubl-deleted Rad23 protein alone. This shows that the increased UV sensitivity of Δrad23 mutants additionally defective in the Rad7 E3 ubiquitin ligase activity (psocs) is specifically related to the loss of the Ubl domain of Rad23 protein.
Figure 5.
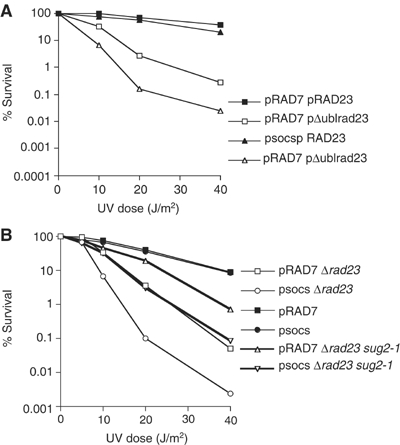
(A) UV survival of the Sc507-derived strains transformed with a centromeric vector expressing either full-length Rad23 from its endogenous promoter (pRAD23) or Rad23 deleted of the Ubl domain (pΔublrad23). (B) UV survival of the Sc507-derived strains listed.
The Rad7 E3 ligase functions in a pathway different from the Rad23/19S RC activity
Mutations in specific 19S RC subunits including sug2-1 can rescue the defect in lesion removal caused by the loss of the ubl domain of Rad23 protein (Gillette et al, 2001). Since our observations show that the roles of Rad23 and the Rad7 E3 ligase functionally overlap, we reasoned that, in the absence of Rad23, ubiquitination of Rad4 could compensate for the loss of the Rad23 ubl domain, either by recruiting the 19S RC or functionally influencing the 19S RC activity. We tested this hypothesis by adding the sug2-1 mutation to the UV-sensitive Δrad23, psocs, strain. Adding the 19S RC mutation to the Δrad23, psocs strain could result in a number of different outcomes. Conceivably, it could have no effect on UV survival of the Δrad23, psocs strain (if the function of the E3 ligase was to recruit the 19S RC), or it could rescue UV sensitivity to the same level as the Δrad23, sug2-1 mutant strain (if the E3 ligase functionally affected the 19S activity). Finally, it could result in an intermediate UV phenotype (which would be consistent with a function that was independent of the 19S RC activity). Figure 5B shows that the sug2-1 mutation displays an intermediate UV phenotype, partially rescuing the psocs, Δrad23 double mutant, but to a level less than the Δrad23, sug2-1 strain. This observation is consistent with the Rad7-containing E3 ligase activity functioning in a pathway different from the Rad23/19S activity. An alternative interpretation is that the 19S mutant, which partially rescues the Δrad23 phenotype, can no longer rescue the Δrad23 phenotype in the background of a pSOCS strain, but now instead partially rescues the loss of Rad7 E3 ligase function. We do not favour this interpretation.
NER in Δrad23 cells requires de novo protein synthesis
Since the Rad7-containing E3 ligase activity functions in a pathway different from the Rad23/19S RC activity, we next turned our attention to exploring how these two pathways might differ. In the absence of Rad23 protein, the rate of NER is markedly reduced (Mueller and Smerdon, 1996). Optimal rates of NER in yeast cells are determined by two components, one that requires de novo protein synthesis and the other that does not (Waters et al, 1993; Al-Moghrabi et al, 2003). We speculated that the slow rate of NER observed in Δrad23 cells might result from the loss of one of these components. It has been shown previously that blocking protein synthesis with cycloheximide reduces the efficiency of NER in yeast (Al-Moghrabi et al, 2003). We confirmed this observation by pretreating cells with cycloheximide prior to, and following exposure to, UV radiation, and measuring cyclobutane pyrimidine dimer (CPD) removal from the genome overall in a WT strain (Figure 6). We confirmed the inhibition of de novo protein synthesis by cycloheximide by examining cyclophilin levels as described earlier (Figure 1). Cycloheximide reduced the efficiency of CPD removal during NER in response to UV radiation over the 3-h period examined, confirming earlier studies (Al-Moghrabi et al, 2003). In contrast to WT cells, when Δrad23 cells were treated with cycloheximide prior to UV irradiation, the rate of lesion removal was reduced to almost undetectable levels (Figure 6). Pretreating Δrad23 cells with cycloheximide does not affect Rad4 protein levels (Figure 1A). These results demonstrate that NER in Δrad23 cells is dependent on de novo protein synthesis.
Figure 6.
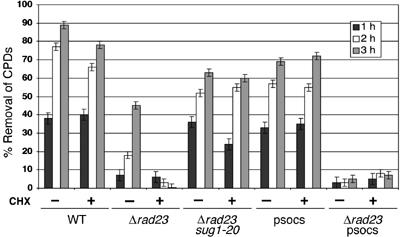
Time course of CPD removal as measured by slot blot assay (see Materials and methods). Strains indicated were treated with cycloheximide (+) or untreated (−). Times indicated refer to recovery times post-UV (h).
The Rad23-mediated nonproteolytic activity of the 19S RC regulates NER pathway I
Mutations in the 19S RC accelerate the rate of lesion removal in Δrad23 cells (Gillette et al, 2001). To determine whether de novo protein synthesis is necessary for this phenomenon, we examined repair in Δrad23, sug1-20 double-mutant cells in the presence and absence of cycloheximide. Figure 6 shows that lesion removal in Δrad23 cells is rescued by specific mutations in the 19S RC even in the presence of cycloheximide, indicating that this component of the NER response occurs independently of de novo protein synthesis.
Collectively, these observations indicate that there are two components to the NER response to UV damage: pathway I occurs independently of de novo protein synthesis; pathway II is dependent on de novo protein synthesis. Pathway I is coregulated by Rad23, which functions, in part, by attenuating the inhibitory effect of the 19S RC on the rate of lesion removal (see Figure 7; Gillette et al, 2001).
Figure 7.
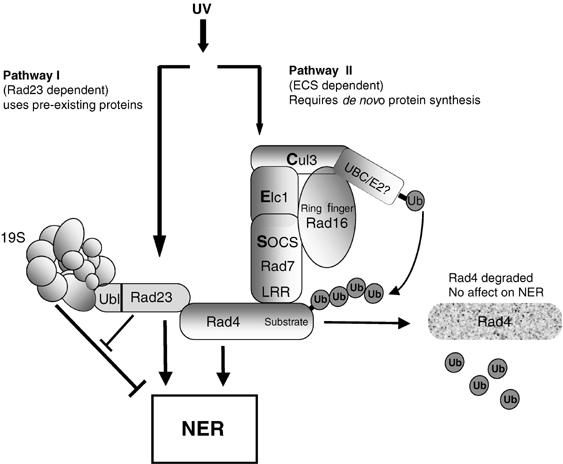
Regulation of the parallel pathways that regulate the NER response to UV. Pathway I is regulated by Rad23 and operates independently of de novo protein synthesis. Rad23 functions, in part, by attenuating the inhibitory effect of the 19S proteasome on the rate of lesion removal. Pathway II relies on de novo protein synthesis and the ubiquitination of Rad4 via the action of the ECS ligase, depicted in the centre of the figure. Subsequently, Rad4 is degraded via the 26S proteasome, with de novo synthesis eventually restoring Rad4 to its pre-UV levels.
The Rad7 E3 ubiquitin ligase regulates NER pathway II
The dependence of pathway II on de novo protein synthesis suggests that it may be part of a transcriptional response that operates following UV radiation. To examine whether the Rad7-containing E3 ligase regulates the cycloheximide-dependent pathway (pathway II), we examined the rate of UV lesion removal in the Rad7-containing E3 ligase-mutated strain. While UV survival is normal in the psocs strain, careful examination of UV lesion removal shows a slight decrease in the amount of lesion removal at later time points (Figure 6). In contrast to the WT strain, the (reduced) level of lesion removal observed in the psocs strain is not affected by cycloheximide. This suggests that the psocs strain is specifically defective in pathway II that requires de novo protein synthesis. Consistent with this interpretation, NER in the psocs, Δrad23 double mutant is almost undetectable, and is not affected by the addition of cycloheximide (Figure 6).
Discussion
We have demonstrated that, after exposing cells to UV radiation, the NER response is comprised of two components, based on their requirement for de novo protein synthesis. Pathway I requires the interaction of Rad23 protein and the 19S RC, and functions independently of de novo protein synthesis. Pathway II requires the novel Rad7-containing E3 ubiquitin ligase and depends on de novo protein synthesis. Both pathways require distinct nonproteolytic activities of the UPP system (see Figure 7). Our observations show that the NER defect associated with Δrad23 cells is related to the rate at which DNA damage is removed after UV, which likely accounts for the moderate UV sensitivity of the Δrad23 strain. Adding cycloheximide to Δrad23 cells severely reduces repair, showing that NER in these cells is dependent on de novo protein synthesis. In contrast, a strain expressing a Rad7 protein mutated in the SOCS box domain relies on pathway I for NER. The rate of repair in this mutant strain is reduced at later times after UV radiation, and is not affected by the addition of cycloheximide. No significant effect on UV survival is observed in the Rad7-containing E3 ligase mutant strain (rad7, socs), suggesting that, under the conditions tested, pathway I primarily influences UV survival. A strain lacking both Rad23 and the Rad7-containing ECS E3 ligase activity shows a decrease in UV survival and almost no lesion removal in the time periods examined. UV survival in this strain is not as sensitive as a strain mutated in a core NER factor. This suggests that, while these pathways regulate the rate of NER, a low level of NER still occurs.
The NER and the UPPs combine to promote efficient DNA repair and UV survival (Schauber et al, 1998; Russell et al, 1999; Bertolaet et al, 2001b; Gillette et al, 2001; Lommel et al, 2002; Ng et al, 2002; Sweder and Madura, 2002; Ramsey et al, 2004). Rad23 protein coordinates these two systems. Our earlier work revealed a nonproteolytic activity of the 19S RC that functioned in NER. Significantly, inhibition of proteolysis both in vivo and in vitro had no effect on NER even in the absence of Rad23 (Russell et al, 1999; Gillette et al, 2001). However, specific defects in the 19S RC can partially rescue the loss of NER function of a strain lacking Rad23 (Gillette et al, 2001). Here we show that this rescue occurs independently of de novo protein synthesis, suggesting that the nonproteolytic role of the 19S RC specifically functions in pathway I.
An understanding of the relationship between NER and the proteasome has been made difficult following reports suggesting that the levels of the mouse XPC protein and its yeast homolog Rad4 protein are modulated by proteolysis (Lommel et al, 2002; Ng et al, 2003). It was also suggested that the stabilization of both proteins in response to DNA damage regulated NER and UV survival. In both cases, these studies involved the use of tagged versions of XPC and Rad4 proteins. In both instances, the epitope-tagged proteins have reduced half-lives compared to the endogenous proteins. Exposure of cells containing these tagged proteins to UV radiation results in what is interpreted to be a transient stabilization of XPC/Rad4 protein. These results led to a model in which proteolysis by the 26S proteasome was responsible for the continual turnover of Rad4/XPC protein. The model suggested that, following UV damage, proteolytic degradation of Rad4/XPC protein by the 26S proteasome was attenuated, resulting in accumulation of this repair factor and enhancement of NER (Lommel et al, 2002; Ng et al, 2003).
In contrast, our studies show that this is not the case when antibodies against endogenous Rad4 protein are used. We show that native Rad4 protein is stable, with a half-life of around 4 h (Figure 1A and Supplementary data S3). Upon UV exposure, steady-state levels of Rad4 protein rapidly decrease, due to increased proteolysis by the proteasome. It is known that the Ubl domain and the Rad4 interacting domain of Rad23 protein are functionally distinct (Bertolaet et al, 2001a, 2001b; Gillette et al, 2001), and it has been suggested that the Rad4-binding domain of Rad23 protected Rad4 from degradation by the 26S proteasome. It is clear that, in the absence of Rad23, steady-state levels of Rad4 are reduced, but the cause of reduced Rad4 levels and whether they directly affect NER have not been established. We show that the reduced levels of Rad4 in Rad23-deleted cells are not due to increased proteolysis of Rad4 in these cells, but rather due to reduced production of Rad4 transcript. Our results reveal the mechanism of reduced Rad4 levels in these cells. Although Rad4 protein levels are reduced in Δrad23 cells, stabilization of Rad4 in proteolytic defective mutants has no effect on the NER defective phenotype of a Δrad23 strain. Others have shown that overexpressing Rad4 in a Δrad23 background has only a minor affect in rescuing UV survival (Xie et al, 2004). These observations suggest that stabilization of Rad4 per se plays no significant role in UV survival. The relationship between steady-state levels of Rad23 and its binding partner is also observed in mammalian cells, where the lack of hHR23A/B results in lowered steady-state levels of XPC (Ng et al, 2003; Okuda et al, 2004). However, unlike yeast, in mammalian cells the half-life of XPC decreases (Okuda et al, 2004). At present it is unclear whether proteolysis plays a role in mammalian NER.
We identified Rad7, Rad16, Elc1, and Cul3 as part of an ECS-type E3 ubiquitin ligase that ubiquitinates Rad4 protein following exposure of cells to UV radiation. This complex ubiquitinates Rad4 protein in vitro. We do not observe a ladder of ubiquitination in the in vitro reaction, nor in vivo. However, Rad4 is degraded by the 26S proteasome in vivo, suggesting that polyubiquitination does occur. Specific point mutations within the conserved SOCS domain of Rad7 protein inhibit the UV-induced ubiquitination of Rad4 protein in vivo. Our results show that the rad7, psocs mutation increases the UV sensitivity of a Δrad23 strain, even though Rad4 protein is stabilized. In contrast, stabilization of Rad4 protein in the Δrad23 strain, by direct inhibition of 26S proteasome proteolytic activity, has no effect on NER (see Figure 4; Gillette et al, 2001). These observations show that the changes observed in the stability of Rad4 protein in response to UV do not influence NER and UV survival. Therefore, it is possible to differentiate between the effect of Rad4 protein ubiquitination and Rad4 protein degradation on NER and UV survival. Ubiquitination of Rad4 protein in response to UV specifically regulates NER via a pathway that requires de novo protein synthesis. This event directly influences NER and UV survival. Rad4 protein is subsequently degraded by the UPP, and this event does not directly influence NER or UV survival. We suggest that this mechanism restores cellular Rad4 protein levels to the un-ubiquitinated, pre-UV state.
Speculation
The two major NER pathways, transcription-coupled repair (TCR) and GGR are functionally conserved in yeast and human cells (Hoeijmakers, 1993). In yeast, Rad7 and Rad16 specifically function in the GGR pathway (Verhage et al, 1996). No structural homologues for these proteins have been found in mammalian cells. Recently, however, Groisman et al (2003) have isolated two distinct protein complexes that differ only in that one contains the DDB2 protein and the other CSA. The DDB1/2 complex functions in GGR and the DDB1/CSA complex in TCR. Both of these complexes contain a cullin (Cull4a) and a RING-finger protein (Roc1), and both are components of cullin-based E3 ubiquitin ligases (Pintard et al, 2004; Willems et al, 2004). It has been shown that this novel E3 ligase ubiquitinates XPC, the mammalian homologue of Rad4 protein (Sugasawa et al, 2005). The authors demonstrate that it is the ubiquitination of XPC protein that influences NER rather than degradation of the protein. XPC protein ubiquitination alters the DNA damage binding characteristics of the XPC/Rad23/centrin complex, and this may alter the way in which NER promotes lesion recognition. These observations suggest striking parallels between the regulation of NER in the yeast and human systems. It appears that functional conservation of the major NER pathways may have been maintained.
Materials and methods
Yeast strains and plasmids
The yeast strains used in this study are WCG4a (WT) and YH129/14 (pre1-1 pre4-1) (Gillette et al, 2001). Research Genetics parental strain BY4741, 2782 (Δelc1), 1982 (Δela1), 4633 (Δcul3), Sc507 (WT), Sc507R7 (Δrad7) and Sc507R23 (Δrad23) were described (Gillette et al, 2001). The deletion of RAD23 in the Research Genetics strains were constructed as described (Gillette et al, 2001). Creation of the Rad7 SOCS box mutation was achieved by site-directed mutagenesis of the WT RAD7 gene cloned in pRS314 as described (Reed et al, 1998). Two point mutations were made, resulting in the amino-acid substitutions, L168A and C172A, within the conserved SOCS box domain. Creation of the Rad16 RING-finger domain mutation was achieved by site-directed mutagenesis of the WT RAD16 gene cloned in pRS314 as described (Reed et al, 1998; data not shown). Two point mutations were made, resulting in the amino-acid substitutions, C552A and H554A, within the RING-finger domain. Strains expressing Ubl-deleted versions of Rad23 were created as described (Russell et al, 1999).
In vitro expression of Rad4 protein
The following primers were used to generate the Rad4 coding sequence.
FWD: 5′AAGAGTACTTAATACGACTCACTATAGGAACAG CCACCATGAATGAAGACCTGCCCAA3′/REV: 5′TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTAA TCAGTCTGATTCCTCTGACATCT3′. PCR was performed using genomic DNA as the template. The purification of PCR product is performed as described (TNT, Promega), with the following exceptions. A total of 10 tubes of PCR product are pooled and precipitated with the same volume of 99% alcohol (−20°C) overnight (spin down at 13 000 r.p.m. for 15 min). The pellet was dried for 30 min. The pellet was redissolved with ddH2O (pH 7–8) for at least 30 min with occasional gentle vortexing. Short-term storage was at −20°C.
Expression of Rad4 protein via coupled in vitro transcription/translation
The TNT® T7 PCR Quick Master Mix was thawed rapidly and placed on ice along with the other components. The reaction components were assembled in a 0.5 ml thin-wall tube according to the manufacturer's protocol (TNT, Promega). After addition of all the components, they were gently mixed by pipetting. The reaction was incubated at 30°C for 60–70 min. The results of the coupled in vitro transcription/translation reaction were analysed by Western blotting following SDS–PAGE gel electrophoresis.
Ubiquitylation reaction of Rad4 protein
The ubiquitylation reaction contains 1 μg E1 (Yeast), 1 μg hUbc4a, 1 μg hUbc4b, 13 μg hUbc4c, 0.1 μg GGR complex, 6 μl 5 × reaction buffer (20 mM MgCl2, 200 mM KCl 180 mM Tris, pH 8.0), 6 μl ATP (20 mM), 2 μl of TNT-expressed Rad4 protein, or an empty vector control. The ubiquitylation reaction is initiated by adding 5 μl ubiquitin and then ddH2O to a total volume of 30 μl. It is incubated at 28°C for 60 min. After the reaction is finished, the product is run on SDS–PAGE gels immediately.
Analysis of Rad4 protein stability and UV survival
Yeast protein extracts were prepared by bead beating in yeast dialysis buffer as described (Reed et al, 1999). Western analysis and immunoprecipitation of Rad4 protein was performed using standard methods, using the antibodies mentioned in the Results. Anti-Rad4 antibody was raised against a specific Rad4 peptide (Affiniti Research Products Limited). Anti-ubiquitin antibody was generated by genetic immunization as described (Barry and Johnston, 1997). Pulse-chase experiments were performed by growing cells to a density of about 1–2 × 107 cells/ml, pelleted, and washed twice with PBS. Cells were resuspended in appropriate media and a pulse of 25 μCi 35S-methionine per ml was applied at 30°C for 1 h. Cells were then pelleted, washed, and resuspended in YPD for the times indicated. Post-UV cellular survival was measured by plating yeast cells onto the appropriate growth medium prior to UV irradiation at the doses stated. The precise details of the UV survival experiments are described elsewhere (Gillette et al, 2001).
Analysis of CPD removal following UV radiation
The slot blot analysis of lesion removal by NER in genomic DNA has been described in detail previously (Reed et al, 1999). Where appropriate, cycloheximide was added to a final concentration of 2 μg/ml.
Northern analysis
The hot phenol method was used for RNA isolation and Northern blotting was described previously (Teng et al, 2002). The probes used are shown below: Upper_Rad4(NTS25): biotin-ACTGATCGAAGTTTTTGCACCAACGATGATA Lower_Rad4(TS25): CATAAAATCCGAATAATCCTCCCCG Actin_NTS: biotin-GCCGGTTTTGCCGGTGACG Actin_TS: CCGGCAGATTCCAAACCCAAAA
Supplementary Material
Supplementary data, S1
Supplementary data, S2
Supplementary data,S3
Supplementary data, S4
Acknowledgments
We thank Yue Xiong for the gift of the Cul3 antibody. This research was supported by MRC CDA and CEG awards to SHR. The work was conducted in the CBI at UT Southwestern during a sabbatical period supported by an MRC CDA to SHR, and at the School of Medicine, Cardiff University.
References
- Al-Moghrabi NM, Al-Sharif IS, Aboussekhra A (2003) UV-induced de novo protein synthesis enhances nucleotide excision repair efficiency in a transcription-dependent manner in S. cerevisiae. DNA Repair (Amst) 2: 1185–1197 [DOI] [PubMed] [Google Scholar]
- Barry MA, Johnston SA (1997) Biological features of genetic immunization. Vaccine 15: 788–791 [DOI] [PubMed] [Google Scholar]
- Bertolaet BL, Clarke DJ, Wolff M, Watson MH, Henze M, Divita G, Reed SI (2001a) UBA domains mediate protein–protein interactions between two DNA damage-inducible proteins. J Mol Biol 313: 955–963 [DOI] [PubMed] [Google Scholar]
- Bertolaet BL, Clarke DJ, Wolff M, Watson MH, Henze M, Divita G, Reed SI (2001b) UBA domains of DNA damage-inducible proteins interact with ubiquitin. Nat Struct Biol 8: 417–422 [DOI] [PubMed] [Google Scholar]
- Chambers RS, Johnston SA (2003) High-level generation of polyclonal antibodies by genetic immunization. Nat Biotechnol 21: 1088–1092 [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Ben-Saadon R (2004) N-terminal ubiquitination: more protein substrates join in. Trends Cell Biol 14: 103–106 [DOI] [PubMed] [Google Scholar]
- Dupre S, Urban-Grimal D, Haguenauer-Tsapis R (2004) Ubiquitin and endocytic internalization in yeast and animal cells. Biochim Biophys Acta 1695: 89–111 [DOI] [PubMed] [Google Scholar]
- Ferdous A, Gonzalez F, Sun L, Kodadek T, Johnston SA (2001) The 19S regulatory particle of the proteasome is required for efficient transcription elongation by RNA polymerase II. Mol Cell 7: 981–991 [DOI] [PubMed] [Google Scholar]
- Ferdous A, Kodadek T, Johnston SA (2002) A nonproteolytic function of the 19S regulatory subunit of the 26S proteasome is required for efficient activated transcription by human RNA polymerase II. Biochemistry 41: 12798–12805 [DOI] [PubMed] [Google Scholar]
- Friedberg EC (2001) How nucleotide excision repair protects against cancer. Nat Rev Cancer 1: 22–33 [DOI] [PubMed] [Google Scholar]
- Gillette TG, Gonzalez F, Delahodde A, Johnston SA, Kodadek T (2004) Physical and functional association of RNA polymerase II and the proteasome. Proc Natl Acad Sci USA 101: 5904–5909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette TG, Huang W, Russell SJ, Reed SH, Johnston SA, Friedberg EC (2001) The 19S complex of the proteasome regulates nucleotide excision repair in yeast. Genes Dev 15: 1528–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A (2002) The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82: 373–428 [DOI] [PubMed] [Google Scholar]
- Gonzalez F, Delahodde A, Kodadek T, Johnston SA (2002) Recruitment of a 19S proteasome subcomplex to an activated promoter. Science 296: 548–550 [DOI] [PubMed] [Google Scholar]
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y (2003) The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113: 357–367 [DOI] [PubMed] [Google Scholar]
- Hanawalt PC (2001) Controlling the efficiency of excision repair. Mutat Res 485: 3–13 [DOI] [PubMed] [Google Scholar]
- Heessen S, Masucci MG, Dantuma NP (2005) The UBA2 domain functions as an intrinsic stabilization signal that protects Rad23 from proteasomal degradation. Mol Cell 18: 225–235 [DOI] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C, Dewar D, Lin Z, Michalickova K, Willems AR, Sassi H, Nielsen PA, Rasmussen KJ, Andersen JR, Johansen LE, Hansen LH, Jespersen H, Podtelejnikov A, Nielsen E, Crawford J, Poulsen V, Sorensen BD, Matthiesen J, Hendrickson RC, Gleeson F, Pawson T, Moran MF, Durocher D, Mann M, Hogue CW, Figeys D, Tyers M (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180–183 [DOI] [PubMed] [Google Scholar]
- Hochstrasser M, Ellison MJ, Chau V, Varshavsky A (1991) The short-lived MAT alpha 2 transcriptional regulator is ubiquitinated in vivo. Proc Natl Acad Sci USA 88: 4606–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH (1993) Nucleotide excision repair. II: From yeast to mammals. Trends Genet 9: 211–217 [DOI] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL (1996) Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J Biol Chem 271: 27280–27284 [DOI] [PubMed] [Google Scholar]
- Lommel L, Ortolan T, Chen L, Madura K, Sweder KS (2002) Proteolysis of a nucleotide excision repair protein by the 26 S proteasome. Curr Genet 42: 9–20 [DOI] [PubMed] [Google Scholar]
- Mueller JP, Smerdon MJ (1996) Rad23 is required for transcription-coupled repair and efficient overall repair in Saccharomyces cerevisiae. Mol Cell Biol 16: 2361–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng JM, Vermeulen W, van der Horst GT, Bergink S, Sugasawa K, Vrieling H, Hoeijmakers JH (2003) A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of Xeroderma pigmentosum group C protein. Genes Dev 17: 1630–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng JM, Vrieling H, Sugasawa K, Ooms MP, Grootegoed JA, Vreeburg JT, Visser P, Beems RB, Gorgels TG, Hanaoka F, Hoeijmakers JH, van der Horst GT (2002) Developmental defects and male sterility in mice lacking the ubiquitin-like DNA repair gene mHR23B. Mol Cell Biol 22: 1233–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda Y, Nishi R, Ng JM, Vermeulen W, van der Horst GT, Mori T, Hoeijmakers JH, Hanaoka F, Sugasawa K (2004) Relative levels of the two mammalian Rad23 homologs determine composition and stability of the Xeroderma pigmentosum group C protein complex. DNA Repair (Amsterdam) 3: 1285–1295 [DOI] [PubMed] [Google Scholar]
- Ortolan TG, Chen L, Tongaonkar P, Madura K (2004) Rad23 stabilizes Rad4 from degradation by the Ub/proteasome pathway. Nucleic Acids Res 32: 6490–6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM (2004) Back to the future with ubiquitin. Cell 116: 181–190 [DOI] [PubMed] [Google Scholar]
- Pintard L, Willems A, Peter M (2004) Cullin-based ubiquitin ligases: Cul3–BTB complexes join the family. EMBO J 23: 1681–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KL, Smith JJ, Dasgupta A, Maqani N, Grant P, Auble DT (2004) The NEF4 complex regulates Rad4 levels and utilizes Snf2/Swi2-related ATPase activity for nucleotide excision repair. Mol Cell Biol 24: 6362–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SH, Akiyama M, Stillman B, Friedberg EC (1999) Yeast autonomously replicating sequence binding factor is involved in nucleotide excision repair. Genes Dev 13: 3052–3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SH, You Z, Friedberg EC (1998) The yeast RAD7 and RAD16 genes are required for postincision events during nucleotide excision repair. In vitro and in vivo studies with rad7 and rad16 mutants and purification of a Rad7/Rad16-containing protein complex. J Biol Chem 273: 29481–29488 [DOI] [PubMed] [Google Scholar]
- Russell SJ, Johnston SA (2001) Evidence that proteolysis of Gal4 cannot explain the transcriptional effects of proteasome ATPase mutations. J Biol Chem 276: 9825–9831 [DOI] [PubMed] [Google Scholar]
- Russell SJ, Reed SH, Huang W, Friedberg EC, Johnston SA (1999) The 19S regulatory complex of the proteasome functions independently of proteolysis in nucleotide excision repair. Mol Cell 3: 687–695 [DOI] [PubMed] [Google Scholar]
- Salghetti SE, Caudy AA, Chenoweth JG, Tansey WP (2001) Regulation of transcriptional activation domain function by ubiquitin. Science 293: 1651–1653 [DOI] [PubMed] [Google Scholar]
- Schauber C, Chen L, Tongaonkar P, Vega I, Lambertson D, Potts W, Madura K (1998) Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 391: 715–718 [DOI] [PubMed] [Google Scholar]
- Strous GJ, Govers R (1999) The ubiquitin–proteasome system and endocytosis. J Cell Sci 112 (Part 10): 1417–1423 [DOI] [PubMed] [Google Scholar]
- Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Hanaoka F (2005) UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 121: 387–400 [DOI] [PubMed] [Google Scholar]
- Sweder K, Madura K (2002) Regulation of repair by the 26S proteasome. J Biomed Biotechnol 2: 94–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Yu Y, Waters R (2002) The Saccharomyces cerevisiae histone acetyltransferase Gcn5 has a role in the photoreactivation and nucleotide excision repair of UV-induced cyclobutane pyrimidine dimers in the MFA2 gene. J Mol Biol 316: 489–499 [DOI] [PubMed] [Google Scholar]
- Verhage RA, van Gool AJ, de Groot N, Hoeijmakers JH, van de Putte P, Brouwer J (1996) Double mutants of Saccharomyces cerevisiae with alterations in global genome and transcription-coupled repair. Mol Cell Biol 16: 496–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wei S, Reed SH, Wu X, Svejstrup JQ, Feaver WJ, Kornberg RD, Friedberg EC (1997) The RAD7, RAD16, and RAD23 genes of Saccharomyces cerevisiae: requirement for transcription-independent nucleotide excision repair in vitro and interactions between the gene products. Mol Cell Biol 17: 635–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters R, Zhang R, Jones NJ (1993) Inducible removal of UV-induced pyrimidine dimers from transcriptionally active and inactive genes of Saccharomyces cerevisiae. Mol Gen Genet 239: 28–32 [DOI] [PubMed] [Google Scholar]
- Watkins JF, Sung P, Prakash L, Prakash S (1993) The Saccharomyces cerevisiae DNA repair gene RAD23 encodes a nuclear protein containing a ubiquitin-like domain required for biological function. Mol Cell Biol 13: 7757–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems AR, Schwab M, Tyers M (2004) A hitchhiker's guide to the cullin ubiquitin ligases: SCF and its kin. Biochim Biophys Acta 1695: 133–170 [DOI] [PubMed] [Google Scholar]
- Xie Z, Liu S, Zhang Y, Wang Z (2004) Roles of Rad23 protein in yeast nucleotide excision repair. Nucleic Acids Res 32: 5981–5990 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data, S1
Supplementary data, S2
Supplementary data,S3
Supplementary data, S4