

Abstract
We have previously used the asialoglycoprotein receptor system to elucidate the pathway of hepatocytic processing of ligands such as asialoorosomucoid (ASOR). These studies suggested that endocytic vesicles bind to and travel along microtubules under the control of molecular motors such as cytoplasmic dynein. We now report reconstitution of this process in vitro with the use of a microscope assay to observe the interaction of early endocytic vesicles containing fluorescent ASOR with fluorescent microtubules. We find that ASOR-containing endosomes bind to microtubules and translocate along them in the presence of ATP. This represents the first time that mammalian endosomes containing a well-characterized ligand have been directly observed to translocate on microtubules in vitro. The endosome movement does not require cytosol or exogenous motor protein, is oscillatory, and is directed toward the plus and minus ends at equal frequencies. We also observe endosomes being stretched in opposite directions along microtubules, suggesting that microtubules could provide a mechanical basis for endocytic sorting events. The movement of endosomes in vitro is consistent with the hypothesis that microtubules actively participate in the sorting and distribution of endocytic contents.
INTRODUCTION
Receptor-mediated endocytosis represents a major pathway whereby nutrients, hormones, enzymes, and viruses enter cells. Such ligands bind to receptors at the cell surface and are internalized and eventually sorted to specific destinations (Evans, 1985; Bu and Schwartz, 1994). This sorting of endocytic material is achieved along a pathway of semistable tubulo-vesicular membranous structures that display characteristic intracellular localization and appearance. Previous studies suggest a relationship between these structures and microtubules, which play a critical but not well-defined role in endocytosis and endocytic processing. Transport from early to late endosomes is microtubule dependent, and microtubules have been shown to promote fusion of endocytic vesicles, whereas drugs affecting microtubule polymerization have been shown to alter endocytosis (Oka and Weigel, 1983; Wolkoff et al., 1984; Bomsel et al., 1990; Aniento et al., 1993; Harada et al., 1995). Phagosomes from a macrophage cell line as well as endosomes from the Dictyostelium slime mold have been shown to translocate on microtubules in vitro (Blocker et al., 1997; Pollock et al., 1998). However, the microtubule-based motility of endosomes is not well characterized. We now provide the first report of an in vitro system in which movement of specific endocytic vesicles has been reconstituted. This system, which uses the asialoglycoprotein receptor and its endocytic ligand asialoorosomucoid (ASOR), allows the observation of both endocytic processing and microtubule movement. It provides a means to test the role of microtubules and specific cofactors in these processes.
Using this system, we demonstrate that ASOR-containing endosomes move on microtubules in vitro, and we show that the velocity of this movement is oscillatory. In addition, we demonstrate the direction of this motility with respect to microtubule polarity and discuss its implications toward the process of endosome sorting. We also provide evidence that microtubules provide a supporting matrix along which endosomes may undergo fission.
MATERIALS AND METHODS
Chemicals
Protein concentration was measured with the use of bicinchoninic acid (Pierce, Rockford, IL) and Coomassie Plus protein assays (Pierce). Immunoblots were performed as described by Oda et al. (1995). Anti-dynein intermediate chain antibody was from Sigma (St. Louis, MO). Anti-kinesin polyclonal antibody was the generous gift of Dr. Mark McNiven (Mayo Clinic, Rochester, MN) (Marks et al., 1995). All reagents were from Sigma unless noted otherwise. Male Sprague-Dawley rats (200–250 g; Taconic Farms, Germantown, NY) were used for isolation of endosomes and liver motor proteins and for primary culture of hepatocytes. All procedures were performed after being approved by the university animal use committee. Human orosomucoid (Sigma) was desialylated to ASOR by acid hydrolysis (Stockert et al., 1980). ASOR was labeled with Texas red sulfonyl chloride, 5- (and 6)-carboxyfluorescein succinimidyl ester (Molecular Probes, Eugene, OR), or Cy5 (Amersham, Arlington Heights, IL) according to the manufacturers' protocols. The resulting fluorescent ASOR generally had 1–2 mol of dye per mol of protein, as measured spectrophotometrically.
Endosome Isolation
After administration of appropriate anesthesia, rats were injected intravenously with 200–300 μl of 10 mg/ml fluorescent ASOR in PBS. Laparotomy was performed at 4.5 min, and at 5 min, the hepatic vein was transected and the liver was blanched by perfusion of ice-cold PBS though the portal vein. The liver was removed, washed in MEPS buffer [35 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (Pipes), 5 mM EGTA, 5 mM MgSO4, 0.2 M sucrose, pH 7.1], and diced with a razor. Three to four milliliters of MEPS plus protease inhibitors (2 mM PMSF, 20 μg/ml N-α-benzoyl-l-arginine methyl ester, 20 μg/ml soybean trypsin inhibitor, 20 μg/ml p-tosyl-l-arginine methyl ester, and 2 mg/ml leupeptin) and 4 mM DTT (lysis buffer) were added, and the liver was homogenized by 25 strokes in a tight Dounce. Lysate was centrifuged twice at 12,500 rpm (tabletop microfuge model Z230m, Hermle, Gosheim, Germany) at 4°C for 1 min. Protease inhibitors and DTT were readded to the resulting postnuclear supernatant (PNS), and this was applied to a 12- × 1.25-cm Sephacryl S200 column (Pharmacia, Uppsala, Sweden) equilibrated in MEPS buffer. Fractions were collected and assayed for vesicles and protein concentration. The initial, highly absorbing peak was pooled, protease inhibitors and DTT were readded, and the pool was adjusted to 1.4 M sucrose with 2.5 M sucrose in MEP buffer. This was loaded on the bottom of a sucrose step gradient consisting of 1.4, 1.2, and 0.25 M sucrose and centrifuged in a SW41 rotor (Beckman, Fullerton, CA) for 135 min at 39,000 rpm with the slowest acceleration and deceleration parameters. After centrifugation, the 1.2/0.25 M cloudy interface was collected, divided into aliquots, and stored in liquid nitrogen. The specifics of this gradient were suggested by Drs. Carol Harley and Duncan Wilson (Albert Einstein College of Medicine), who have observed that the endocytic marker rab5 floats to the 1.2/0.25 M interface in similar gradients (personal communication). Endosomes were counted with the aid of NIH Image particle-counting software (developed by Wayne Rasband at the U.S. National Institutes of Health [http://rsb.info.nih.gov/nih-image/]). Threshold and minimum and maximum particle size were chosen to give linear values when diluted. Each data point represents an average of six microscope fields.
Motor Protein Isolation
A PNS was prepared as described above except that the lysis buffer was PMEE (35 mM Pipes, 5 mM MgSO4, 1 mM EGTA, 0.5 mM EDTA, pH 7.4) plus 0.25 M sucrose, 4 mM DTT, and protease inhibitors. This was centrifuged twice for 5 min each at 60,000 rpm in a TLA 100 rotor (Beckman) at 4°C. Twenty micromolar Taxol, 1 mM GTP, and 1 mg/ml polymerized bovine brain tubulin (Cytoskeleton, Denver, CO) were added to this supernatant, which was then incubated at 37°C for 30 min. The mixture was then centrifuged through 40% glycerol (in PMEE plus 20 μM Taxol and 4 mM DTT) for 5 min at 60,000 rpm in the TLA 100 rotor at 25°C to pellet microtubules and associated proteins. The pellet was resuspended in PMEE containing 20 μM Taxol, 4 mM DTT, and 4 mM ATP in the presence of an ATP-regenerating system (0.16 mg/ml creatine phosphokinase, 8 mM phosphocreatine) and incubated at 37°C for 5 min. One millimolar ATP was added, and microtubules were pelleted in a Beckman airfuge at 15 psi for 5 min. The supernatant was collected and applied to a 6- × 0.7-cm Sephadex G25 column (Pharmacia) equilibrated in PMEE. The peak eluting in the void volume was pooled, divided into aliquots, and frozen in liquid nitrogen.
Motility Assay
Motility assays were performed in a chamber consisting of two pieces of double-stick tape (12 mm × 6.35 mm; Scotch 3M, St. Paul, MN) sandwiched between optical glass, creating an internal volume of 3–5 μl (Figure 1). Assays were performed at 35°C with the use of degassed “motility buffer” (PMEE, 20 μM Taxol, 2 mg/ml BSA, 4 mM DTT, containing an oxygen-scavenging system consisting of 0.3 mg/ml catalase, 28 U/ml glucose oxidase, 10 mM glucose [Bouin et al., 1976; Kishino and Yanagida, 1988]). For motility assays, the following ingredients were added sequentially: 5 μl of motor protein (3-min incubation, three 15-μl motility buffer washes), 5 μl of microtubules (3-min incubation, three 15-μl washes), and 5 μl of endosomes (5-min incubation, wash). Motility was initiated with the addition of 4 mM ATP and the ATP-regenerating system. In some experiments, endosomes were added to microtubules in the presence of ATP. In other experiments, 10 mg/ml DEAE-dextran (Pharmacia) rather than motor proteins was used to adhere microtubules to glass.
Figure 1.

Motility chamber and experimental design. The movement of fluorescent endocytic vesicles and microtubules was monitored with an inverted microscope with the use of a motility chamber (A) constructed with double-stick tape sandwiched between a large coverslip and glass cut from a microscope slide. In the gliding assay, motor protein is perfused into the chamber, followed by microtubules, which bind to the motor proteins. Microtubule movement was observed after ATP addition. In the vesicle motility assay (B), motor protein is perfused into the chamber, followed by microtubules, followed by vesicles. The vesicles bind microtubules and are assayed for motility upon addition of ATP.
Imaging Microtubule-based Motility
Imaging was performed at the Analytical Imaging Facility (Albert Einstein College of Medicine) with the use of a 60×, numerical aperture 1.4 planapo objective combined with a Nikon (Garden City, NY) Diaphot inverted microscope connected to a silicon-intensified tube camera (Hamamatsu, Bridgewater, NJ) to record directly to videotape. Alternatively, an Olympus (Melville, NY) 1X70 inverted microscope containing automatic excitation and emission filter wheels (Ludl Electronics, Hawthorne, NY) connected to a Photometrics (Tucson, AZ) cooled charge-coupled device camera run by I.P. Lab Spectrum software (Scanalitics, Fairfax, VA) running on a Power Macintosh computer (Apple Computer, Cupertino, CA) was used. I.P. Lab Spectrum scripting software was used to collect images rapidly and to switch between fluorescent channels. Additionally, the computer monitor recorded on videotape. Microscope stages were maintained at 35°C with the use of a thermal stage or hot-air apparatus.
Determination of Vesicle and Microtubule Velocities
Videos of moving vesicles and microtubules were digitized with the use of the NIH Image movie-making macro (1 frame/s), saved as QuickTime movies (Apple Computer, Cupertino, CA), and either traced with the use of DIAS 2.0 software (Soll Tech, Iowa City, IA) or noted by mouse pixel position. The velocity of a vesicle was measured only when the end of the underlying microtubule was stationary. The end of a gliding microtubule and the center of a vesicle were used to mark their positions with time. For velocity plots, the raw data were smoothed twice with the use of the default Tukey window. The percentage of moving vesicles (Table 1) was determined by making a printout of a microscope field at the time of the addition of ATP, circling vesicles that were attached to microtubules, and then looking at the video to determine whether each vesicle moved or remained stationary.
Table 1.
Influence of microtubule attachment method on parameters of endocytic vesicle motility
| Microtubule attachment method | Motile vesicles (% of total) | Minus-end-directed movement (% of total movement) | Velocity (μm/s) |
|---|---|---|---|
| DEAE-dextran | 33 (174) | 49 (39) | 0.71 ± 0.44 (10) |
| DEAE-dextran plus vesicle preincubation with exogenous motor proteins | 39 (122) | 45 (31) | 0.71 ± 0.36 (10) |
| Motor proteins | 27 (199) | 47 (43) | 0.55 ± 0.23 (21) |
The number of vesicles counted is in parentheses.
Fluorescent Microtubules
Rhodamine- and fluorescein-labeled tubulins were purchased from Cytoskeleton. A total of 3 mg/ml fluorescent tubulin was polymerized in BRB80 buffer (80 mM Pipes, 1 mM EGTA, 1 mM MgCl2, 1 mM GTP, pH 6.8 [Howard and Hyman, 1993]) plus 10% glycerol at 37°C for 30 min and then stabilized with 20 μM Taxol. Microtubules were pelleted to remove nonpolymerized tubulin by centrifugation through BRB80 plus 60% glycerol, 20 μM Taxol at 15 psi in a Beckman airfuge. Polarity-marked microtubules were prepared essentially as described by Matteoni and Kreis (1987). Rhodamine- or fluorescein-labeled tubulin was polymerized into “seeds” in BRB80 plus 60% glycerol at 37°C for 15 min. Ten micromolar Taxol was added, and this was centrifuged through 60% glycerol (in BRB80, no Taxol) in an airfuge, as described above. The seeds were resuspended in BRB80 plus 60% glycerol and sheered by pipetting up and down five times with a gel-loading pipette. Tubulin was polymerized off the seeds (“growth tubulin”) at concentrations of ∼2.5 mg/ml growth tubulin to 1 mg/ml seeds for 30 min. BRB80, 20 μM Taxol was then added to stabilize the microtubules.
ATPase Assays
The appearance of free phosphate and the corresponding decrease in ATP signature was quantified with the use of nuclear magnetic resonance (NMR) spectroscopy with the assistance of Dr. Raj Gupta (Albert Einstein College of Medicine), as described by Dowd and Gupta (1995). PNS made as described above (68 mg/ml protein) was diluted 61-fold into PMEE at 37°C, 4 mM ATP was added, and NMR scans were captured at 5-min intervals. To measure the ATPase activity of fractions from the endosome purification, malachite green assays were used in the manner of Chan et al. (1986) and Henkel et al. (1988). The fractions were diluted 150- to 500-fold, and 4 mM ATP was used as the substrate.
RESULTS
Motility Assays
A motility chamber similar to that described previously (Hyman and Mitchison, 1993) was used to monitor microtubule–endosome interactions in vitro. Figure 1 depicts the motility chamber and the experimental design. Motor protein that flows into the chamber by capillary action adheres to glass in a biologically active state (Vale et al., 1992). After incubation, excess motor protein is washed from the chamber and fluorescent microtubules are introduced. The microtubules bind to the glass-attached motor protein, excess microtubules are washed out, ATP is then added, and the movement of microtubules is observed (the gliding assay). Motor protein from rat liver was isolated and found to be active in microtubule gliding assays.
ASOR, which binds to the asialoglycoprotein receptor on hepatocytes, was used to label endosomes. The uptake of fluorescent ASOR was confirmed by observing primary cultured rat hepatocytes under a fluorescent microscope (not shown). To determine if hepatocyte endosomes would translocate on microtubules in vitro, PNSs containing fluorescent ASOR–labeled endosomes were perfused into motility assay chambers containing microtubules bound to the glass through DEAE-dextran. Although endosomes bound to the microtubules, no motility was observed upon addition of ATP.
Endosome motility was further investigated by adding endosome-containing PNS into microtubule gliding assays. Unexpectedly, the presence of the PNS caused microtubule gliding to stop. Neither endosomes nor microtubules moved under these conditions. The gliding-arrest activity of PNS was concentration dependent, and gliding activity required several minutes to recover after washout of PNS.
ATPase Activity in PNS
A potential cause of gliding arrest in the experiments described above is ATPase activity that may be present in the PNS. To determine the level of ATPase activity in PNS, NMR spectroscopy was used. The phosphate peaks of ATP and ADP were quantified. Figure 2A shows an NMR scan of a PNS preparation that was diluted 61-fold before incubation with 4 mM ATP (assay concentration) for 60 min. After this incubation, peaks corresponding to free phosphate, AMP, and ADP were substantial. At zero time, only ATP phosphate peaks were visible (not shown). A plot of free phosphate versus time (Figure 2B) revealed an initial PNS ATP hydrolysis rate of 0.03 mM·min−1·(mg/ml total protein)−1. Because the motility assays used undiluted, highly concentrated PNS (8–70 mg/ml), 0.24–2.1 mM ATP would be hydrolyzed during the first minute of the motility assay. This ATPase activity would rapidly deplete the 4 mM ATP present in the reaction as well as generate ADP, which is known to inhibit motor protein activity (Moss et al., 1992; Johnson and Gilbert, 1995). An ATP-regenerating system would be overwhelmed under these conditions.
Figure 2.

NMR spectroscopy demonstrates that the endosome-containing PNS hydrolyzes ATP to ADP and Pi. (A) ATP was incubated for 10 min with PNS, and 31P-NMR spectra were determined as described by Dowd and Gupta (1995). The data are presented as intensity versus parts per million. Peaks for α-, β-, and γ-phosphates on ATP were observed and were the only peaks seen before the addition of PNS (data not shown). Ten minutes after addition of PNS, peaks for α- and β-phosphates on ADP were observed along with free phosphate (Pi), indicating the presence of ATPase activity. (B) The rate of ATP hydrolysis by PNS was determined by following the formation of Pi over time. The peak for free Pi was recorded at regular intervals and converted to millimolar free phosphate by integrating the normalized, calibrated peak. A plot of Pi versus time was linear, with a slope of 0.03 mM·min−1·(mg/ml total protein)−1. Therefore, undiluted PNS as used in the motility assays would hydrolyze as much as 2 mM phosphate per minute, a rate that would quickly deplete the 4 mM ATP used in the assay, despite the presence of an ATP-regenerating system.
Purification of Endosomes with Sucrose Flotation
We reasoned that because the PNS inhibited microtubule gliding, it would also inhibit vesicle motility. This necessitated purification of the fluorescent ASOR-containing endosomes by means of the scheme shown in Figures 3 and 4. The PNS was applied to an S200 gel filtration column (Figure 3), and the void volume was pooled and loaded onto the bottom of a sucrose gradient (Figure 4) in which low-density endosomes floated to the 0.25/1.2 M interface, where the major portion of endocytic vesicles (71% of the starting material) was recovered. This gradient was designed so that soluble proteins and high-density organelles such as lysosomes and mitochondria remained at the bottom.
Figure 3.

Gel filtration of PNS containing fluorescent endosomes. As a first step in the purification of fluorescent endosomes, the PNS was applied to an S200 gel filtration column. The OD280 is shown for the eluting fractions (0.8 ml each). The excluded volume eluted as a highly absorbing, light-scattering peak that contained the fluorescent endosomes. Fractions 7–11, indicated by the solid line, were pooled and loaded onto the bottom of a sucrose step gradient.
Figure 4.

Sucrose step gradient purification of fluorescent endosomes. The pool from the S200 gel filtration column was made 1.4 M (1.18 g/ml) sucrose and loaded onto the bottom of a sucrose step gradient containing three layers as shown. The gradient was centrifuged to equilibrium, and the fluorescent endosomes (vesicles) (A) floated to the 0.25/1.2 M interface while the bulk of the protein (B) remained at the bottom in fractions 9–16. (C) The specific activity highlights the enrichment of endosomes at this interface. The cloudy layer at fractions 2–4 constituted the final endosome pool and contained the sufficiently high number of endosomes that was required to observe their interaction with microtubules under a microscope.
Figure 4 shows that the gel filtration and sucrose gradient procedure led to a significant purification of endosomes in terms of vesicles per (milligram per milliliter protein) (Figure 4A). The sucrose gradient concentrated the endosomes at the 0.25/1.2 M interface, which facilitated their observation by microscopy. The purified endosomes also contained 40-fold less ATPase activity compared with the PNS (Figure 5). Immunoblot analysis revealed that dynein and kinesin were both present in the endosome fraction at approximately the same concentrations as in the PNS when normalized for total protein (data not shown).
Figure 5.

ATPase assays of fractions from the endosome purification demonstrate that the final pool has low ATPase activity. The PNS, sucrose gradient load, and sucrose gradient fractions were assayed for ATPase activity, and the results are shown. The final endosome fraction had 40-fold lower ATPase activity but contained an approximately equal number of vesicles as the PNS (see Figure 4A). Most of the ATPase activity remained with the bulk of the total protein, where the gradient was loaded (fractions 9–16) (see Figure 4B).
Motility of Liver Endosomes
Endosomes isolated by this protocol exhibited microtubule-based motility. In the experiment illustrated in Figure 6 and in the movie (Figure 6.mov), microtubules were adhered to the glass via motor protein. Subsequently, endosomes were added in the presence of 4 mM ATP and an ATP-regenerating system. Figure 6 shows five frames taken from a video and displayed at ∼10-s intervals, as indicated in the upper left of each frame. Two vesicles (ves 1 and ves 2) are seen to move over distances of 5 μm or more. This experiment demonstrates conclusively that isolated liver endosomes containing the ligand ASOR are capable of microtubule-based movement in vitro. No vesicle motility was observed in the absence of ATP.
Figure 6.
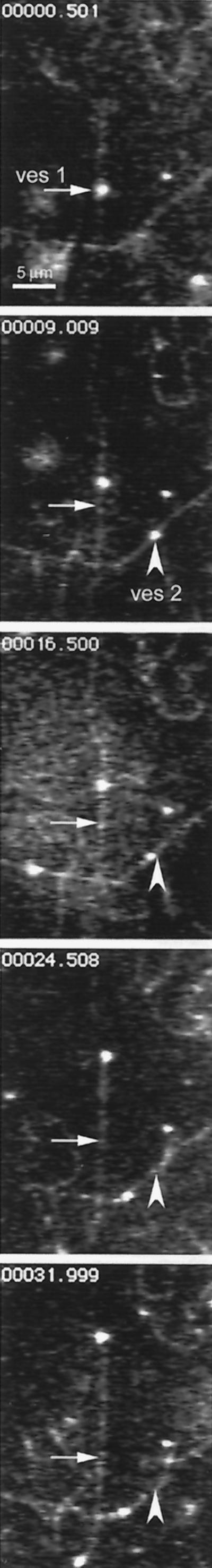
Purified endocytic vesicles exhibit microtubule-based, ATP-dependent motility (see movie Figure 6.mov). Five video frames are shown at ∼10-s intervals, as indicated in the upper left of each panel. Two vesicles (ves 1 and ves 2) can be seen translocating upon microtubules for distances of 5 μm or more. Arrows and arrowheads mark the initial positions of the vesicles.
In these experiments, motor protein was used to adhere microtubules to glass, and microtubule movement served as a monitor of motor protein activity. Endosome movement was clearly distinguishable from microtubule gliding. Many endosomes also bound to the glass despite the inclusion of motor protein and BSA as blocking agents. Other potential blocking agents, such as DEAE-dextran, aspartate, polysiloxane (Sigmacote), and γ-globulin, proved even less effective than the combination of BSA and motor protein.
The velocities of both vesicle motility and microtubule gliding were determined. The microtubule gliding rate of 1.42 ± 0.42 μm/s (n = 17) was fast compared with published reports (e.g., 0.44 μm/s [Vale et al., 1985] and 1.09 μm/s [Nakajima et al., 1995]), whereas the vesicle motility of 0.55 ± 0.23 μm/s (n = 21) was somewhat slower (e.g., 0.87 μm/s [Gilbert and Sloboda, 1989], 1.4 μm/s [Clague, 1998], and 1.2–0.9 μm/s [Blocker et al., 1997]). The significance of these differences is unclear, but the differences may be related in part to differences in the methods used to calculate velocity (see below).
Polarity of Microtubule and Endosome Motilities
To determine the polarity of both microtubule gliding and endosome translocation, we used a protocol similar to that used by Howard and Hyman (1993) to label the pointed ends of microtubules. Dim fluorescent tubulin was polymerized from bright fluorescent (rhodamine or fluorescein) microtubule seeds, resulting in polarity-marked microtubules. Figure 7A shows several frames taken from a video (Figure 7A.mov) that demonstrates the translocation of polarity-marked microtubules driven by motor proteins isolated from liver. The numbers 1–4 are provided to aid in distinguishing individual microtubules. Most of the microtubules moved with a minus-end motor direction. It should be noted that we always refer to the direction in which the putative motor is moving. For example, microtubules gliding with the minus end at the front are scored as plus-end directed, because the motor, attached to the glass, is pulling toward the plus end of the microtubule. Microtubule 1 moves toward the lower right with its bright end trailing, indicating minus-end-directed motor activity. The same direction of movement is seen for microtubules 3 and 4. Microtubule 2 moves with its seed at the leading end, indicating plus-end motor activity.
Figure 7.


Gliding of polarity-marked microtubules demonstrates their direction of movement (see movie Figure 7A.mov). (A) Polarity-marked microtubules are seen gliding across the coverslip powered by motor protein from rat liver. Numbers and arrows are presented to aid in following individual microtubules. Time in seconds is indicated in the upper left of each panel. Direction is noted with reference to the direction that the motor is moving, as described in RESULTS. Microtubules 1, 3, and 4 are powered in the minus-end (dynein-like) direction; microtubule 2 is powered in the plus-end (kinesin-like) direction. (B) Overall, the gliding was 77% minus-end directed and 23% plus-end directed (n = 65).
As seen in Figure 7B, microtubule gliding powered by liver motor protein showed 23% plus-end-directed (kinesin-like) and 77% minus-end-directed (dynein-like) movement (n = 65). We had no reason to expect either plus- or minus-end-directed movement in gliding assays, although our previous studies suggested that a larger percentage of liver dynein bound to and released from microtubules compared with kinesin (Oda et al., 1995). A single microtubule, as a rule, moved in only one direction while it traversed large regions of the coverslip. Occasionally, examples were seen of microtubules that moved in one direction, paused, and then moved in the other direction.
An example of a vesicle moving on a polarity-marked microtubule is shown in Figure 8A (see Figure 8A.mov). To avoid confusing the bright fluorescence of vesicles with microtubule polarity marks, we used fluorescein polarity marks and Texas red–labeled endocytic vesicles, rapidly switching between fluorescence filters during the experiment. Time (in seconds) is indicated in the upper left corner of each frame. At 0.5 and 5.5 s, a vesicle that is bound to a microtubule and moving toward one of its ends is seen. At 6 s, a switch to the fluorescein channel reveals a polarity mark near the end of the microtubule in the direction the vesicle is moving, thus indicating minus-end-directed movement of the vesicle. Texas red fluorescence associated with the translocating vesicle and a nearby glass-attached vesicle can also be seen bleeding through into the fluorescein channel. In addition, a nearby microtubule that was not scored in this assay can be seen with at least two seed marks, indicated by the asterisks.
Figure 8.

The movement of an endocytic vesicle on a polarity-marked microtubule (see movie Figure 8.mov). (A) Eleven frames taken from a video are shown. Time is indicated in the upper left of each panel. The rhodamine channel, showing endocytic vesicles and microtubules, is in red. The fluorescein channel, showing seed marks for polarity determination, is in green. An endocytic vesicle attached to a microtubule is indicated at 0.5 s. At 5.5 s, the vesicle has moved to the right, and at 6.0 s, a switch to the fluorescein channel reveals a seed mark near the end of the microtubule, indicating that the vesicle is traveling toward the minus end. Bright fluorescence from the vesicle can be seen bleeding through into the fluorescein channel. In subsequent frames, the vesicle can be seen moving to the end of the microtubule. A nearby microtubule containing two seed marks (*) can also be seen. This microtubule was not scored in the assay. (B) Overall vesicle movement was 47% minus-end directed and 53% plus-end directed (n = 43).
Forty-seven percent of the vesicles were found to move toward the minus end and 53% moved toward the plus end (n = 43) (Figure 8B). This essentially unbiased direction of movement implies that such endosomes would not localize to the cell center over time in the absence of additional regulation of their motility. Instead, the unbiased direction of movement would result in dispersal of endosomes throughout the cytosol. To confirm that we were scoring motility correctly, microtubule gliding was scored in the presence of motor protein inhibitors. One millimolar 5′adenylylimido-diphosphate (AMP-PNP) is known to preferentially inhibit kinesins, whereas low concentrations of vanadate inhibit dyneins (Brady, 1991; Harrison and Huebner, 1997). As can be seen in Figure 9, AMP-PNP reduced the plus-end motility (−80% change) but had a much smaller effect on the minus-end motility (−20%). On the other hand, vanadate (5 μM) caused the reverse effect, in which minus-end movement was reduced by 83% and plus-end-directed movement was reduced by 26%.
Figure 9.

Studies in the presence of inhibitors confirm that the plus- and minus-end directions have been scored correctly. The microtubule gliding assay was analyzed in the presence of known inhibitors of kinesin and dynein. One millimolar AMP-PNP was used to preferentially inhibit kinesin; 5 μM vanadate was used to preferentially inhibit cytoplasm dynein. The direction of movement (or lack of movement) was scored, and the percentage of total microtubules was plotted as a pie graph as indicated. The difference, before and after the inhibitor, of each pie slice is presented at the bottom as percentage change [(a2 − a1)/a1 × 100%]. Number was 54, 24, 35, and 51 for the experiments + ATP, 1 mM AMP-PNP, + ATP, and 5 μM vanadate, respectively.
To ensure that motor protein that was used to attach the microtubules to glass did not affect vesicle motility, we measured motility with the use of DEAE-attached microtubules. Furthermore, we determined whether preincubation of vesicles with motor protein, which caused predominantly minus-end-directed motility in gliding assays (Figure 7B), would alter the directionality or velocity of vesicle movement.
As seen in Table 1, the percentage of moving vesicles was similar when DEAE instead of motor proteins was used to attach the underlying microtubules to the glass coverslips (33% versus 27%). Preincubation of vesicles with motor proteins caused only a minor increase in motility (33% versus 39%). The directionality was unchanged by the presence or absence of motor protein and remained between 45% and 49% minus-end directed. Velocity was slightly increased (from 0.55 to 0.71 μm/s) when DEAE-dextran was used. However, we observed that endosome velocity was not constant but rather was oscillatory in nature, as described below.
Oscillation of Motility
The motility of microtubules as well as vesicles had distinct oscillations. Figure 10 shows representative plots of instantaneous velocity from two gliding microtubules and two moving vesicles. It is clear from these plots and from reviewing movies that the movement was not continuous but occurred in spurts. Vesicles, in contrast to microtubules, tended to stop altogether or release from microtubules after only one or two oscillations. Vesicle motility had lower amplitudes and shorter peak-to-peak intervals compared with gliding microtubules. Also, plus-end-directed gliding showed increased pausing times and slightly decreased amplitudes than minus-end gliding. These observations indicate that the velocity parameter does not adequately describe microtubule-based motility and that duration of movement (tendency to oscillate), maximal velocity, and tendency to release from microtubules also should be considered.
Figure 10.

Microtubule gliding and endocytic vesicle translocation are oscillatory events. The instantaneous velocities from two representative microtubules and two vesicles are shown. Microtubule gliding was powered by motor protein isolated from rat liver, and endocytic translocation was powered by endosomes isolated from rat liver (without exogenous motor protein). The gliding of minus-end-directed microtubules (A) showed larger velocity amplitudes with shorter pauses compared with plus-end gliding (B). Translocating vesicles (C and D) showed smaller velocity amplitudes and shorter peak-to-peak intervals compared with gliding microtubules (A and B).
Bidirectional Behavior of Endosomes
Occasionally, we observed endocytic vesicles that switched direction of movement. An example of this behavior is shown in Figure 11A (see Figure 11A.mov). Here, a vesicle (arrow) moving toward the left side of the frame is seen. The microtubule contains a seed mark near its center (X) prohibiting scoring of vesicle direction. At 5.8 s, the vesicle stops moving and appears stretched (*) as a result of forces acting in opposite directions. The vesicle then proceeds in the other direction, translocating nearly 5 μm. We found that this type of behavior often led to detachment or fission of vesicles. Figure 11B (see Figure 11B.mov) shows an example of vesicle fission. At 0 s, a single vesicle is visible with indications of intravesicle separation of the fluorescence. The vesicle moves slightly to the lower right by 5.9 s and then divides into separate vesicles by 11.9 s.
Figure 11.

Endosome stretching and fission along microtubules (see movies Figure 11A.mov and Figure 11B.mov). (A) An endosome is stretched (*) as it is pulled in opposite directions, demonstrating that opposing motors are active on individual endosomes. Note that the deformed shape is maintained for some seconds after the stretching event. The X indicates a seed mark (not used in this assay). (B) An endosome undergoes a fission event along a microtubule. Arrows indicate the initial positions of the vesicles. Time (in seconds) is listed at the upper left of each frame.
DISCUSSION
We have presented the characterization of an in vitro system to observe directly the microtubule-based transport of individual endocytic vesicles that had been isolated from rat hepatocytes. This represents an important step in elucidating the role of microtubules in endocytic processing. Previous reports on in vitro microtubule-based vesicular movement include studies on phagocytosed fish-gelatin–coated latex beads in macrophages and pinocytosed dextran in cellular slime molds (Blocker et al., 1997; Pollock et al., 1998). The system presented here is the first to study the in vitro movement of endosomes that contain a receptor–ligand pair whose endocytic uptake and processing have been well characterized in differentiated mammalian cells (Stockert, 1995). We have used this system to describe the movement of endosomes on microtubules, to present a means by which this movement may be inhibited, to indicate the direction of this movement with respect to microtubule polarity, and to provide evidence that endosomes may be pulled apart along microtubules.
Endosomes Move on Microtubules In Vitro
Although evidence for the movement of endocytic vesicles on microtubules has been growing for some time (Pastan and Willingham, 1981; Matteoni and Kreis, 1987; Goltz et al., 1992; Oda et al., 1995), this report shows definitively that vesicles containing the endocytosed ligand ASOR can translocate on microtubules for distances of 20 μm or more. In previous studies, we showed that vesicles containing ASOR associate with microtubules and that a significant fraction of vesicles can be released upon addition of ATP (Oda et al., 1995). We have confirmed these results in this report with the use of a direct microscopic assay. ASOR-containing vesicles bound to microtubules, and a significant portion began translocating after the addition of ATP, often leading to their release from the microtubule. Immunoblot analysis revealed that the amount of dynein and kinesin in the endosome fraction is equivalent to the amount that is present in the PNS when lanes are loaded with the same amount of total protein. That is, dynein and kinesin are not enriched in the endosome fraction. Presumably, endosomes contain many proteins but require only a limited number of motor molecules to be motile. This point is in agreement with our findings that endosome motility is not affected by the addition of exogenous motor protein (Table 1). These observations suggest that endosomes are saturated with motor proteins at normal cytosolic concentrations. It is known that only small concentrations of motors are required for motility, and in fact, larger concentrations can reduce motility (Bohm et al., 1997).
Hepatocytes contain a large amount of soluble and membrane-associated ATPases. NMR spectroscopy confirmed that the PNS membrane fraction converted ATP to ADP and Pi in quantities that would be expected to inhibit motility in our assays. Purification of endosomes through gel filtration and sucrose gradients reduced the amount of contaminating ATPase activity 40-fold while maintaining a high concentration of endosomes. This led to a population of endosomes that moved on microtubules and required no addition of cytosol or motor protein to stimulate microtubule-based movement.
Endosomes Showed No Bias of Movement with Respect to Microtubule Polarity
Endocytosed asialoglycoprotein undergoes a net progression in hepatocytes from the basolateral cell periphery to the cell interior, and many investigators have predicted that cytoplasmic motors that propel organelles inward would be important for endocytic processing. We and others provided evidence that cytoplasmic dynein, a minus-end and hence inward-directed motor, is involved in the transport of endosome/lysosome/phagosomes (Aniento et al., 1993; Oda et al., 1995; Blocker et al., 1997). However, some observations on the movement of endosomes within cells show that endosomes move toward and away from the cell center at similar frequencies and that localization at the cell center is achieved through small biases in movement or through anchoring of endosomes/lysosomes at the cell center (Matteoni and Kreis, 1987; De Brabander et al., 1988; Suomalainen et al., 1999).
The early endosomes in this study had no bias in their direction of movement, consistent with the hypothesis that endosomes at this stage of their processing do not make net progress toward the center of a cell but instead move in a manner that facilitates their distribution throughout the cytoplasm. This has the potential to aid in the separation of ligand from vesicle, because it has been suggested that microtubules may allow pulling of receptor-containing vesicle membrane away from ligand-containing vesicle contents (Goltz et al., 1992; Satir, 1994; Oda et al., 1995). Figure 11A (see Figure 11A.mov) shows an endosome being pulled toward both ends of a microtubule, and Figure 11B (see Figure 11B.mov) shows that this can lead to vesicle fission. It is possible that later endosomes may exhibit more unidirectional movement, although this has not been examined in the present study.
Blocker et al. (1997) report that phagosomes (after cytosol has been added) move with a 70% bias in favor of minus movement. Pollock et al. (1998) report that “most” of their fluorescent dextran-containing endosomes from Dictyostelium, obtained after 20 min of dextran uptake, moved toward the minus end. The different result in our study is potentially due to the stage of endocytic processing, because our endosomes were isolated after 5 min of uptake and have a low buoyant density, i.e., they are likely to be early endosomes.
Endosomes and Microtubules Move in an Oscillatory Manner in the Presence of Competing Motors
The velocity of gliding microtubules and translocating endosomes is oscillatory (Figure 10). Microtubule gliding exhibited longer spurts of motility with higher amplitude compared with the movement of vesicles. In addition, microtubule gliding showed longer pauses in the plus-end versus the minus-end direction. This finding demonstrates that the oscillations are characteristic for particular motility, and we propose that they offer specific “signatures” for motile events.
The oscillations may derive from competition between motors (in motion and in rigor as well as from opposite directions), inherent bursts of enzymatic activity, and/or local variation in ATP concentration. In vivo, the movement of organelles is oscillatory (Herman and Albertini, 1982; Pryer et al., 1986; Rogers et al., 1997), although the details of these oscillations have not been reported. Suomalainen et al. (1999) report that the movement of adenovirus within endosomes is oscillatory. Vale et al. (1992) report motility oscillations when dynein and kinesin are used together during in vitro motility assays, and these generally are accompanied by a change in direction. We also observed changes in direction, and these occurred during pauses in motility (e.g., Figure 11, A and B, and corresponding movies). Competition between motors, therefore, remains the prime candidate for the source of the oscillations.
ATP as a Regulator of Motility
Because the movement of endosomes is dependent on ATP concentration (Bohm et al., 1997), it is possible that competition between motors and membrane-associated ATPases could regulate the motility of endosomes within cells. In this hypothesis, ATP that is present at the surface of an endosome could be used by either motors or endosome-associated ATPases. Such ATPases function to acidify vesicles at the expense of ATP. As the vesicle reaches proper pH, the ATPase would be inhibited via mass action, causing a local increase in ATP concentration that could then be used by the vesicular motor proteins. This highly speculative hypothesis is supported by observations that ATP depletion inhibits endocytic vesicle processing and motility (Tolleshaug et al., 1985).
CONCLUSION
We have presented a system whereby endocytic sorting and microtubule-based motility can be observed directly and in real time. Endosome motility is ATP dependent, and the effect of local concentrations of ATP on endocytic movement and processing may deserve consideration. We have shown that endosomes move in both directions along microtubules and propose that their eventual localization within cells involves an additional element, either regulation or an anchoring system. Individual vesicles may move in both directions, and control over directionality remains unknown. Oscillation in the velocity of moving vesicles may provide a clue, because pauses in vesicular movement appear to provide an opportunity for vesicles to switch direction. Furthermore, the opposing forces from vesicular motors provide a means to pull endocytic receptors away from their dissociated ligands. Separation of receptor from ligand along intracellular endocytic tubules was observed many years ago (Geuze et al., 1983), and our observations are consistent with the possibility that microtubules and associated motors provide the mechanical basis for this separation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Carol Harley and Duncan Wilson for suggestions about the sucrose gradient, Dr. Mark McNiven for kinesin antibodies, Dr. Raj Gupta for the NMR studies, Drs. Peter Satir, Richard Stockert, and Dennis Shields for critical reading of the manuscript, and the Analytical Imaging Facility for providing the microscopy resources. This work was supported by National Institutes of Health grants DK41918 and DK41296.
Footnotes
REFERENCES
- Aniento F, Emans N, Griffiths G, Gruenberg J. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J Cell Biol. 1993;123:1373–1387. doi: 10.1083/jcb.123.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blocker A, Severin FF, Burkhardt JK, Bingham JB, Yu H, Olivo JC, Schroer TA, Hyman AA, Griffiths G. Molecular requirements for bidirectional movement of phagosomes along microtubules. J Cell Biol. 1997;137:113–129. doi: 10.1083/jcb.137.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm KJ, Stracke R, Unger E. Factors determining kinesin-driven microtubule motility in vitro. Cell Biol Int. 1997;21:854–857. [Google Scholar]
- Bomsel M, Parton R, Kuznetsov SA, Schroer TA, Gruenberg J. Microtubule- and motor-dependent fusion in vitro between apical and basolateral endocytic vesicles from MDCK cells. Cell. 1990;62:719–731. doi: 10.1016/0092-8674(90)90117-w. [DOI] [PubMed] [Google Scholar]
- Bouin JC, Atallah MT, Hultin HO. The glucose oxidase–catalase system. Methods Enzymol. 1976;44:478–488. doi: 10.1016/s0076-6879(76)44034-x. [DOI] [PubMed] [Google Scholar]
- Brady ST. Molecular motors in the nervous system. Neuron. 1991;7:521–533. doi: 10.1016/0896-6273(91)90365-7. [DOI] [PubMed] [Google Scholar]
- Bu G, Schwartz AL. Receptor-mediated endocytosis. In: Arias IW, Boyer JL, Fausto N, Jakoby WB, Schachter D, Sharfritz DA, editors. The Liver: Biology and Pathobiology. New York: Raven Press; 1994. pp. 259–274. [Google Scholar]
- Chan KM, Delfert D, Junger KD. A direct colorimetric assay for Ca2+-stimulated ATPase activity. Anal Biochem. 1986;157:375–380. doi: 10.1016/0003-2697(86)90640-8. [DOI] [PubMed] [Google Scholar]
- Clague MJ. Molecular aspects of the endocytic pathway. Biochem J. 1998;336:271–282. doi: 10.1042/bj3360271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Brabander M, Nuydens R, Geerts H, Hopkins CR. Dynamic behavior of the transferrin receptor followed in living epidermoid carcinoma (A431) cells with nanovid microscopy. Cell Motil Cytoskeleton. 1988;9:30–47. doi: 10.1002/cm.970090105. [DOI] [PubMed] [Google Scholar]
- Dowd TL, Gupta RK. NMR studies of the effect of Mg2+ on postischemic recovery of ATP and intracellular sodium in perfused kidney. Biochim Biophys Acta. 1995;1272:133–139. doi: 10.1016/0925-4439(95)00077-1. [DOI] [PubMed] [Google Scholar]
- Evans WH. Preparation of low-density “endosome” and “endosome”-depleted Golgi fractions from rat liver. Methods Enzymol. 1985;109:246–257. doi: 10.1016/0076-6879(85)09090-5. [DOI] [PubMed] [Google Scholar]
- Geuze HJ, Slot JW, Strous GJ, Lodish HF, Schwartz AL. Intracellular site of asialoglycoprotein receptor-ligand uncoupling: double-label immunoelectron microscopy during receptor-mediated endocytosis. Cell. 1983;32:277–287. doi: 10.1016/0092-8674(83)90518-4. [DOI] [PubMed] [Google Scholar]
- Gilbert SP, Sloboda RD. A squid dynein isoform promotes axoplasmic vesicle translocation. J Cell Biol. 1989;109:2379–2394. doi: 10.1083/jcb.109.5.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goltz JS, Wolkoff AW, Novikoff PM, Stockert RJ, Satir P. A role for microtubules in sorting endocytic vesicles in rat hepatocytes. Proc Natl Acad Sci USA. 1992;89:7026–7030. doi: 10.1073/pnas.89.15.7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada M, Sakisaka S, Yoshitake M, Ohishi M, Itano S, Shakado S, Mimura Y, Noguchi K, Sata M, Yoshida H. Role of cytoskeleton and acidification of endocytic compartment in asialoglycoprotein metabolism in isolated rat hepatocyte couplets. Hepatology. 1995;21:1413–1421. [PubMed] [Google Scholar]
- Harrison RE, Huebner E. Unipolar microtubule array is directly involved in nurse cell-oocyte transport. Cell Motil Cytoskeleton. 1997;36:355–362. doi: 10.1002/(SICI)1097-0169(1997)36:4<355::AID-CM5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Henkel RD, VandeBerg JL, Walsh RA. A microassay for ATPase. Anal Biochem. 1988;169:312–318. doi: 10.1016/0003-2697(88)90290-4. [DOI] [PubMed] [Google Scholar]
- Herman B, Albertini DF. The intracellular movement of endocytic vesicles in cultured granulosa cells. Cell Motil. 1982;2:583–597. doi: 10.1002/cm.970020607. [DOI] [PubMed] [Google Scholar]
- Howard J, Hyman AA. Preparation of marked microtubules for the assay of the polarity of microtubule-based motors by fluorescence microscopy. Methods Cell Biol. 1993;39:105–113. doi: 10.1016/s0091-679x(08)60164-8. [DOI] [PubMed] [Google Scholar]
- Hyman AA, Mitchison TJ. An assay for the activity of microtubule-based motors on the kinetochores of isolated Chinese hamster ovary chromosomes. Methods Cell Biol. 1993;39:267–276. doi: 10.1016/s0091-679x(08)60176-4. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Gilbert SP. Pathway of the microtubule-kinesin ATPase. Biophys J. 1995;68:173S–176S. [PMC free article] [PubMed] [Google Scholar]
- Kishino A, Yanagida T. Force measurements by micromanipulation of a single actin filament by glass needles. Nature. 1988;334:74–76. doi: 10.1038/334074a0. [DOI] [PubMed] [Google Scholar]
- Marks DL, LaRusso NF, McNiven MA. Isolation of the microtubule-vesicle motor kinesin from rat liver: selective inhibition by cholestatic bile acids. Gastroenterology. 1995;108:824–833. doi: 10.1016/0016-5085(95)90457-3. [DOI] [PubMed] [Google Scholar]
- Matteoni R, Kreis TE. Translocation and clustering of endosomes and lysosomes depends on microtubules. J Cell Biol. 1987;105:1253–1265. doi: 10.1083/jcb.105.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss AG, Gatti JL, Witman GB. The motile beta/IC1 subunit of sea urchin sperm outer arm dynein does not form a rigor bond. J Cell Biol. 1992;118:1177–1188. doi: 10.1083/jcb.118.5.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Komatsu M, Toyoshima I, Kuramitsu T, Ono T, Funaoka M, Kato J, Masamune O. Purification and characterization of cytoplasmic dynein of rabbit liver. J Hepatol. 1995;23:66–70. doi: 10.1016/0168-8278(95)80312-2. [DOI] [PubMed] [Google Scholar]
- Oda H, Stockert RJ, Collins C, Wang H, Novikoff PM, Satir P, Wolkoff AW. Interaction of the microtubule cytoskeleton with endocytic vesicles and cytoplasmic dynein in cultured rat hepatocytes. J Biol Chem. 1995;270:15242–15249. doi: 10.1074/jbc.270.25.15242. [DOI] [PubMed] [Google Scholar]
- Oka JA, Weigel PH. Microtubule-depolymerizing agents inhibit asialo-orosomucoid delivery to lysosomes but not its endocytosis or degradation in isolated rat hepatocytes. Biochim Biophys Acta. 1983;763:368–376. doi: 10.1016/0167-4889(83)90098-8. [DOI] [PubMed] [Google Scholar]
- Pastan IH, Willingham MC. Journey to the center of the cell: role of the receptosome. Science. 1981;214:504–509. doi: 10.1126/science.6170111. [DOI] [PubMed] [Google Scholar]
- Pollock N, Koonce MP, de Hostos EL, Vale RD. In vitro microtubule-based organelle transport in wild-type Dictyostelium and cells overexpressing a truncated dynein heavy chain. Cell Motil Cytoskeleton. 1998;40:304–314. doi: 10.1002/(SICI)1097-0169(1998)40:3<304::AID-CM8>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Pryer NK, Wadsworth P, Salmon ED. Polarized microtubule gliding and particle saltations produced by soluble factors from sea urchin eggs and embryos. Cell Motil Cytoskeleton. 1986;6:537–548. doi: 10.1002/cm.970060602. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Tint IS, Fanapour PC, Gelfand VI. Regulated bidirectional motility of melanophore pigment granules along microtubules in vitro. Proc Natl Acad Sci USA. 1997;94:3720–3725. doi: 10.1073/pnas.94.8.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satir P. Motor molecules of the cytoskeleton: possible functions in the hepatocyte. In: Arias IW, Boyer JL, Fausto N, Jakoby WB, Schachter D, Shafritz DA, editors. The Liver: Biology and Pathobiology. New York: Raven Press; 1994. pp. 45–52. [Google Scholar]
- Stockert RJ. The asialoglycoprotein receptor: relationships between structure, function, and expression. Physiol Rev. 1995;75:591–609. doi: 10.1152/physrev.1995.75.3.591. [DOI] [PubMed] [Google Scholar]
- Stockert RJ, Haimes HB, Morell AG, Novikoff PM, Novikoff AB, Quintana N, Sternlieb I. Endocytosis of asialoglycoprotein-enzyme conjugates by hepatocytes. Lab Invest. 1980;43:556–563. [PubMed] [Google Scholar]
- Suomalainen M, Nakano MY, Keller S, Boucke K, Stidwill RP, Greber UF. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J Cell Biol. 1999;144:657–672. doi: 10.1083/jcb.144.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolleshaug H, Kolset SO, Berg T. The influence of cellular ATP levels on receptor-mediated endocytosis and degradation of asialo-glycoproteins in suspended hepatocytes. Biochem Pharmacol. 1985;34:1639–1645. doi: 10.1016/0006-2952(85)90628-8. [DOI] [PubMed] [Google Scholar]
- Vale RD, Malik F, Brown D. Directional instability of microtubule transport in the presence of kinesin and dynein, two opposite polarity motor proteins. J Cell Biol. 1992;119:1589–1596. doi: 10.1083/jcb.119.6.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Schnapp BJ, Reese TS, Sheetz MP. Organelle, bead, and microtubule translocations promoted by soluble factors from the squid giant axon. Cell. 1985;40:559–569. doi: 10.1016/0092-8674(85)90204-1. [DOI] [PubMed] [Google Scholar]
- Wolkoff AW, Klausner RD, Ashwell G, Harford J. Intracellular segregation of asialoglycoproteins and their receptor: a prelysosomal event subsequent to dissociation of the ligand-receptor complex. J Cell Biol. 1984;98:375–381. doi: 10.1083/jcb.98.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.