

Abstract
The pH-induced conformational change of influenza virus hemagglutinin (HA) has been investigated by calculating the change of electrostatic energy of the fragment of HA2 upon pH change. The average charge and electrostatic free energy are calculated as a function of pH for the fusion peptide (residues 1–20 of HA2) and the polypeptide of residues 54–77 of HA2 by using the finite difference Poisson-Boltzmann method. It is found that as pH decreases from 8 to 5, the electrostatic free energy of the fusogenic state is lowered by ∼2 kcal/mol and the fusogenic state is less ionized compared to that of the native state for both polypeptides. For the fusion peptide at the fusogenic state, most of ionizable residues are neutral at acidic pH except Glu-11. For the polypeptide of residues 54–77 at the fusogenic state, most of residues except Glu-74 and His-64 are fully charged between pH 5 and pH 8.
INTRODUCTION
Infection of cell by influenza viruses is initiated by the fusion between viral and cellular membranes. It has been reported that the hemagglutinin (HA) protein mediates both binding of the virus to the cell surface and the subsequent fusion between viral and cellular membranes in the fusion process (1,2). HA is a homotrimeric type I transmembrane protein (3). Each 70-kDa subunit of the trimer structure is composed of two disulfide-linked polypeptide chains, a receptor-binding chain (HA1) and a fusogenic chain (HA2). The native HA1/HA2 complex found on the surface of the native virus does not have a fusion activity. Once the virus binds to its receptor on the cell surface, it is endocytosed by the cell. The endosome produced by the process provides the acidic environment (pH 5–6) for the virus, which can initiate the fusion activity of the virus. After the condition within the endosome reaches pH 5, a large conformational change occurs in HA and then the viral membrane fuses with the cellular membrane. Therefore, it is generally accepted that acidic pH is the trigger of the HA fusion activity (2).
The pH-induced conformational change of HA has been studied in detail by solving the crystal structure of HA in the native state and the low-pH state (4–7). In the native state, as shown in Fig. 1 A, the HA2 N-terminal fusion peptide (residues 1–20) is buried in the hydrophobic core of the protein and fastens two helices together in a helical-hairpin structure, which contains a short helix (residues 38–55) connected to a long helix (residues 77–125) by the loop region (residues 56–75). The pH-induced and irreversible change of conformation in HA2 exposes the fusion peptide to the target membrane, while directing the receptor binding domain HA1 away from the fusion site. In the low-pH conformation of the so-called fusogenic state, as shown in Fig. 1 B, the residues 56–75 corresponding to the loop region in the native state form an α-helix and the N-terminal residues are placed toward the target membrane. The structure of the fusogenic state was first anticipated in the spring-loaded mechanism for activation of membrane fusion (8), in which a region folded as a long loop in the native HA converts to a three-stranded coiled-coil in the low-pH state. The helical residues 105–113 form a reverse turn and then the helical residues 114–129 are jackknifed back. These two helices and the β-sheet (residues 130–133 and 137–140) generate a new hydrophobic core at one end of the coiled-coil in the HA (6). It has been suggested that the reversal in direction may displace the HA1 from the site of membrane fusion (1).
FIGURE 1.

(A) Crystal structure of the native state of HA (1hgd.pdb). (B) Crystal structure of the fusogenic state of HA (1htm.pdb). The structures of a HA2 segment at both states are also shown, where the numbers indicate the residues of HA2 segment.
There have been many studies on the origin of the pH-induced and large conformational change of HA. It has been proposed that the native HA is a metastable state (2,8), which means that the fusogenic state of HA is thermodynamically more stable than the native state of HA. According to this suggestion, the kinetic barrier for changing the conformation of HA from the native state to the fusogenic state (lower energy state) should be lowered to induce the conformational change from the native state to the fusogenic state. It has been also suggested that the interaction between HA1 and HA2 can act as the kinetic barrier (1–3,9). Since HA is synthesized as a precursor HA0 in the cell and then undergoes proteolytic cleavage to generate the two disulfide-linked subunits HA1 and HA2 (1,2), the kinetic barrier is naturally imposed during the folding of HA. Therefore, it is believed that the drop in pH within the endosome can weaken the interaction between HA1-HA2 and hence induce the large conformational change of HA. Recently, it has been reported that the full-length HA2 chain expressed in Escherichia coli can be refolded to the fusogenic conformation at neutral pH in the absence of HA1, which provides further evidence for the notion that the native state of HA is metastable and that the drop in pH lowers the kinetic barrier for rearrangement to the fusogenic conformation (3).
Although the proposition about the origin of the pH-induced conformational change seems reasonable, there are still unresolved questions related to the mechanism of the conformational change, i.e., how the change of pH can lower the kinetic barrier and what the exact nature of the kinetic barrier is. Here, we attempt to elucidate the origin of the conformational change upon pH change by investigating the change of protonation state of amino acid residues of HA2 as well as the electrostatic free energy. The difference of electrostatic free energy between the native state and the fusogenic state are calculated as a function of pH using the finite difference Poisson-Boltzmann method. The average charges of ionizable residues are also calculated as a function of pH. To reduce an enormous computational cost of the electrostatic calculation for the whole protein, we select the fusion peptide of residues 1–20 and the peptide of residues 54–77 of HA2 as model polypeptides, since they undergo the largest conformational change upon pH change and then form the central part of the coiled-coil structure of HA before the membrane fusion.
METHODS
The generation of model polypeptides
The polypeptides of residues 1–20 and 54–77 of HA2 at the native and the low-pH states are generated using the coordinates in the crystal structures of HA at the native (1hgd.pdb) and low-pH (1htm.pdb) states, respectively (5,6), and then hydrogen atoms are added to the model polypeptides using the CHARMM program (10). The values of atom radii and charges are taken from the all-hydrogen PARAM 22 force filed (11). The trimer structures of model polypeptides are also generated based on the crystal structures except the trimer of residues 1–20 at the low-pH state. Since the crystal structure of the trimer of residues 1–20 is not determined at low pH, it is generated using the CHARMM program (10) in such a way that the packing between helices is maximally stabilized, where the hydrophilic residues are positioned to form the inner face of the trimer. Although it is assumed in this study that the peptide of residues 1–20, the fusion peptide, is perfectly helical at pH 5, the helical model of the fusion peptide has been reported for several studies (12–14). Han et al. showed that the fusion peptide is almost helical at pH 5 except a kink near Asp-12 (12). Efremov et al. showed that the lowest energy state of the fusion peptide was α-helical and was absorbed on the bilayer in oblique orientation with its N-terminus immersed in the hydrocarbon region of the membrane (13). Bechor and Ben-Tal used the helical model of the fusion peptide for calculating the interactions of the fusion peptide with lipid bilayers (14). Up to date, there have been no structural data for the trimer structure of the fusion peptide at pH 5 before insertion into lipid bilayer. However, it should be noted here that the fusion peptide forms the helical structure at pH 5 and that it is the part of the coiled-coil of the fusogenic HA2 at pH 5. Therefore, it is likely that the trimer of the fusion peptide also forms a coiled-coil structure, as suggested by this study.
Hence, the peptide trimer generated by a perfect helical model of fusion peptide is used for calculation of electrostatic energy in this study. The model peptides used in this study are shown in Fig. 2.
FIGURE 2.

Monomer and trimer structures of the model peptides. The side chain of acidic residue is colored as red, and the side chain of basic residue is colored as blue. (A) The structure of fusion peptide (residues 1–20). The structures of monomer and trimer of the native state are based on the crystal structure (1hgd.pdb). The structure of monomer of the fusogenic state is first generated as a perfect helix and then the trimer structure is generated by maximizing the interaction between three helices with three ionizable residues (Glu-11, Glu-15, and Asp-19) directing inward. (B) The structures of the polypeptide of residues 54–77: The structures of monomer and trimer of the native and fusogenic states are based on the crystal structure (1htm.pdb).
The calculation of electrostatic free energy and average charge
The change of electrostatic free energy as a function of pH is calculated according to the method developed by Yang et al. (15,16). If a protein has N ionizable residues, a given ionization state n, where n = 1 to 2N, can be defined in terms of the vector δn(i) with i = 1 to N, where δn(i) is 0 when the group i is neutral and 1 when it is charged. A reference state of zero free energy is defined as the state corresponding to all ionizable groups in their neutral form. The pH-dependent free energy of the nth state in the protein ΔGn is given as
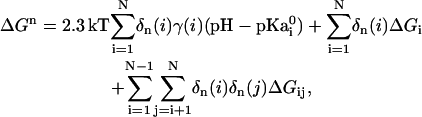 |
(1) |
where 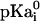 is pKa of an isolated amino acid in solution, ΔGi is the change in electrostatic energy of charging an amino acid in the protein when all other ionizable residues are in their neutral state relative to the same charging process of the isolated amino acid in water, ΔGij is the electrostatic interaction between residues i and j in their charged state, and γ(i) = −1 or 1 for an acidic and basic residue, respectively. The total free energy of ionizable residues of a folded protein can be obtained from the statistical mechanical expression
is pKa of an isolated amino acid in solution, ΔGi is the change in electrostatic energy of charging an amino acid in the protein when all other ionizable residues are in their neutral state relative to the same charging process of the isolated amino acid in water, ΔGij is the electrostatic interaction between residues i and j in their charged state, and γ(i) = −1 or 1 for an acidic and basic residue, respectively. The total free energy of ionizable residues of a folded protein can be obtained from the statistical mechanical expression
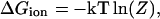 |
(2) |
where Z is the partition function for 2N ionization states of the folded protein
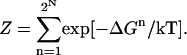 |
(3) |
Therefore, the difference of free energy between the native state and the fugogenic state can be written as
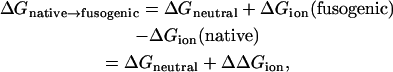 |
(4) |
where ΔGneutral is the free energy difference between the native state and the fusogenic state when all residues are in neutral form. Only ΔΔGion is pH-dependent. The more details are well described in the references by Yang et al. (15,16). The electrostatic energies in the above equations are calculated using the finite difference Poisson-Boltzmann method implemented in the PBEQ of CHARMM (10).
RESULTS
First, the difference of electrostatic free energy between the native state and the fusogenic state as well as the average charge of ionizable residue are calculated as a function of pH for the fusion peptide trimer of residues 1–20. As shown in Fig. 3, the polypeptide trimer of residues 1–20 shows a sharp transition of ΔΔGion between pH 5 and pH 8. ΔΔGion decreases sharply as pH changes from 8 to 5, which demonstrates that the fusogenic state favors a low pH condition, whereas the fusogenic state is less ionized between pH 5 and pH 8, as shown in the profile of total charge of Fig. 3 A. When the average charges of each residue of the fusogenic state and the native state are plotted against pH in Fig. 3, B and C, respectively, it is realized that ionizable residues show different ionization behavior depending upon their environment. In the helical structure of the fusogenic state, most of ionizable residues are neutral at acidic pH except Glu-11. Glu-11 is ionized in entire pH range examined because the ionized form of Glu-11 is stabilized by the attractive interaction with N-terminal. Since Glu-11, Glu-15, and Asp-19 are located in the same face of helix, as shown in Fig. 2 A, the electrostatic repulsion between them becomes larger when they are all ionized. Therefore, it seems that Glu-15 and Asp-19 are not ionized to avoid the repulsive interaction. In the native state where the fusion peptide has looser structure than the helical structure of the fusogenic state, Glu-11 and Glu-15 become partially ionized at neutral pH, whereas Asp-19 is not ionized in all pH range examined.
FIGURE 3.

Electrostatic free energy and charge of the fusion peptide trimer of residues 1–20 as a function of pH. (A) The difference of electrostatic free energy between the fusogenic and native state (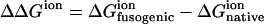 ) and the total charges of the fusogenic and native state as a function of pH. (B) The charges of ionizable residues at the fusogenic state as a function of pH. (C) The charges of ionizable residues at the native state as a function of pH.
) and the total charges of the fusogenic and native state as a function of pH. (B) The charges of ionizable residues at the fusogenic state as a function of pH. (C) The charges of ionizable residues at the native state as a function of pH.
For the polypeptide trimer of residues 54–77, the difference of electrostatic free energy between the native state and the fusogenic state as well as the average charge of ionizable residue are also calculated as a function of pH, as shown in Fig. 4. ΔΔGion decreases sharply as pH decreases from 8 to 5, whereas the fusogenic state is less ionized between pH 5 and pH 8, as can be seen in the profile of total charge of Fig. 4 A. In the fusion peptide, there are only three acidic residues and they are positioned repeatedly every fourth residue, which makes a hydrophilic face in the helical structure. However, in the polypeptide of residues 54–77, there are six acidic residues and six basic residues, and, moreover, hydrophobic and hydrophilic residues are positioned in a repeating heptad pattern (8). Because the different types of negatively or positively charged residues are positioned less periodically in the polypeptide of residues 54–77 than in the fusion peptide, it is difficult to predict the ionization behavior of ionizable residues in the polypeptide of residues 54–77 compared to the fusion peptide. As shown in Fig. 2 B, both acidic and basic residues make an outer hydrophilic surface in the fusogenic state of the polypeptide of residues 54–77, and besides, the acidic and basic residues make extensive contact with each other, which seems to stabilize the ionized form of the residues due to the attractive electrostatic interaction. In the fusogenic structure of the polypeptide of residues 54–77, Glu-74 is not ionized in the entire pH range examined in this study, whereas His-64 becomes neutral at pH 9 as pH increases, as can been in Fig. 4 B. This is because there is no positively charged basic residue near Glu-74, as shown in Fig. 2 B, which may stabilize the charged form of Glu-74, and because Glu-74 may have repulsive interaction with Glu-69 of the nearest helix in the trimer structure, as shown in Fig. 4 B, if Glu-74 is charged. In the native structure of the polypeptide of residues 54–77, only Glu-72 shows a change of ionization state with pH, as can been in Fig. 4 C. Interestingly, when the above calculation is performed for the polypeptide monomer of residues 54–77 (data not shown), the polypeptide monomer of the fusogenic state shows an ionization behavior totally different from the polypeptide trimer, i.e., the polypeptide monomer of the fusogenic state does not favor a low pH condition, whereas there is no significant difference of ionization behavior between the polypeptide monomer and the trimer at the native state. Therefore, it is inferred that the electrostatic interaction between different helices in the polypeptide trimer may also play an important role to induce the low-pH-induced transition of HA2 conformation. It should be noted here that electrostatic free energies in Figs. 3 and 4 are positive. However, considering that ΔGneutral is negative, an unfavorable electrostatic contribution to the total free energy can be compensated. Although an exact calculation of ΔGneutral is not easy, one can estimate the value of ΔGneutral approximately by calculating nonpolar potential energies of bond stretching, angle bending, dihedral, and van der Waals energies. When nonpolar potential energies are calculated by using the CHARMM22 force field, it is found that ΔGneutral for the trimers of residues 1–20 and residue 54–77 are −117.72 kcal/mol and −213.44 kcal/mol, respectively.
FIGURE 4.

Electrostatic free energy and charge of the polypeptide trimer of residues 54–77 as a function of pH. (A) The difference of electrostatic free energy between the fusogenic and native state (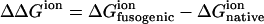 ) and the total charges of the fusogenic and native state as a function of pH. (B) The charges of His-64, Glu-69, and Glu-74 at the fusogenic state as a function of pH. Other ionizable residues are fully charged in the pH range examined in this study. The image shows that Glu-74 in one helix is very close to Glu-69 of the nearest helix, where carbond, hydrogen, nitrogen, and oxygen are colored as gray, white, blue, and red, respectively. (C) The charges of ionizable residues at the native state as a function of pH. All the residues except Glu-72 are not represented in this figure since the residues are negatively or positively charged in the whole pH range examined in this study.
) and the total charges of the fusogenic and native state as a function of pH. (B) The charges of His-64, Glu-69, and Glu-74 at the fusogenic state as a function of pH. Other ionizable residues are fully charged in the pH range examined in this study. The image shows that Glu-74 in one helix is very close to Glu-69 of the nearest helix, where carbond, hydrogen, nitrogen, and oxygen are colored as gray, white, blue, and red, respectively. (C) The charges of ionizable residues at the native state as a function of pH. All the residues except Glu-72 are not represented in this figure since the residues are negatively or positively charged in the whole pH range examined in this study.
Here, it is interesting to compare the results of the current calculation with the experimental results of Carr and Kim (8). We calculated the difference of stability between the native state and the fusogenic state of HA2 fragment based on the known three-dimensional structure of HA2, whereas Carr and Kim synthesized peptides of LOOP-36 (residues 54–89 of HA2) and LOOP-52 (residues 38–89) and analyzed thermodynamic properties of peptides at various pHs and temperatures. According to their results, LOOP-36 is highly helical and trimeric at pH 4.8, whereas it is random coiled and monomeric at pH 7.2. Furthermore, a longer peptide, LOOP-52, forms a trimer more readily than does LOOP-36 and is more stable at pH 4.8 than at pH 7.0 (a transition midpoint, Tm = 52° at pH 7.0 and Tm = 72° at pH 4.8). Our calculation also shows that the helical state of monomer of residues 54–77 is not favored at lower pH. For the trimer structure, however, the fusogenic state of residues 54–77 becomes more stable as pH decreases.
DISCUSSION
Recently, it has been suggested that HA2 has the fusogenic conformation at neutral pH in the absence of HA1 and that a low pH is only required to remove the kinetic constraint preventing the HA2 from forming the fusogenic structure (3,9). When the crystal structure of native HA is compared with that of the fusogenic HA, it seems that an extensive contact between HA1 and HA2 acts as the kinetic constraint. Especially, in the structure of the native state, extensive interactions are apparent between the loop region of HA2 and its corresponding HA1 subunit, and, moreover, the native HA may be stabilized by the fusion peptide, which makes significant hydrophobic interactions in the core of the native structure (8). Then, one may have a question: How does a low-pH affect the kinetic constraint? Chen et al. (17) answered this question by analyzing the crystal structure of uncleaved precursor HA0 and by comparing it with the cleaved HA (HA1/HA2). According to their explanation, there is a cavity in HA0 consisting of ionizable residues (His-17 of HA1; Asp-109, Asp-112, and Lys-117 of HA2), and after the cleavage of HA0 into HA (HA1/HA2), the fusion peptide moves into the cavity due to attractive interactions between newly formed N-terminal of the fusion peptide and negatively charged residues in the cavity. Consequently, they proposed that the movement of the fusion peptide into the cavity containing ionizable residues may set a low-pH trigger, since pH can change the ionization state of ionizable residues in the cavity and thereby the fusion peptide is destabilized in the cavity at low pH due to both the charge-charge repulsion between positively charged residues and N-terminal of the fusion peptide and the loss of hydrogen bond between ionizable residues and backbone of the fusion peptide.
It should be noted here that the displacement of HA1 from the fusion site is accompanied with the conformational change from native HA2 to fusogenic HA2, which means that these two changes are coupled (1). When pH in endosome drops abruptly from 7 to 5, it is likely that the native HA2 is destabilized significantly due to unfavorable electrostatic interaction, whereas the interaction between HA1 and HA2 is also largely weakened. Our results show that the electrostatic free energy of the fusogenic state of HA2 fragments is lower than that of the native state of HA2 fragments at low pH. Although the effect of pH on the stability of HA2 is considered only for the fragment of HA2 in this study, the free energy difference between the native state and the fusogenic state may become more important if the entire structure of hemagglutinin is considered. When potential energies of native and fusogenic hemagglutinin are calculated using the CHARMM 22 force field by setting dielectric constant at unity and by assuming that all ionizable residues are charged at high pH and that only Glu and Asp residues are neutral at low pH, it reveals that the potential energy of the native HA2 is higher at low pH than at high pH by 6000 kcal/mol and the interaction energy between HA1 and HA2 is weaker at low pH than at high pH by 2000 kcal/mol. This provides a qualitative estimation of the effect of pH on the free energy of the entire hemagglutinin. If full electrostatic calculation for the entire hemagglutinin is performed, the exact charge distributions at high and low pH can be obtained and therefore the effect of pH on the conformational change of hemagglutinin can be explained more clearly.
Acknowledgments
The authors thank the Korea Science and Engineering Foundation (KOSEF) for financial support through the Hyperstructured Organic Materials Research Center (HOMRC).
References
- 1.Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 76:531–569. [DOI] [PubMed] [Google Scholar]
- 2.Carr, C. M., C. Chaudhry, and P. Kim. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. USA. 76:14306–14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swalley, S. E., B. M. Baker, L. J. Calder, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 2004. Full-length linfluenza hemagglutinin HA2 refolds into the trimeric low-pH-induced conformation. Biochemistry. 43:5902–5911. [DOI] [PubMed] [Google Scholar]
- 4.Wilson, I. A., J. J. Skehel, and D. C. Wiley. 1981. Structure of the hemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature. 289:366–373. [DOI] [PubMed] [Google Scholar]
- 5.Sauter, N. K., J. E. Hanson, G. D. Glick, J. H. Brown, R. L. Crowther, S. Park, J. J. Skehel, and D. C. Wiley. 1992. Binding of influenza virus hemagglutinin to analogs of its cell-surface receptor, sialic acid: analysis by proton nuclear magnetic resonance spectroscopy and x-ray crystallography. Biochemistry. 31:9609–9621. [DOI] [PubMed] [Google Scholar]
- 6.Bullough, P. A., F. M. Hughson, J. J. Skehel, and D. C. Wiley. 1994. Structure of influenza hemagglutinin at the pH of membrane fusion. Nature. 371:37–43. [DOI] [PubMed] [Google Scholar]
- 7.Chen, J., J. J. Skehel, and D. C. Wiley. 1999. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA2 subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. USA. 96:8967–8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carr, C. M., and P. S. Kim. 1993. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 73:823–832. [DOI] [PubMed] [Google Scholar]
- 9.Chen, J., A. W. Stephen, W. Weissenhorn, L. J. Calder, F. M. Hughson, J. J. Skehel, and D. C. Wiley. 1995. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc. Natl. Acad. Sci. USA. 92:12205–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a programmfor macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 11.MacKerell, A. D., Jr., D. Bashford, M. Bellot, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher III, B. Roux, B. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 12.Han, X., J. H. Bushweller, D. S. Cafiso, and L. K. Tamm. 2001. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 8:715–720. [DOI] [PubMed] [Google Scholar]
- 13.Efremov, R. G., D. E. Nolde, P. E. Volynsky, A. A. Chernyasky, P. V. Dubovskii, and A. S. Arseniev. 1999. Factors important for fusogenic activity of peptides: molecular modeling study of analogs of fusion peptide of influenza hemagglutinin. FEBS Lett. 462:205–210. [DOI] [PubMed] [Google Scholar]
- 14.Bechor, D., and N. Ben-Tal. 2001. Implicit solvent model studies of the interactions of the influenza hemagglutinin fusion peptide with lipid bilayers. Biophys. J. 80:643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang, A. S., M. R. Gunner, R. Sampogna, K. Sharp, and B. Honig. 1993. On the calculation of pKas in proteins. Proteins. 15:252–265. [DOI] [PubMed] [Google Scholar]
- 16.Yang, A. S., and B. Honig. 1993. On the pH dependence of protein stability. J. Mol. Biol. 231:459–474. [DOI] [PubMed] [Google Scholar]
- 17.Chen, J., K. H. Lee, D. A. Steinhauer, D. J. Stevens, J. J. Skehel, and D. C. Wiley. 1998. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell. 95:409–417. [DOI] [PubMed] [Google Scholar]