

Abstract
Cell response to extracellular cues is often driven by gradients of morphogenetic and chemotactic proteins, and therefore descriptions of how such gradients arise are critical to understanding and manipulating these processes. Many of these proteins are secreted in matrix-binding form to be subsequently released proteolytically, and here we explore how this feature, along with small dynamic forces that are present in all tissues, can affect pericellular protein gradients. We demonstrate that 1), pericellular gradients of cell-secreted proteins can be greatly amplified when secreted by the cell in matrix-binding form as compared to a nonmatrix-interacting form; and 2), subtle flows can drive significant asymmetry in pericellular protein concentrations and create transcellular gradients that increase in the direction of flow. This study thus demonstrates how convection and matrix-binding, both physiological characteristics, combine to allow cells to create their own autologous chemotactic gradients that may drive, for example, tumor cells and immune cells into draining lymphatic capillaries.
INTRODUCTION
It is well established that many extracellular mediators induce signaling via their transcellular gradients rather than their absolute amounts, such as morphogen gradients driving morphogenetic processes (e.g., cellular differentiation, cell polarization, organization, and tissue remodeling (1,2)) and chemokine gradients directing leukocyte migration (3). Furthermore, cells can sense exquisitely small transcellular gradients, some as low as 1% (4). Predicting and understanding such cell behaviors therefore requires knowledge of the factors at play in controlling pericellular protein gradients (herein referred to simply as morphogen gradients, which will include chemokines). Gradients can arise from coupled diffusion-reaction mechanisms that were described by Turing (5), who showed that stochastic perturbations in diffusion or reaction rates between competing morphogens could grow into large-scale asymmetry. The resulting heterogeneous spatial patterns, which are necessary for the development of complex organisms, can therefore occur despite homogeneous initial conditions and the symmetric nature of diffusion. Although this paradigm forms the basis of nearly all morphogenesis models and is useful for describing static systems with morphogens that are cell-secreted in a well-defined environment, there are other biophysical factors that can affect pericellular morphogen distribution in a 3D tissue environment, such as matrix interactions and subtle interstitial flows. The effects of such biophysical factors on morphogen gradients are poorly understood.
For example, it is now appreciated that many morphogens are secreted in precursor forms that contain specific motifs that bind to components of the extracellular matrix (ECM) such as collagen, fibrin and glycoaminoglycans (GAGs) (6–9) to be later released by cell-mediated proteolysis (6,10,11). Native morphogens such as VEGF165, for example, have proteolytically cleavable sites that separate the matrix-binding portion of the protein from the cell-signaling portion (11). Similar proteins have also been engineered with a matrix-binding site such that liberation occurs simply by substrate degradation, resulting in a protein with a small ECM fragment attached (12,13). Thus, the ECM is an important source of many morphogens and chemokines, and this is likely to affect the gradients of active morphogens that can become established around cells.
In addition to matrix-binding properties of proteins, the biomechanical environment may also affect pericellular morphogen distribution. Living biological tissues are dynamic, and physical movements such as ambulation, breathing, and cardiac rhythms as well as pressure differentials within tissue result in interstitial fluid displacement within the ECM (14). The lymphatic system drains much of this displaced fluid, and it is estimated that it processes up to 8 liters of lymph per day in the adult human (15), with interstitial flow velocities on the order of 0.1–1.0 μm/s (16–19). Additionally, in vitro 3D perfused tissue constructs with interstitial velocities up to 10 μm/s have shown enhanced morphogenetic responses (20–23). This convection, however slow, is likely to affect the gradients of proteins with small diffusion coefficients, even (as we will show) when diffusion dominates the overall transport distances.
Indeed, we recently found that interstitial flow synergizes with the matrix-bound growth factor VEGF to drive capillary formation from a single-celled suspension of endothelial cells in vitro (21). We proposed a novel mechanism to explain this effect: an amplification and biasing of free VEGF gradients that guides cell-cell communication and eventual network formation. Here we generalize this mechanism and explore its limits and robustness. Specifically, we model gradients of cell-secreted versus ECM-liberated morphogens (via cell-secreted proteases) under a variety of conditions. We show that although interstitial flow itself creates substantial asymmetry in the pericellular concentration profile of a secreted morphogen, the combination of flow plus matrix-binding of morphogens enables the formation of transcellular gradients. This has important and novel implications for directed chemotaxis and morphogenesis.
METHODS
Governing equations
The model involved two simulation steps: first, establishing hypothetical pericellular gradients of a cell-secreted protease, such as a matrix metalloproteinase, and second, using the protease gradient from step 1 to model the liberation and gradient formation of morphogen from the ECM. The first step was also performed for the cell-secreted morphogen rather than the protease, to compare relative gradients of cell-secreted versus matrix-released morphogens under otherwise equivalent conditions. For both of these models, we used a steady-state convection-diffusion mass balance:
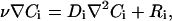 |
(1) |
where v is the velocity field around the cell, Ci is the concentration of solute i (either protease, Cp, or morphogen, Cm), Di is the diffusion coefficient of i (assumed isotropic and homogeneous), and Ri is generation or disappearance of i in the matrix. In the case of cell-secreted protease, Rp accounts for degradation in protease, and in the case of the ECM-released morphogen, Rm is the generation of morphogen from the bulk according to the proteolytic liberation of ECM-bound morphogen into a soluble form.
Defining 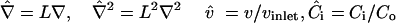 , and Co = maximum concentration, Eq. 1 can be nondimensionalized:
, and Co = maximum concentration, Eq. 1 can be nondimensionalized:
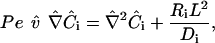 |
(2) |
where the dimensionless Peclet number (Pe) represents the ratio of convective and diffusive transport: Pe = Lvinlet/Di, L = characteristic length (here, cell radius).
The fluid velocity profile was modeled using Brinkman's equation for flow through porous media (24):
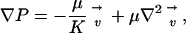 |
(3) |
where P is the pressure, μ is the solute viscosity, and K is the Darcy permeability of the ECM. Barman and colleagues solved the Brinkman velocity field for a sphere (25), and we have adapted this solution for use in our model. The resulting component velocities in cylindrical coordinates are:
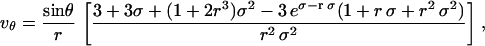 |
(4) |
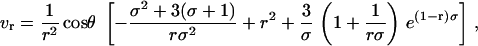 |
(5) |
where r is the radial distance (normalized by the cell radius a) and 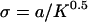 . K varies by many orders of magnitude depending on tissue type (14,26) and state of remodeling or pathology, and in general, K values in vitro are much higher than those seen in vivo. For these simulations we set K = 10−12 cm2, a value between that for in vitro collagen gels (which is on the order of 10−10 to 10−9 cm2 (27)) and real tissues (which generally range from 10−11 to 10−15 cm2, depending on the tissue); however, it should be noted that the lowest expected K value in vivo (10−15 cm2) resulted in velocity values that were within 2% of that for K = 10−12 cm2 (data not shown), whereas for the highest expected in vitro value (10−9 cm2) the maximum velocity differences were 30% but only within a few microns of the cell. Velocities at coordinates in a plane bisecting the sphere and parallel to flow were calculated and converted into component velocities and coordinates in a 2D Cartesian plane for use in the calculation.
. K varies by many orders of magnitude depending on tissue type (14,26) and state of remodeling or pathology, and in general, K values in vitro are much higher than those seen in vivo. For these simulations we set K = 10−12 cm2, a value between that for in vitro collagen gels (which is on the order of 10−10 to 10−9 cm2 (27)) and real tissues (which generally range from 10−11 to 10−15 cm2, depending on the tissue); however, it should be noted that the lowest expected K value in vivo (10−15 cm2) resulted in velocity values that were within 2% of that for K = 10−12 cm2 (data not shown), whereas for the highest expected in vitro value (10−9 cm2) the maximum velocity differences were 30% but only within a few microns of the cell. Velocities at coordinates in a plane bisecting the sphere and parallel to flow were calculated and converted into component velocities and coordinates in a 2D Cartesian plane for use in the calculation.
We selected inlet velocities ranging from 0.1 to 6.0 μm/s to cover a range of published in vivo and in vitro interstitial flow velocities (16–23). Diffusion coefficients Di (Table 1) were calculated by first using the relationship Do = 3600(MW)(−0.34), where Do is the diffusion coefficient in free solution at 23°C and MW is the protein molecular weight (28), then adjusting to 37°C using the Stokes-Einstein relationship, and further adjusting to 70% of that predicted in free solution to account for matrix interactions (28–31). For the sake of comparison, we considered the diffusion coefficients of the matrix-binding ligand and cell-secreted protease to be 120 μm2/s and 80 μm2/s, respectively, according to the ranges given in Table 1 for each. The modeled situations were then classified according to Peclet number.
TABLE 1.
Properties of selected morphogens, chemokines, and proteases
| Morphogen or chemokine | Molecular mass (kDa) | Binding substrate | Cellular effect | Diffusion coefficient (μm2/s) |
|---|---|---|---|---|
| VEGF165 | 38.2 (dimer) | Proteoglycans (6) Fibrin (53) | Vascular permeability and angiogenesis | 92 |
| bFGF | 18 | Fibrin (9) | Upregulation of u-PA, u-PAR, and u-PAI | 120 |
| CCL5CCL17CCL21 | 7.5–14 | Sulfated GAGs (7) | Leukocyte recruitment | 130–160 |
| Interleukin-1B | 17 | Fibrin (8) | Upregulation of NO and chemokines | 121 |
| VonWillebrand factor | 20 | Fibrin (54)Collagens and glycoproteins (55) | Platelet adhesion and storage of factor VIII | 115 |
| Endostatin | 20 | GAGs (56) | Angiogenesis inhibitor | 115 |
| PEDF | 50 | GAGs/Collagen (57) | Neurotrophic and antiangiogenic | 84 |
| Protease | Molecular mass(kDa) | Specificity | Diffusion coefficient (μm2/s) | |
| u-PA | 31–50 | Plasminogen | 84–97 | |
| t-PA | 70 | Plasminogen | 75 | |
| Plasmin | 92 | Fibrin | 68 | |
| MMP2 | 64–72 | Collagen/gelatin | 74–77 | |
| MMP3 | 43–52 | Fibrin collagen/gelatin | 83–89 | |
| MMP9 | 84–92 | Collagen | 68–71 |
Steady-state assumption
Our goal was to seek “snapshots” of typical pericellular morphogen concentrations rather than record the temporal evolution of such patterns. In justifying the steady-state treatment of the problem we compared the characteristic time constants inherent in the model. Here, the characteristic times for solute diffusion tdiff and convection tconv over a length scale 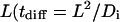 and
and 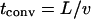 ) are both on the order of seconds, whereas the characteristic time for cell movement over the same length (
) are both on the order of seconds, whereas the characteristic time for cell movement over the same length (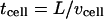 ) is at least an order of magnitude larger for cells such as fibroblasts, tumor cells, and endothelial cells (32–34). Thus, we considered the evolution of the gradients to occur on a short timescale compared to that of the temporal change of such gradients due to cell movement, and thus the steady-state assumption is valid for comparing gradient shapes in cell-secreted versus matrix-released morphogens, or in static versus flow conditions, at a given instant in time. Of course, true steady state in any biological system would be difficult to realize because of specific and dynamic cell response to receptor-ligand interactions (e.g., receptor internalization and recycling, etc.); our goal here was to seek generalizable phenomena valid across a broad range of situations.
) is at least an order of magnitude larger for cells such as fibroblasts, tumor cells, and endothelial cells (32–34). Thus, we considered the evolution of the gradients to occur on a short timescale compared to that of the temporal change of such gradients due to cell movement, and thus the steady-state assumption is valid for comparing gradient shapes in cell-secreted versus matrix-released morphogens, or in static versus flow conditions, at a given instant in time. Of course, true steady state in any biological system would be difficult to realize because of specific and dynamic cell response to receptor-ligand interactions (e.g., receptor internalization and recycling, etc.); our goal here was to seek generalizable phenomena valid across a broad range of situations.
Cell release of proteases and morphogens
Cell secretion of any protein is a complex and highly regulated process, but here we chose two simplified limiting cases in specifying the cell-secreted protease boundary conditions. The first condition was a constant surface concentration, and the other was a constant surface flux. Additionally, the cell surface was made impermeable to both convective and diffusive fluxes of the proteases, which were considered noninteracting with the matrix. Morphogens were considered either noninteracting or subject to binding at the cell surface, allowing us to explore the effects of cell consumption.
Protease decay
Many proteases such as MMPs are heavily regulated posttranslationally by interactions with tissue inhibitors of metalloproteinases, enzymatic processing, and endocytosis after binding to chaperone molecules (reviewed in Sternlicht and Werb (35)). However, there is little information about the physiological rates at which these inactivation mechanisms take place, and furthermore, these interactions are likely to be highly specific to the particular ECM and protease in question. We assumed the protease degradation rate was first-order such that 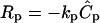 (36). To explore and generalize the effects of decaying protease on relative gradient shapes between flow versus static conditions and secreted versus matrix-liberated morphogens, we examined two conditions, kp = 0 s−1 and 0.2 s−1, the larger value yielding degradation on the same order of magnitude as the diffusion and convection terms in Eq. 1.
(36). To explore and generalize the effects of decaying protease on relative gradient shapes between flow versus static conditions and secreted versus matrix-liberated morphogens, we examined two conditions, kp = 0 s−1 and 0.2 s−1, the larger value yielding degradation on the same order of magnitude as the diffusion and convection terms in Eq. 1.
Matrix-released morphogens
We considered that the morphogen was stored bound to the ECM and liberated to a soluble form only through proteolysis. Under this assumption, the generation term Rm was used to account for this release and was assumed to be linearly proportional to the concentration of proteases 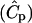 that were generated by the cell:
that were generated by the cell: 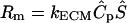 , where Ŝ is the dimensionless concentration of bound morphogen and kECM is the rate constant for liberation of morphogen from the ECM. We first assumed that Ŝ was uniform throughout the ECM and we assigned the product
, where Ŝ is the dimensionless concentration of bound morphogen and kECM is the rate constant for liberation of morphogen from the ECM. We first assumed that Ŝ was uniform throughout the ECM and we assigned the product 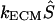 a value of 1.0 s−1, making all terms in Eq. 1 of equal importance. The concentration field
a value of 1.0 s−1, making all terms in Eq. 1 of equal importance. The concentration field 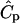 used for the ECM-released morphogen calculation was the steady-state solution calculated for a cell-secreted protease at the same flow conditions. For these calculations, the cell was not a source of morphogen and was impermeable to convective and diffusive fluxes of the released morphogen. Additional calculations were performed to explore the cell boundary condition of morphogen consumption; these are discussed below. The effects of morphogen decay were also explored through the addition of a decay term
used for the ECM-released morphogen calculation was the steady-state solution calculated for a cell-secreted protease at the same flow conditions. For these calculations, the cell was not a source of morphogen and was impermeable to convective and diffusive fluxes of the released morphogen. Additional calculations were performed to explore the cell boundary condition of morphogen consumption; these are discussed below. The effects of morphogen decay were also explored through the addition of a decay term 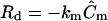 in the same manner as detailed for the decay of proteases listed above.
in the same manner as detailed for the decay of proteases listed above.
Nonuniform distribution of ECM-bound morphogen
In many cases, the matrix-binding form of a morphogen would be secreted into the ECM by the same cell that would later proteolytically release it, and thus the initial pericellular distribution of bound morphogen would be spatially nonuniform. In this case the distribution would depend on the flow velocity and morphogen diffusion coefficient, and would be essentially identical (in dimensionless terms) to the distribution of the cell-secreted proteases under the same flow conditions. The effects of this nonuniform distribution were examined using the same assumptions as before, i.e., 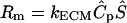 , with the only difference being that Ŝ , the matrix-bound morphogen concentration, was in this case nonuniform and assigned the same numerical values as the protease concentrations under the various flow conditions (i.e.,
, with the only difference being that Ŝ , the matrix-bound morphogen concentration, was in this case nonuniform and assigned the same numerical values as the protease concentrations under the various flow conditions (i.e., 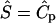 ), whereas kECM was assigned a value of 1.0 s−1.
), whereas kECM was assigned a value of 1.0 s−1.
Consumption of ECM-released morphogens
Although many soluble proteases are readily degraded in the extracellular environment, some ECM-released morphogens such as VEGF have been reported to be fairly stable in the ECM (11,36). However, the morphogen may be consumed through cell binding and internalization. Though the kinetics of receptor-ligand interactions are highly specific and dynamic, we examined the general effects of such cell surface consumption on final morphogen distribution by comparing three different scenarios: no binding, baseline binding, and exaggerated binding cases. The kinetics were simplified such that the binding and cell internalization were modeled according to 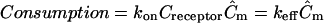 . For the baseline binding case, we used a specific example from the literature for VEGF, where keff = 0.4 s−1 (37), and assumed that all bound ligands would be internalized to maximize the effects. For the exaggerated case keff was increased to 1.0 s−1.
. For the baseline binding case, we used a specific example from the literature for VEGF, where keff = 0.4 s−1 (37), and assumed that all bound ligands would be internalized to maximize the effects. For the exaggerated case keff was increased to 1.0 s−1.
Solution algorithm
Equation 1 was numerically solved in two dimensions using a finite-volume simulation employing the tridiagonal matrix algorithm. The algorithm was contained in custom code written for MATLAB 6.5.1 (The MathWorks, Natick, MA) and was executed on a personal computer. Due to symmetry, the solution was sought only in a half-domain with the cell being represented by a semicircular disk 10 μm in radius. There were 43,624 control volumes in the solution domain (133 in the vertical and 328 in the horizontal) of uniform 1-μm2 size. For all solutions, fluid entering the solution domain did not contain any of the solute. The border of symmetry through the cell centerline had a no-flux boundary, whereas the other three boundaries were zero diffusive flux boundaries. A maximum change of calculated concentration of <0.25% for any given control volume between iterations was used as the convergence criterion.
RESULTS
Directional bias in cell-secreted proteins
For secreted proteins such as proteases or morphogens with diffusion coefficients in the range of 0–120 μm2/s, we found that the relevant ranges of interstitial flow of 0.1–6.0 μm/s can significantly bias the pericellular distributions of such cell-secreted proteins, even with very low Peclet numbers (Fig. 1). Although many metabolic compounds such as glucose, vitamins, and CO2 have large enough diffusion coefficients to be unaffected by physiological flows, most proteins are large enough (Table 1) that even subtle flows can cause noticeable biasing in their pericellular distributions.
FIGURE 1.

Physiological ranges of flow velocities and diffusion coefficients result in biased cell-secreted morphogen gradients. Typical concentration contours show the trend toward biased gradients as either velocity is increased or the diffusion coefficient of the secreted protein is decreased. This biasing occurs for low Peclet numbers (Pe < 1).
Creation of transcellular gradients of matrix-released morphogens
Our data show that the distributions of morphogens that are proteolytically released from the ECM under flow conditions exhibit amplified gradients, with greater asymmetry and increased downstream ranges than if the same molecules had been secreted in soluble form by the cell under the same flow conditions. The two-step calculations whereby proteases secreted by the cell in turn liberate morphogens from their ECM-bound form was examined for a physiological range of flow conditions (Fig. 2). Transcellular gradients refer to the percent differences between upstream and downstream morphogen concentrations across the cell (Fig. 2 F), which determines the extent to which the cell can sense and respond to the gradient; positive numbers refer to gradients that increase in the direction of flow across the cell. The degree of amplification by matrix-binding was striking: for example, whereas the transcellular gradient at the highest flow velocity for the cell-secreted morphogen was only 5%, the corresponding value for the ECM-released morphogen was 133%, a 26-fold increase. Even the slowest velocity tested (0.12 μm/s) resulted in a marked amplification of the transcellular morphogen gradient when compared to static case (2% vs. 0%), and is between the values of 1%, shown to be adequate for neutrophil sensing (4), and 10%, suggested to be adequate for fibroblast sensing (38).
FIGURE 2.

Creation and amplification of autologous morphogen gradients by subtle physiological flows and matrix-binding properties of morphogen. (A) Dimensionless concentration gradients of cell-released proteases calculated using a constant surface concentration are increasingly skewed in the direction of flow with increasing flow velocities. Red, 1 (maximum concentration); dark blue, 0. (B) Dimensionless concentration gradients of liberated morphogen are released from the ECM through the action of the cell-secreted protease whose profiles are shown in A. (C) Distributions of cell-released morphogen demonstrate, when compared with the corresponding profiles in B, the marked gradient amplification effect in matrix-released versus cell-secreted morphogen properties under otherwise identical conditions. (D) Cell-secreted morphogen concentrations as calculated along a line parallel to flow and passing through the cell midlines. All flow conditions result in cell concentrations that decrease with increasing distance from the cell. (E) ECM-released morphogen concentrations show greater asymmetry compared to those of cell-secreted morphogens for the same flow conditions. Interestingly, the higher Peclet numbers (0.25 and 0.5) show increasing concentration gradients with increasing distance downstream from the cell. (F) Calculated transcellular gradients (percentage difference between the downstream and upstream sides of the cell) reveal the degree of gradient amplification that is achieved when morphogen is secreted into matrix-binding form and demonstrate the potential for autologous chemotaxis gradients.
This autologous gradient generation can provide the cell with chemotactic cues and is important, given that chemotactic responses typically occur according to a direction of increasing chemokine concentration relative to the cell (39). It is generally assumed that other cells are necessarily the source of such chemokine gradients. Our results, however, introduce and demonstrate a mechanism by which a cell can create an autologous chemotactic gradient even at relatively low but physiological Peclet numbers. This phenomenon is not possible at physiological flow rates when the chemokines are directly cell-secreted in their active forms.
Effect of protease and morphogen decay
Soluble proteases can be inactivated in the pericellular environment, and ECM-released morphogens are also susceptible to inactivation or rebinding to the matrix in the pericellular environment. For this reason we explored the effects of bulk degradation terms (Rp, and Rd) for the Pe = 0.25 case, comparing the effects of decay of cell-secreted protease, decay of ECM-released morphogen, and decay of both (Fig. 3). In all cases, “decay” refers to any mechanism of inactivation of the soluble form of the protein, whether by chemical inhibitors, further cleavage, matrix binding, or other mechanisms.
FIGURE 3.

Effect of protein decay on protease and morphogen gradients for Pe = 0.25. Pericellular distributions of cell-secreted protease (top row) and resulting ECM-released morphogen (bottom row) with (A) no decay, (B) morphogen decay of km= 0.2 s−1, (C) protease decay of kp = 0.2 s−1, and (D) both protease and morphogen decay kp and km = 0.2 s−1. In all cases the ECM-released morphogen concentrations were normalized: dark red, 1 (maximum concentration); dark blue, 0. (E) Concentrations of ECM-released morphogen as measured along a line passing through the cell midline parallel to flow in each of the conditions (A–D), each with and without inclusion of a cell consumption term. keff was varied between 0 s−1 (solid lines), 0.4 s−1 (thick dotted lines), and 1.0 s−1 (thin dotted lines). (F) Calculated transcellular gradients were reduced in magnitude by the decay considerations, and cell consumption affected the gradients by negligible amounts; however, in all cases the transcellular gradients still increased in the direction of flow, and remained well above physiologically detectable levels of 1–10%.
We found that adding these decay terms reduced both the final ECM-released morphogen concentrations and the transcellular morphogen gradients that could be formed (Fig. 3). However, these decreases were not drastic (<50%), and the general result that subtle flow and matrix-binding properties of morphogen combined to facilitate autologous transcellular gradients still remained robust. It should be noted that a decay rate constant of 0.2 s−1 was used for both protease and morphogen decay so that the effect would be large enough to observe, and although exact values for the decay constants would vary between individual proteases and morphogens, the value we used would likely overestimate the effects for ECM-released morphogens. For example, decay rates for basic fibroblast growth factor have been estimated at ∼4 orders of magnitude lower than that used here (36), and a reduction of km for the ECM-released morphogen from 0.2 to 0.01 s−1 yielded results that differed from the nondecay case by <1% (data not shown). These results show that decay terms can act to diminish the amplification effects of matrix binding, but that autologous transcellular gradient formation is still a robust phenomenon.
Effects of cell consumption
In addition to protein decay, loss of morphogen may occur by receptor binding and endocytosis, so we examined the effects of cell consumption on the overall morphogen concentration for Pe = 0.25. At the baseline consumption rate our results showed little effect of cell consumption on the overall transcellular morphogen gradients (Fig. 3). The consumption calculations were performed both with and without decay terms and although generally minimal, the effects of consumption were the most pronounced when no other decay terms were included (Fig. 3 E). In the higher binding case, which was performed to better determine the qualitative response of this consumption term, the transcellular gradients were again not markedly different compared to the baseline consumption case. The results did suggest, however, that cell consumption can actually aid in the formation of downstream gradients that increase with increasing distance from the cell due to the loss of ligands at the cell surface. In summary, cell consumption has negligible effects on transcellular gradients for the conditions tested, but in general it can increase downstream morphogen gradients to make them positive.
Effects of boundary conditions on computed gradients
To determine the effects of boundary conditions on pericellular morphogen profiles, both constant surface concentration and constant flux boundary conditions were compared in calculating cell-released protease gradients for Pe = 0.25. Likewise, for ECM-released morphogen gradients, the initial conditions were varied between uniform and nonuniform matrix-bound morphogen (see Methods). These two different boundary conditions were used to compare two limiting cases rather than to mimic the actual physiological condition. Given the complexity of the inherent regulation mechanisms, either choice of boundary condition is a gross simplification of what is likely in reality a spatially inhomogeneous phenomenon (and dependent on the details of the particular protease being secreted), but we considered these two cases to represent limits or boundaries. In one extreme (constant-flux BC), the cell would be secreting protease at a constant rate without any feedback of the external concentration, so that no matter what the external concentration, the same amount is constantly being secreted. In the other extreme, the cell is acutely sensitive to the external concentration of the secreted protein and it autoregulates secretion according to what it senses externally.
We saw that the final morphogen distribution was relatively insensitive to the choice of boundary conditions (Fig. 4). Thus, we concluded that the general phenomenon of flow-biased and flow-amplified morphogen gradient formation is relatively insensitive to what is actually happening at the cell surface with regard to secretion or receptor ligation.
FIGURE 4.

Effects of boundary and initial conditions on morphogen gradients for Pe = 0.25. (A) Cell-secreted protease distribution assuming a constant surface concentration (top row) and corresponding ECM-released morphogen distribution (bottom row) serve as a control for evaluating effects of changing boundary conditions. Dark red, 1 (maximum concentration); dark blue, 0. (B) The constant surface flux protease condition resulted in a slightly altered cell-released protease distribution (top row) when compared to the constant concentration boundary case (A); however, the dimensionless ECM-released morphogen profiles shown on the bottom row appear nearly identical. (C) Protease distribution of A, with the resulting ECM-released morphogen profile calculated assuming that the ECM-bound morphogen was nonuniformly distributed. The ECM-released morphogen gradient (bottom row) shares the same qualitative shape as for the previous two conditions. (D) Dimensionless ECM-released concentration profiles along the bisecting midline of the three cases shown in A–C are similar.
DISCUSSION
Asymmetry in morphogen gradients is typically described using diffusion-reaction models under static conditions, but living systems are dynamic, low levels of interstitial flow are ubiquitous, and many secreted morphogens are bound to the matrix to be later released. Furthermore, nature hints at a role of flow in directing morphogenesis; for example, convection of amniotic fluid has been shown to help determine left/right asymmetry in developing mouse embryos (40), and interstitial fluid flow was demonstrated to guide lymphangiogenesis in a wound-healing model (41) and drive blood and lymphatic capillary morphogenesis in vitro (20,21). With these examples as guides, we have developed a model that shows how the combination of ECM-binding morphogens and interstitial flow can provide cells with developmental cues in the form of strong autologous transcellular gradients. In its general form, our model is not used to predict specific gradients that would arise from any particular morphogen situation, but rather to demonstrate the general phenomenon of autologous morphogen and chemokine gradient formation and amplification via matrix-binding morphogen properties coupled with very subtle dynamic forces. The minimum transcellular difference in morphogen concentration required for cell recognition varies by cell type and morphogen/receptor system. Morphogens can signal through several families of receptors such as tyrosine kinases and G-protein coupled receptors and each involves its own cascade and intercellular amplification system (reviewed in Kholodenko (42)). These intracellular amplification systems allow cells to create large internal gradients of response molecules such as AKT protein kinase in response to much shallower extracellular signaling gradients. Minimum required external gradient thresholds range from as low as 1% for neutrophil sensing (4) to levels on the order of 10% or more for fibroblasts (38). Although 1% autocrine morphogen gradients are possible due to interstitial flow alone (Fig. 2 F), a transcellular gradient of 10% or more is not possible for a cell-secreted morphogen under physiological flow rates alone, but rather only when combined with matrix-binding characteristics of the morphogen. Whatever the minimum threshold required for a given cell/morphogen pair may be, ECM binding can result in up to 25-fold amplification over directly secreted morphogens.
Our results have intriguing implications for chemotactic processes. It is well known that both metastatic tumor cells and activated immune cells traffic in the lymphatic system (43,44), but exactly how these cells efficiently migrate through the tissue matrix toward the nearest lymphatic vessel is not known. Both cell types migrate up gradients of the chemokines CCL21 and CCL19 to reach the lymphatics (45,46); both of these molecules are matrix-binding (7), and the net direction of interstitial flow is always and necessarily toward the nearest draining lymphatic vessel. Interestingly, dendritic cells themselves secrete CCL19 (46), and it is not known whether tumor cell secretion of chemokines affects their migration toward lymphatics, but recent work in our laboratory has shown a connection between autocrine CCL21 and tumor cell migration (Shields, J. D., M. E. Fleury, C. Yong, G. J. Randolph, M. A. Swartz, unpublished material). Thus, one might speculate that autologous chemotaxis is a mechanism whereby immune cells and tumor cells migrate toward draining lymphatics. Additionally, although the calculations have been developed with matrix-binding morphogens in mind, ECM fragments themselves have also been shown to serve as a chemotactic factor—for example, as in the case of fibrin degradation products (48,49)—and our model predictions are valid for comparing pericellular gradients of such matrix fragments as well.
In addition to shedding light on a basic phenomenon, this research may be useful in tissue engineering, whose primary goal is to recapitulate certain aspects of tissue architecture function in vitro. Much research has been devoted to specifying cell patterns within a matrix, for example by layered two-dimensional films (50), laser-guided “cell writing” (51), or cell dielectrophoresis (52). In contrast to such prescriptive designs, our results demonstrate the potential to engineer 3D tissues using an appropriate ECM (i.e., one that is growth-factor laden or rich in binding sites) and introducing physiological dynamic forces such as interstitial flow to permit synergistic self-organization to occur. The work presented here may thus serve as a guide for the rational use of flow and matrix-binding in controlling morphogen patterning.
In conclusion, our results show that interstitial flow and matrix-binding morphogens, both physiological conditions, combine to robustly create asymmetric pericellular morphogen gradients and to amplify them over static conditions in which the cell secretes active morphogens directly. This mechanism may not only help to explain developmental asymmetry, but it may also 1), serve as an alternative mechanosensing mechanism for the cell to gather information about the dynamic status of its environment, and 2), drive autologous chemotaxis to help direct migrating cells into tissue-draining lymphatics.
Acknowledgments
The authors are grateful to Mr. John Pedersen for helpful discussions and to the National Institutes of Health/National Heart, Lung, and Blood Institute (R01HL075217-01), the U.S. National Science Foundation (BES-0134551), the Swiss National Science Foundation (107602), and the EPFL for funding.
References
- 1.Gurdon, J. B., and P. Y. Bourillot. 2001. Morphogen gradient interpretation. Nature. 413:797–803. [DOI] [PubMed] [Google Scholar]
- 2.Ruhrberg, C., H. Gerhardt, M. Golding, R. Watson, S. Ioannidou, H. Fujisawa, C. Betsholtz, and D. T. Shima. 2002. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 16:2684–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manes, S., C. Gomez-Mouton, R. A. Lacalle, S. Jimenez-Baranda, E. Mira, and C. Martinez-A. 2005. Mastering time and space: immune cell polarization and chemotaxis. Semin. Immunol. 17:77–86. [DOI] [PubMed] [Google Scholar]
- 4.Zigmond, S. H. 1977. Ability of polymorphonuclear leukocytes to orient in gradients of chemotactic factors. J. Cell Biol. 75:606–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turing, A. M. 1952. The chemical basis of morphogenesis. Philos. Trans. R. Soc. Lond. B. 237:37–72. [Google Scholar]
- 6.Park, J. E., G. A. Keller, and N. Ferrara. 1993. Vascular Endothelial growth-factor (VEGF) isoforms. Differential deposition into the subepithelial extracellular-matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell. 4:1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel, D. D., W. Koopmann, T. Imai, L. P. Whichard, O. Yoshie, and M. S. Krangel. 2001. Chemokines have diverse abilities to form solid phase gradients. Clin. Immunol. 99:43–52. [DOI] [PubMed] [Google Scholar]
- 8.Sahni, A., M. Guo, S. K. Sahni, and C. W. Francis. 2004. Interleukin-1 β but not IL-1 α binds to fibrinogen and fibrin and has enhanced activity in the bound form. Blood. 104:409–414. [DOI] [PubMed] [Google Scholar]
- 9.Sahni, A., T. Odrljin, and C. W. Francis. 1998. Binding of basic fibroblast growth factor to fibrinogen and fibrin. J. Biol. Chem. 273:7554–7559. [DOI] [PubMed] [Google Scholar]
- 10.Houck, K. A., D. W. Leung, A. M. Rowland, J. Winer, and N. Ferrara. 1992. Dual regulation of vascular endothelial growth-factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 267:26031–26037. [PubMed] [Google Scholar]
- 11.Lee, S., S. M. Jilani, G. V. Nikolova, D. Carpizo, and M. L. Iruela-Arispe. 2005. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J. Cell Biol. 169:681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hall, H., T. Baechi, and J. A. Hubbell. 2001. Molecular properties of fibrin-based matrices for promotion of angiogenesis in vitro. Microvasc. Res. 62:315–326. [DOI] [PubMed] [Google Scholar]
- 13.Zisch, A. H., U. Schenk, J. C. Schense, S. E. Sakiyama-Elbert, and J. A. Hubbell. 2001. Covalently conjugated VEGF-fibrin matrices for endothelialization. J. Controlled Release. 72:101–113. [DOI] [PubMed] [Google Scholar]
- 14.Levick, J. R. 1987. Flow through interstitium and other fibrous matrices. Q. J. Exp. Physiol. 72:409–437. [DOI] [PubMed] [Google Scholar]
- 15.Renkin, E. M. 1986. Some consequences of capillary-permeability to macromolecules. Starlings hypothesis reconsidered. Am. J. Physiol. 250:H706–H710. [DOI] [PubMed] [Google Scholar]
- 16.Chary, S. R., and R. K. Jain. 1989. Direct measurement of interstitial convection and diffusion of albumin in normal and neoplastic tissues by fluorescence photobleaching. Proc. Natl. Acad. Sci. USA. 86:5385–5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dafni, H., T. Israely, Z. M. Bhujwalla, L. E. Benjamin, and M. Neeman. 2002. Overexpression of vascular endothelial growth factor 165 drives peritumor interstitial convection and induces lymphatic drain: Magnetic resonance imaging, confocal microscopy, and histological tracking of triple-labeled albumin. Cancer Res. 62:6731–6739. [PubMed] [Google Scholar]
- 18.Jain, R. K. 1999. Transport of molecules, particles, and cells in solid tumors. Ann. Rev. Biomed. Eng. 1:241–263. [DOI] [PubMed] [Google Scholar]
- 19.Leu, A. J., D. A. Berk, F. Yuan, and R. K. Jain. 1994. Flow velocity in the superficial lymphatic network of the mouse tail. Am. J. Physiol. 36:H1507–H1513. [DOI] [PubMed] [Google Scholar]
- 20.Ng, C. P., C. L. E. Helm, and M. A. Swartz. 2004. Interstitial flow differentially stimulates blood and lymphatic endothelial cell morphogenesis in vitro. Microvasc. Res. 68:258–264. [DOI] [PubMed] [Google Scholar]
- 21.Helm, C. L. E., M. E. Fleury, A. H. Zisch, F. Boschetti, and M. A. Swartz. 2005. Synergy between interstitial flow and VEGF directs capillary morphogenesis in vitro through a gradient amplification mechanism. Proc. Natl. Acad. Sci. USA. 102:15779–15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semino, C. E., R. D. Kamm, and D. A. Lauffenburger. 2006. Autocrine EGF receptor activation mediates endothelial cell migration and vascular morphogenesis induced by VEGF under interstitial flow. Exp. Cell Res. 312:289–298. [DOI] [PubMed] [Google Scholar]
- 23.Hosseinkhani, H., Y. Inatsugu, Y. Hiraoka, S. Inoue, and Y. Tabata. 2005. Perfusion culture enhances osteogenic differentiation of rat mesenchymal stem cells in collagen sponge reinforced with poly(glycolic acid) fiber. Tissue Eng. 11:1476–1488. [DOI] [PubMed] [Google Scholar]
- 24.Brinkman, H. C. 1947. A calculation of the viscous force exerted by a flowing fluid on a dense swarm of particles. Appl. Sci. Res. Sect. A. Mech. 1:27–34. [Google Scholar]
- 25.Barman, B. 1996. Flow of a Newtonian fluid past an impervious sphere embedded in a porous medium. Indian J. Pure Appl. Math. 27:1249–1256. [Google Scholar]
- 26.Anand, S., J.-H. Wu, and S. L. Diamond. 1995. Enzyme-mediated proteolysis of fibrous biopolymers: dissolution front movement in fibrin or collagen under conditions of diffusive or convective transport. Biotechnol. Bioeng. 48:89–107. [DOI] [PubMed] [Google Scholar]
- 27.Ng, C. P., and M. A. Swartz. 2003. Fibroblast alignment under interstitial fluid flow using a novel 3-D tissue culture model. Am. J. Physiol. 284:H1771–H1777. [DOI] [PubMed] [Google Scholar]
- 28.Berk, D. A., F. Yuan, M. Leunig, and R. K. Jain. 1993. Fluorescence photobleaching with spatial Fourier analysis: measurement of diffusion in light-scattering media. Biophys. J. 65:2428–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berk, D. A., F. Yuan, M. Leunig, and R. K. Jain. 1997. Direct in vivo measurement of targeted binding in a human tumor xenograft. Proc. Natl. Acad. Sci. USA. 94:1785–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pluen, A., P. A. Netti, R. K. Jain, and D. A. Berk. 1999. Diffusion of macromolecules in agarose gels: comparison of linear and globular configurations. Biophys. J. 77:542–552 [comment]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramanujan, S., A. Pluen, T. D. McKee, E. B. Brown, Y. Boucher, and R. K. Jain. 2002. Diffusion and convection in collagen gels: implications for transport in the tumor interstitium. Biophys. J. 83:1650–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raeber, G. P., M. P. Lutolf, and J. A. Hubbell. 2005. Molecularly engineered PEG hydrogels: a novel model system for proteolytically mediated cell migration. Biophys. J. 89:1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolf, K., I. Mazo, H. Leung, K. Engelke, U. H. von Andrian, E. I. Deryugina, A. Y. Strongin, E. B. Brocker, and P. Friedl. 2003. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 160:267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stokes, C. L., D. A. Lauffenburger, and S. K. Williams. 1991. Migration of individual microvessel endothelial cells: stochastic model and parameter measurement. J. Cell Sci. 99:419–430. [DOI] [PubMed] [Google Scholar]
- 35.Sternlicht, M. D., and Z. Werb. 2001. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 17:463–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong, S., and F. Yuan. 2001. Numerical simulations of angiogenesis in the cornea. Microvasc. Res. 61:14–27. [DOI] [PubMed] [Google Scholar]
- 37.Mac Gabhann, F., and A. S. Popel. 2005. Differential binding of VEGF isoforms to VEGF receptor 2 in the presence of neuropilin-1: a computational model. Am. J. Physiol. 288:H2851–H2860. [DOI] [PubMed] [Google Scholar]
- 38.Schneider, I. C., and J. M. Haugh. 2005. Quantitative elucidation of a distinct spatial gradient-sensing mechanism in fibroblasts. J. Cell Biol. 171:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Devreotes, P., and C. Janetopoulos. 2003. Eukaryotic chemotaxis: distinctions between directional sensing and polarization. J. Biol. Chem. 278:20445–20448. [DOI] [PubMed] [Google Scholar]
- 40.Nonaka, S., Y. Tanaka, Y. Okada, S. Takeda, A. Harada, Y. Kanai, M. Kido, and N. Hirokawa. 1998. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 95:829–837. [DOI] [PubMed] [Google Scholar]
- 41.Boardman, K. C., and M. A. Swartz. 2003. Interstitial flow as a guide for lymphangiogenesis. Circ. Res. 92:801–808. [DOI] [PubMed] [Google Scholar]
- 42.Kholodenko, B. 2006. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 7:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Randolph, G. J., V. Angeli, and M. A. Swartz. 2005. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev.Immunol. 5:617–628. [DOI] [PubMed] [Google Scholar]
- 44.Stacker, S. A., M. G. Achen, L. Jussila, M. E. Baldwin, and K. Alitalo. 2002. Lymphangiogenesis and cancer metastasis. Nat. Rev. Cancer. 2:573–583. [DOI] [PubMed] [Google Scholar]
- 45.Forster, R., A. Schubel, D. Breitfeld, E. Kremmer, I. Renner-Muller, E. Wolf, and M. Lipp. 1999. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 99:23–33. [DOI] [PubMed] [Google Scholar]
- 46.Ohl, L., M. Mohaupt, N. Czeloth, G. Hintzen, Z. Kiafard, J. Zwirner, T. Blankenstein, G. Henning, and R. Forster. 2004. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity. 21:279–288. [DOI] [PubMed] [Google Scholar]
- 47.Reference deleted in proof.
- 48.Kodama, M., M. Naito, H. Nomura, A. Iguchi, W. D. Thompson, C. M. Stirk, and E. B. Smith. 2002. Role of D and E domains in the migration of vascular smooth muscle cells into fibrin gels. Life Sci. 71:1139–1148. [DOI] [PubMed] [Google Scholar]
- 49.Naito, M., C. M. Stirk, E. B. Smith, and W. D. Thompson. 2000. Smooth muscle cell outgrowth stimulated by fibrin degradation products: the potential role of fibrin fragment E in restenosis and atherogenesis. Thromb. Res. 98:165–174. [DOI] [PubMed] [Google Scholar]
- 50.Klebe, R. J. 1988. Cytoscribing: a method for micropositioning cells and the construction of two-dimensional and 3-dimensional synthetic tissues. Exp. Cell Res. 179:362–373. [DOI] [PubMed] [Google Scholar]
- 51.Nahmias, Y., R. E. Schwartz, C. M. Verfaillie, and D. J. Odde. 2005. Laser-guided direct writing for three-dimensional tissue engineering. Biotechnol. Bioeng. 92:129–136. [DOI] [PubMed] [Google Scholar]
- 52.Alp, B., G. M. Stephens, and G. H. Markx. 2002. Formation of artificial, structured microbial consortia (ASMC) by dielectrophoresis. Enzyme Microb. Technol. 31:35–43. [Google Scholar]
- 53.Sahni, A., S. K. Sahni, P. J. Simpson-Haidaris, and C. W. Francis. 2004. Fibrinogen binding potentiates FGF-2 but not VEGF induced expression of u-PA, u-PAR, and PAI-1 in endothelial cells. J. Thromb. Haemost. 2:1629–1636. [DOI] [PubMed] [Google Scholar]
- 54.Keuren, J. F. W., D. Baruch, P. Legendre, C. V. Denis, P. J. Lenting, J. P. Girma, and T. Lindhout. 2004. Von Willebrand factor C1C2 domain is involved in platelet adhesion to polymerized fibrin at high shear rate. Blood. 103:1741–1746. [DOI] [PubMed] [Google Scholar]
- 55.Sadler, J. E. 1998. Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 67:395–424. [DOI] [PubMed] [Google Scholar]
- 56.Reis, R. C. M., D. Schuppan, A. C. Barreto, M. Bauer, J. P. Bork, G. Hassler, and T. Coelho-Sampaio. 2005. Endostatin competes with bFGF for binding to heparin-like glycosaminoglycans. Biochem. Biophys. Res. Commun. 333:976–983. [DOI] [PubMed] [Google Scholar]
- 57.Yasui, N., T. Mori, D. Morito, O. Matsushita, H. Kourai, K. Nagata, and T. Koide. 2003. Dual-site recognition of different extracellular matrix components by anti-angiogenic/neurotrophic serpin, PEDF. Biochemistry (Mosc.). 42:3160–3167. [DOI] [PubMed] [Google Scholar]