

Abstract
It has been a significant challenge to quantitatively study the dynamic intracellular processes in live cells. These studies are essential for a thorough understanding of the underlying mechanisms regulating the signaling pathways and the transitions between cell cycle stages. Our studies of Cdc20, an important mitotic checkpoint protein, throughout the cell cycle demonstrate that fluorescence correlation spectroscopy is a powerful tool for in vivo quantitative studies of dynamic intracellular processes. In this study, Cdc20 is found to be present primarily in a large complex (>1 Mda) during interphase with a diffusion constant of 1.8 ± 0.1 μm2/s and a concentration of 76 ± 24 nM, consistent with its association with the APC/C. During mitosis, however, a proportion of Cdc20 dissociates from APC/C at a rate of 12 pM/s into a soluble pool with a diffusion constant of 19.5 ± 5.0 μm2/s, whose size is most consistent with free Cdc20. This free pool accumulates to 50% of total Cdc20 (∼40 nM) during chronic activation of the mitotic checkpoint but disappears during mitotic exit at a rate of 31 pM/s. The observed changes in the biochemical assembly states of Cdc20 closely correlate to the known temporal pattern of the activity of APC/CCdc20 in mitosis. Photon counting histograms reveal that both complexes contain only a single molecule of Cdc20. The underlying mechanisms of the activities of APC/CCdc20 throughout the cell cycle are discussed in light of our experimental observations.
INTRODUCTION
Cell cycle progression requires the ordered accumulation and destruction of specific proteins, including the mitotic cyclins that in turn control the activity of their associated cyclin-dependent kinases (CDKs) (1,2). The anaphase-promoting complex/cyclosome (APC/C) is a large, multi-protein complex whose E3-ubiquitin ligase activity is precisely regulated to ensure the timely ubiquitin-mediated proteolysis of cyclins and other key cell cycle regulators. During mitosis, the APC/C is responsible for the irreversible segregation of replicated chromatids to each daughter cell. This action is delayed by the mitotic checkpoint (also known as the spindle assembly checkpoint) until all chromosomes are attached to spindle microtubules at the kinetochores. Central to the checkpoint are the unattached kinetochores which generate a “wait anaphase” signal to prevent premature exit of mitosis (3–6). Cdc20 is an essential activator of the APC/C through its action as a substrate-specific adaptor protein that allows recognition of a destruction box containing proteins by the APC/C for ubiquitination (7,8). Inhibiting the ability of Cdc20 to facilitate APC/C's recognition of substrates such as cyclin B and securin is therefore the primary function of the mitotic checkpoint. For this, unattached kinetochores generate one or more Cdc20 inhibitors that selectively block Cdc20-stimulated APC/C action on these substrates. Proposed Cdc20 inhibitors include phosphorylation of Cdc20 itself by the mitotic kinase Bub1 (9), direct binding by oligomerized Mad2 (10) or BubR1 (11), or a four component complex (named MCC, for mitotic checkpoint complex) comprising BubR1, Mad2, Bub3, and Cdc20 (12).
Although significant progress has been made in understanding the mitotic checkpoint at the molecular level through in vitro studies, much of the dynamic and kinetic information in live cells that is crucial for developing a model for the signaling pathway remains unknown. A key question is whether Cdc20 serves as an essential, stoichiometric component of the APC/C or as a kiss-and-run facilitator that identifies substrates, brings them to the APC/C, and then releases to begin another cycle of substrate binding and recruitment (13). Lack of a clear dynamic picture of the interaction between the two proteins has led to two controversial models on how the mitotic checkpoint regulates APC/CCdc20. The first model suggests that Cdc20 has a dynamic interaction with the APC/C so that it allows Mad2, together with other checkpoint proteins, to inhibit APC/C by sequestering its activator Cdc20 (10,14). On the other hand, the second model suggests that the previously proposed MCC, which contains checkpoint proteins Mad2, BubR1, Cdc20, and Bub3, may directly bind to and inhibit the APC/C, which has itself been sensitized by unattached kinetochores (12,15). Similar questions also arise for APC/CCdc20 inhibition in interphase. Emi1 has been identified as an interphase inhibitor (16,17). However, it is not clear whether Emi1 binds and inhibits Cdc20 already associated with the APC/C or sequesters Cdc20 as it dissociates from the APC/C (16).
Understanding of the dynamic interactions between the checkpoint proteins is important for establishing models that explain the central question of the mitotic checkpoint, i.e., how the “wait anaphase” signal from a single unattached kinetochore is transduced to halt the progression of the mitosis (6,15,18). Definitive answers to these questions, like many others arising from various cellular studies in cell biology, require quantitative understanding of the in vivo dynamic intracellular processes in live cells. This poses significant challenges to the traditional biochemical experimental methods and requires noninvasive in vivo and real time quantitative measurements of the molecular concentrations, the molecular diffusions, and the rates of reactions of protein-protein interactions in live cells.
Fluorescence correlation spectroscopy (FCS) is a powerful tool for studying the molecular dynamics of diffusion, rates of biochemical reactions, as well as absolute concentrations with single-molecule sensitivity (19,20). Recent developments in FCS have demonstrated its potential as a new, enabling technology that allows noninvasive real time quantitative measurements of individual molecules in living cells (21–27). In this study, we demonstrate that we can quantitatively monitor the evolution of the Cdc20-related biochemical reaction processes in live cells with FCS. Throughout the entire cell cycle, we measured the temporal dissociation of a small Cdc20-containing species from a large Cdc20-containing complex (APC/CCdc20), the diffusion constants and absolute concentrations of each Cdc20-containing species at various stages of the cell cycle, the real time rates of the reaction between the two Cdc20-containing complexes, and the temporal degradation of the small Cdc20 species at the exit of the mitosis. The underlying mechanisms of the activities of APC/CCdc20 throughout the cell cycle are also discussed in light of our experimental observations.
MATERIALS AND METHODS
Constructs and cell lines
Cells and methods of cell culture used in these studies were from established sublines of the rat kangaroo Potorous tridactylus, PTK2. PTK2 cells and derived lines were cultured in MEM-Earle's supplemented with 10% fetal bovine serum, sodium pyruvate, penicillin, and streptomycin. Cell lines stably expressing fluorescent protein fusions to human Cdc20 were generated by amphotropic retroviral infection (as described in Shah et al.(28)).
The cDNA for human Cdc20 was excised from a clone provided by Prof. Peter Sorger (Massachusetts Institute of Technology, Cambridge, MA) and ligated into the enhanced cyan fluorescent protein (ECFP)-C1. This fusion cDNA was ligated into the SnaBI/EcoRI sites of pBABEpuro, a retroviral vector.
The retroviral plasmid containing the fluorescent protein fusion was cotransfected using the Fugene transfection reagent (Roche Pharmaceuticals, Indianapolis, IN) into 293-GP cells (a human embryonic kidney cell line harboring a portion of the Murine Moloney Leukemia Virus genome) along with a VSV-G pseudotyping plasmid to generate amphotropic virus. Forty-eight hours after transfection, the culture supernatant was collected, filtered, and mixed with 8 μg/mL hexadimethrine bromide (Polybrene, Sigma, St. Louis, MO), and 10% of the total filtrate was placed onto a subconfluent culture (30–40%) of PTK2 cells in 35-mm dishes; 48 h after infection, cells were split and replated in 10-cm dishes and subjected to selection in 2 μg/mL puromycin for 14 days. High expressors (top 10%) were cloned by fluorescence-activated cell sorting (FACSVantage, Becton Dickinson, San Jose, CA). Cells were maintained as polyclonal lines, with cells expressing varying levels of fluorescent protein fusion.
Instrumentation and measurements
Live cell images were taken on a modified Zeiss Axiovert inverted microscope using a 63× high numerical aperture (NA 1.4) Plan Apochromat objective. Images were collected by a digital camera (C4742-95, Hamamatsu, Hamamatsu City, Japan) and captured to a computer through the use of AxioVision software (Carl Zeiss, Göttingen, Germany).
For flow cytometry analysis, cells from both control and ECFP-Cdc20 cell lines were fixed in 50% ethanol for 20 min on ice before treating with RNase at a final concentration of 50 μg/ml. Propidium iodide was added to the cell suspension at a final concentration of 50 μg/ml for 15 min for cell cycle measurement on BD FACS (Becton, Dickinson and Co., Franklin Lakes, NJ).
FCS with two-photon excitation was performed on a modified Zeiss Axiovert inverted microscope using one of the camera ports for FCS detection (Fig. 1 A and (26)). Briefly, the collimated beam of a mode-locked tunable Coherent (Palo Also, CA) Mira 900 Titanium-Sapphire laser with 76 MHz, 120 fs pulse width was coupled through a Zeiss 63× Plan Apochromat oil immersion objective (NA = 1.4). The fluorescence from ECFP has an emission peak at 477 nm and was collected with a backscattering geometry and passed through a blue interference filter (HQ480/100M, Chroma Tech, Brattleboro, VT). Photon counts were detected with a GaAsP photomultiplier tube (PMT) detector (H7421-40, Hamamatsu). The detector signal was correlated online by a Flex5000/FAST correlator (correlator.com). In the PCH experiments, a Flex02-12D digital correlator was used to record the photon counts with a dwell time of 50 μs and a measurement time of 30 s. The recorded photon counts were stored and then analyzed with LFD Globals Unlimited software (Champaign, IL). A detailed description of the FCS experimental apparatus can be found elsewhere (26).
FIGURE 1.

(A) Experimental setup for two-photon FCS measurements. (B) Flow cytometry cell cycle analysis of the control PTK2 cells and ECFP-Cdc20 expressing PTK2 cells. (C) ECFP-Cdc20 expressing PTK2 cell undergoing mitosis. (D) Average lengths of each phase in mitotic ECFP-Cdc20 expressing PTK2 cells versus that in the control PTK2 cells.
PTK2 cells were seeded into 35-mm coverslip-bottom microwells (MatTek, Ashwell, MA) in Phenol red-free culture medium. The cells were allowed to adhere overnight at 37°C in a 7.5% CO2 incubator. For nocodazole experiments, cells were exposed to 300 nM nocodazole for 2–4 h. Cells in mitosis were identified by phase contrast microscopy for FCS measurements. During the FCS measurements, temperature on the sample stage was 30°C, controlled by an Air Stream Incubator (Nicholson Precision Instruments, Bethesda, MD). The laser intensity was 1.80 mW at the sample to avoid bleaching and photodamage to the cells. Autocorrelation curves measured from cells were averages of 3–4 successive measurements, each 60-s long.
As described in our previous study (26), to evaluate the effect of photobleaching of diffusing ECFP to our recovered diffusion constants, we conducted several FCS measurements in solution with a laser power varying from 1.5 to 5 mW and obtained virtually identical autocorrelation curves. FCS measurements were then performed within a small volume inside the cytosol of a cell with a laser power varying from 1.5 to 2.4 mW. Again we obtained identical autocorrelation curves. The results of the control experiments indicate that there is no detectable photobleaching of diffusing ECFP within the range of incident laser intensity in our experiments.
Data analysis
Any dynamic process that affects the emission of fluorescent molecules in a solution causes fluctuations in the fluorescence signal F(t) that can be characterized by a normalized autocorrelation function:
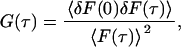 |
(1) |
where F〈t〉 is the fluorescence obtained from the excitation volume at delay time τ, brackets denote ensemble average, and δF(τ) = F(τ) − F〈τ〉. The fitting formula is the standard fit for the three-dimensional multi-component diffusion:
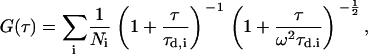 |
(2) |
where Ni is the number of species i, τd,i is the characteristic diffusion time during which an ith species molecule resides in the excitation volume with an axial (z0) to lateral (r0) dimension ratio ω (= z0/r0), and 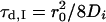 is defined as the average lateral diffusion time under two-photon excitation for an ith species molecule with diffusion coefficient Di through the excitation volume. For a single diffusion species, the average number of molecules N = γ/G(0), with γ of 0.076 (29). Thus the number of photons per molecule per second η (i.e., molecular brightness) can be calculated from the average detected fluorescence intensity with the average number of molecules in the excitation volume. In the presence of the cellular background, the measured correlation function amplitude must be scaled by 〈F(t)〉2/[〈F(t)〉 − 〈FBG〉]2, where 〈FBG〉 is the time-averaged background signal obtained from experiments on nontransfected cells as a control (26,30).
is defined as the average lateral diffusion time under two-photon excitation for an ith species molecule with diffusion coefficient Di through the excitation volume. For a single diffusion species, the average number of molecules N = γ/G(0), with γ of 0.076 (29). Thus the number of photons per molecule per second η (i.e., molecular brightness) can be calculated from the average detected fluorescence intensity with the average number of molecules in the excitation volume. In the presence of the cellular background, the measured correlation function amplitude must be scaled by 〈F(t)〉2/[〈F(t)〉 − 〈FBG〉]2, where 〈FBG〉 is the time-averaged background signal obtained from experiments on nontransfected cells as a control (26,30).
To calculate the rates of changes in the concentration of measured species within mitosis, we recorded the average time span that each phase takes (Fig. 1 D). The rate of changes in concentration is calculated based on the time span of the phases of interest and the corresponding changes in the concentration.
In the case of multiple diffusion species, a photon-counting histogram (PCH) is applied to recover the molecular brightness ηi of each species (31). This method analyzes the probability distribution of the photon counts that is experimentally determined by the PCH, which has the sensitivity to resolve a mixture of monomers and dimers. In practice, PCH represents the probability to detect k photons per sampling time. The probability p(k) to detect k photons from a single diffusing molecule is a weighted average of Poisson distributions, each with the mean value ɛI(r):
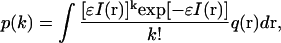 |
(3) |
where I(r) is the point spread function normalized at the origin and q(r) is the probability to find the molecule at the position r. To generalize this equation for N diffusing molecules, I(r) and q(r) must be replaced by 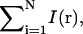 and
and 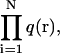 respectively, and the integration is performed over the 3N coordinates of the molecules. Finally, to determine the PCH for an open two-photon excitation volume with a fluctuating number of molecules inside, p(k) is averaged with a Poisson distribution n(N) for the number of molecules:
respectively, and the integration is performed over the 3N coordinates of the molecules. Finally, to determine the PCH for an open two-photon excitation volume with a fluctuating number of molecules inside, p(k) is averaged with a Poisson distribution n(N) for the number of molecules:
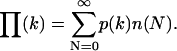 |
(4) |
RESULTS
FCS measurements were conducted within the cytoplasm of interphase and mitotic cells. FCS is able to resolve a mixture of fluorescent species by differences in their diffusion constant especially when the molecular mass between the two species differs more than a factor of 5–8 (32). Therefore, diffusion of the free ECFP-Cdc20 fusion (∼85 kDa) is expected to be much faster than when it is bound to the APC/C (∼1,600 kDa). In addition, the molecular brightness η, another important parameter of the ECFP-Cdc20 fusion, can be determined by FCS and a related technique, PCH (24,31), and allows the determination of the number of Cdc20 molecules per diffusing complex. The knowledge of this is important because, although Cdc20 has been found in the APC/C immunoprecipitates, it has not been quantitatively identified as a stoichiometric component in the APC/C purifications (3).
Lines of PtK2 cells were generated using amphotropic retroviruses to stably introduce a gene encoding ECFP fused in frame with Cdc20. Consistent with previous observations, ECFP-Cdc20 was localized to the cytoplasm and the centrosome of interphase cells (33). As expected from the intermolecular associations of other GFP-Cdc20 fusions that have been shown to retain the functional associations with endogenous partners (33,34), the resulting cells displayed normal cell cycles, advancing through interphase and each step in mitosis with timings that were indistinguishable from the initial cells (Fig. 1, B–D).
Cdc20 is a component of a stable megadalton complex in interphase
Thirteen interphase cells were followed, and FCS measurements were sequentially made on each until it entered mitosis. Autocorrelation functions obtained from cells that entered mitosis within 2 h after FCS measurements were considered to be from cells in the G2 phase of the cell cycle. Other autocorrelation functions obtained between 15 and 6 h before the cells entered mitosis were considered from late G1, S phase or early G2 phase cells, respectively. FCS measurements were also conducted on cells known to be in early G1 since they were made in cells immediately after the abscission of two daughter cells. In all of these interphase cells, Cdc20 behavior could be well modeled by a single three-dimensional diffusion species model. An average cytoplasmic concentration of 76 ± 24 nM was recovered from this analysis, similar to the 100-nM level measured with quantitative immunoblotting for endogenous Cdc20 (11). In addition, an average diffusion constant of 1.8 ± 0.1 μm2/s (Fig. 2) was also obtained which indicates that, in interphase cells, most Cdc20 is stably associated with a large complex. The intracellular diffusion constant of free ECFP (∼30 kDa) was determined to be 21 μm2/s (26). Whereas other FCS measurements have identified complexes in the million dalton (MD) range to have a diffusion constant of 3.2 μm2/s (27), the small 1.8 μm2/s diffusion constant for the ECFP-Cdc20 must indicate that throughout interphase most Cdc20 is found in a large complex with a molecular mass in the range of >1 MDa. Since the APC/C is a high molecular mass complex composed of at least 11 subunits whose molecular mass is estimated to be ∼1.5 MDa (12) and previous studies have demonstrated that Cdc20 binds to interphase APC/C both in vivo and in vitro (10,11), this large Cdc20-containing complex observed in interphase is consistent with its association with a complex containing APC/C, which we will hereinafter refer to as APC/CCdc20.
FIGURE 2.

(A) ECFP-Cdc20 expressing PTK2 cell in early G1 phase. (B) ECFP-Cdc20 expressing PTK2 cell in interphase. (C) The autocorrelation functions from the FCS measurements on interphase cells (G2 and early G1 phase) are fitted with a single species model (bold solid lines), which reveals that Cdc20 is a stable component of a large complex in interphase cells with a diffusion constant of D = 1.8 ± 0.1 μm2/s.
A smaller complex of Cdc20 accumulates early in mitosis and is lost after telophase
FCS measurements were conducted on individual mitotic cells (n = 14) as mitosis progressed. The laser beam was positioned at randomly selected locations in the cytoplasm to avoid chromosomes. In prophase (Fig. 3 A) (before nuclear envelop breakdown), the measured autocorrelation functions were still well fitted by a single species model with an identical diffusion constant to that of interphase cells (∼1.8 μm2/s) (Fig. 3 G), indicating most or all Cdc20 stays associated with a >1 MDa complex consistent with APC/CCdc20. However, as mitosis proceeded beginning with nuclear envelope breakdown (Fig. 3, B–E), the autocorrelation functions exhibited a faster decay (Fig. 3 H), indicating the emergence of a significantly faster diffusing component.
FIGURE 3.

(A) ECFP-Cdc20 cell in prophase. (B) ECFP-Cdc20 cell in prometaphase. (C) ECFP-Cdc20 cell in metaphase. (D) ECFP-Cdc20 cell in anaphase. (E) ECFP-Cdc20 cell in telophase. (F) ECFP-Cdc20 cell in cytokinesis. (G) The autocorrelation functions from the prophase and the cytokinesis phases of mitotic cells indicate that Cdc20 stays bound to the large complex at the beginning and the end of the mitosis, respectively. (H) FCS measurements demonstrate a prominent faster decay of the autocorrelation functions from the onset of prometaphase as a result of the emergence of a faster diffusion component. The autocorrelation functions need to be fitted with a two species model with D1 (D = 19.5 ± 5.1 μm2/s) and D2, the large complex already observed in interphase (D = 1.7 ± 0.2 μm2/s). Starting from the transition between late prometaphase and metaphase, the decay of the autocorrelation functions reverses and continually slows down until cytokinesis, where the autocorrelation functions can be adequately analyzed by a single species model. The gray solid lines show a single species model fit for prophase and cytokinesis, and the colored bold solid lines represent a two species model for other phases in mitosis.
The autocorrelation functions obtained between prophase and cytokinesis could not be fitted with a single species model. A two species model, however, provided an excellent fit to the experimental evidence (Fig. 3 H), with the first species (D1) with a diffusion constant of 19.5 ± 5.0 μm2/s. As before, a larger complex (D2) was also present with a diffusion constant (1.7 ± 0.2 μm2/s) indistinguishable from the large APC/CCdc20 complex seen in interphase. The abundance of the larger complex D2 decreased steadily from prophase to metaphase and then remained at that level from anaphase to cytokinesis (Fig. 4 A). The smaller complex, D1, was essentially absent during interphase, but gradually increased at a rate of 12 pM/s after nuclear envelope breakdown, reaching a maximum in late prometaphase, when it was ∼½ of total Cdc20 (Fig. 4, B and C).
FIGURE 4.

Concentration of Cdc20 complexes is calculated based on the calibrated excitation volume of our system (26) and Ni obtained through the analysis of autocorrelation functions with Eq. 2. (A) Concentration of large Cdc20 complex (D2) across the cell cycle. (B) Concentration of small Cdc20 complex (D1) across the cell cycle. (C) The changes in the total concentration of Cdc20 across the cell cycle. The solid line represents the temporal pattern of APC/CCdc20 activities described in previous review (4). (D) The fluorescence intensity was monitored during the FCS measurements. It shows that in early mitosis the fluorescence intensity is stable, indicating a stable abundance of Cdc20 in the cells. The decrease in the intensity from late prometaphase reflects the degradation of Cdc20 during the mitotic exit.
Constant fluorescence intensity demonstrated that Cdc20 levels were stable from prophase until late prometaphase (Fig. 4 D), indicating that the smaller D1 complex may result from Cdc20 dissociating from the large APC/CCdc20 complex. At late prometaphase/metaphase, however, the fluorescence intensity of ECFP-Cdc20 decreased, reaching a level by cytokinesis that was only half that in early mitosis (Fig. 4 D). This loss was not due to repetitive photobleaching since fluorescence intensity recorded with a 2-s measurement time (compared to 3–4 min for a FCS measurement) in prophase followed by a second measurement at anaphase or telophase revealed the same changes (data not shown). The small complex D1 progressively decreased at a rate of 31 pM/s, becoming undetectable by cytokinesis (Fig. 4, B and C), as manifested by the continuous loss of the fast decay component in the autocorrelation functions until cytokinesis, where the autocorrelation curves were again well fitted with a single species model with a diffusion constant ∼1.8 μm2/s (Fig. 3, G and H). Previous real time imaging had reported Cdc20 proteolysis during mitotic exit (33); our evidence reveals that it is the small (D1) species that is exclusively lost after anaphase onset.
To determine the abundances of the two Cdc20 complexes during maximally activated mitotic checkpoint signaling, microtubules in mitotic cells (n = 4) were disassembled by treatment with nocodazole for 2–4 h, a time sufficient for complete APC/CCdc20-mediated degradation of cyclin A (2–4 times the length required for these cells to normally enter and complete mitosis, as shown in Fig. 1 D). FCS measurements on these cells revealed a significant amount (∼25 nM) of the APC/C-free D1 complex of Cdc20 (16 ± 6 μm2/s) (Fig. 4 B).
Only one ECFP-Cdc20 exists in each Cdc20-containing complex throughout the cell cycle
Recent studies have demonstrated that analysis of the molecular brightness η (number of photons emitted per molecule per second) can identify the oligomerization state of proteins (24,26,27). This is because a dimer appears twice as bright as the monomer so that the molecular brightness of a dimer will be twice that of a monomer.
The molecular brightness η of ECFP-Cdc20 in interphase cells was determined to be 3500 ± 400 cpsm, essentially indistinguishable from our previous determination for monomeric ECFP (3400 ± 100 cpsm) (26). Since each Cdc20 protein is genetically tagged with one ECFP molecule, this indicates that each large complex in interphase carries only one ECFP-Cdc20.
For mitotic cells in which the autocorrelation functions could only be fitted with a two species model, the amplitude of an autocorrelation curve G(0) does not have the simple relationship to the number of diffusing molecules as it does in a single species model (29). To recover the molecular brightness of D1 and D2, we used a related technology-PCH (31). PCH measurements were conducted first on nontransfected control cells to evaluate the effects of autofluorescence of the cells. PCH analysis yielded a molecular brightness of 270 ± 30 cpsm and 7 ± 2 autofluorescent molecules in the excitation volume. The PCH measurements were then conducted on ECFP-Cdc20 cells at each mitotic stage. For the determination of the molecular brightness of D1 and D2, autofluorescence was accounted for by including the autofluorescent molecules in the PCH analysis using the average molecular brightness and number of autofluorescent molecules obtained from uninfected control cells. Although the proportions of the large and small complexes vary significantly at different mitotic stages (Fig. 4, A and B), this analysis recovered a single molecular brightness value of 3400 ± 600 cpsm throughout the mitosis (Fig. 5). Therefore, the transient small complex (D1) and the APC/CCdc20 (D2) complex present throughout the cell cycle contain a single molecule of Cdc20.
FIGURE 5.

PCH of mitotic ECFP-Cdc20 cells. The symbols represent the experimental data; the solid line shows fits with a single component model, indicating that there is a single molecular brightness for ECFP-Cdc20 regardless of the ECFP-Cdc20 complex.
DISCUSSION
In this study, we demonstrated that FCS is an important tool for quantitative investigations of in vivo dynamic intracellular processes via the real time observation of the evolution of the biochemical assembly states of Cdc20 throughout the entire cell cycle. We found that Cdc20 is stoichiometrically complexed to form APC/CCdc20 throughout interphase with a diffusion constant of 1.8 ± 0.1 μm2/s and a concentration of 76 ± 24 nM. After mitotic entry, as much as half of all Cdc20 is released into a much smaller complex (or complexes) at a rate of 12 pM/s, which peaks at metaphase (∼40 nM) before quantitative loss by telophase at a rate of 31 × 10−3 nM/s. This APC/C-free Cdc20 has a much faster diffusion constant (19.5 ± 5.0 μm2/s), consistent with either free monomeric Cdc20 or a small Cdc20-containing complex. Both APC/CCdc20 and the small Cdc20 species contain only a single molecule of Cdc20. The emergence of the smaller species cannot result from photodynamic flickering, a phenomenon in which a faster decay component emerges relative to molecular diffusion due to the proton displacement inside the molecular structure, because ECFP does not exhibit such proton-driven flickering (26). The quantitative information of the dynamic behavior of the Cdc20 species obtained in this study provides significant insights into the underlying mechanisms regulating the activity of APC/CCdc20 throughout the cell cycle.
Inhibition of interphase APC/CCdc20 complex
Our FCS measurements have demonstrated that interphase Cdc20 is in a megadalton complex consistent to the APC/CCdc20. Recent work has identified Emi1/Rcal as a protein that inhibits APC/CCdc20 in interphase (16,17,35,36). However, how Emi1 inhibits APC/CCdc20 in interphase is not well understood. Fractionation experiments have shown separate Emi1-Cdc20 and APC/CCdc20 complexes in egg extracts, suggesting a model in which Cdc20 is sequestered away from the APC/C by Emi1 (16). On the other hand, exogenously added Emi1 can inhibit the APC/C already associated with Cdc20 in mitotic egg extracts, leading to a direct inhibition model (16). The molecular mass of free ECFP-Cdc20 and ECFP-Cdc20-Emi1 is 85 kDa and 135 kDa, respectively, whereas the mass of APC/C is ∼1.5 MD. If the interaction between Cdc20 and APC/C is dynamic to produce a significant steady-state concentration of Cdc20 unbound to APC/C, a much faster and smaller component should have been detected in our analysis of the autocorrelation functions in view of the significant difference in mass between APC/CCdc20 and free Cdc20 or Cdc20-Emi1. Our evidence in interphase indicates that most of the Cdc20 is associated with APC/C to form APC/CCdc20 in interphase. Considering the possibility that small Cdc20 complexes could be present in interphase at concentrations below the detection limit of FCS, we conclude that Emi1 acting primarily by directly inhibiting the APC/C already associated with Cdc20 should be the predominant mechanism of the inhibition of APC/CCdc20.
Mitotic exit switching from APC/CCdc20 to APC/CCdh1
It is accepted that Cdc20 is degraded through APC/CCdh1 ubiquitination at the end of mitosis and thereby APC/CCdc20 is inactivated (37–39). However, details related to this mechanism regulating the switch from APC/CCdc20 to APC/CCdh1 are still not entirely clear (5). For example, biochemical analysis has revealed that a considerable amount of Cdc20 survives this mitotic degradation and enters the next round of the cell cycle (10,40). Our data demonstrate the degradation of the Cdc20 not bound to APC/C at the end of mitosis, indicating that APC/CCdh1 apparently targets only the APC/C-free Cdc20 at mitotic exit.
Implications for the regulation of APC/CCdc20 in mitosis
The temporal pattern of APC/CCdc20 activity has been well established (4,5,41) (also shown in Fig. 4 C). APC/CCdc20 is activated at the onset of prometaphase when it initiates the degradation of cyclin A (42–44) and Nek2A (45). Its activity reaches a maximum, plateauing between late prometaphase and anaphase. Its activity is then gradually reduced, becoming inactivated at the end of mitosis. To this, our data demonstrate the emergence of APC/C-free Cdc20 at the onset of prometaphase, yielding a maximization quantity during late prometaphase and metaphase before its disappearance at the end of mitosis. The presence and the disappearance of this APC/C-free Cdc20 are closely correlated with the temporal pattern of the APC/CCdc20 activity in mitosis (Fig. 4, B and C). This raises the possibility that this APC-free Cdc20 plays a critical role in the regulation of APC/CCdc20 activity.
A central question in mitosis is whether Cdc20 serves as a substrate recruiter while being an essential, stoichiometric component of the APC/C or as a kiss-and-run facilitator that identifies substrates, brings them back to the APC/C, and then releases to begin another cycle of substrate binding and recruitment (13). The concurrence of the appearance of APC/C-free Cdc20 and the activation of APC/CCdc20 suggests that, after the rapid phosphorylation and degradation of Emi1 early in mitosis, this small Cdc20 complex is released from the APC/C for the recruitment of cyclin A for its ubiquitination by APC/C and subsequent degradation.
A second key unresolved issue in mitosis is how APC/CCdc20 is inactivated for securin and cyclin B recognition by actively signaling the mitotic checkpoint when all the while cyclin A is being ubiquitinated. Two competing models have been proposed, namely, sequestration of Cdc20 and direct inhibition of Cdc20 already bound to APC/C (10,12,14,15). Appearance of the small Cdc20 component when the mitotic checkpoint is maximally active is consistent with the recruiter for substrates to the APC/C, due to the dynamic interaction. Our data indicate that the small Cdc20 is neither sequestered from the APC/C nor bound to the APC/C as its stoichiometric component, even when the mitotic checkpoint is maximally signaling. Future studies are needed to elucidate the detailed mechanisms of interaction between checkpoint proteins. This will require a combination of approaches, including FCS dual-color cross correlation spectroscopy as an essential complement to the more frequently used methods focused on biochemistry and molecular biology.
Acknowledgments
The authors thank Dr. Zhongping Chen for his help in developing the FCS system. Z.W. thanks Dr. C. H. Sun for helpful discussion, L. Li and A. Stacy for assistance in the experiments, Dr. J. L. Marsh for his help during the course of this work, and Dr. E. Gratton for providing LFD Global Unlimited software for PCH data analysis. J.V.S. thanks Drs. Robert Hagan and Peter Sorger for providing the human Cdc20 cDNA, Frank Furnari for assistance in developing the retroviral system, and the University of California, San Diego, Cancer Center Flow Cytometry Facility for cell sorting.
This work was supported by National Institutes of Health Grant GM66051 (partially to Z.W.), National Institutes of Health Grant RR-14892 and Air Force Grant F49620 (to M.B.), and National Institutes of Health Grant GM29513 (to D.W.C.). D.W.C. receives salary support from the Ludwig Institute for Cancer Research.
Zifu Wang and Jagesh V. Shah contributed equally to this work.
Jagesh V. Shah's present address is Dept. of Systems Biology, Harvard Medical School, Boston, MA.
References
- 1.Minshull, J., J. J. Blow, and T. Hunt. 1989. Translation of cyclin messenger-RNA is necessary for extracts of activated xenopus eggs to enter mitosis. Cell. 56:947–956. [DOI] [PubMed] [Google Scholar]
- 2.Murray, A. W., and M. W. Kirschner. 1989. Cyclin synthesis drives the early embryonic-cell cycle. Nature. 339:275–280. [DOI] [PubMed] [Google Scholar]
- 3.Page, A. M., and P. Hieter. 1999. The anaphase-promoting complex: new subunits and regulators. Annu. Rev. Biochem. 68:583–609. [DOI] [PubMed] [Google Scholar]
- 4.Peters, J. M. 2002. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell. 9:931–943. [DOI] [PubMed] [Google Scholar]
- 5.Harper, J. W., J. L. Burton, and M. J. Solomon. 2002. The anaphase-promoting complex: it's not just for mitosis any more. Genes Dev. 16:2179–2206. [DOI] [PubMed] [Google Scholar]
- 6.Shah, J. V., and D. W. Cleveland. 2000. Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell. 103:997–1000. [DOI] [PubMed] [Google Scholar]
- 7.Glotzer, M., A. W. Murray, and M. W. Kirschner. 1991. Cyclin is degraded by the ubiquitin pathway. Nature. 349:132–138. [DOI] [PubMed] [Google Scholar]
- 8.Fang, G. W., H. T. Yu, and M. W. Kirschner. 1998. Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell. 2:163–171. [DOI] [PubMed] [Google Scholar]
- 9.Tang, Z., H. Shu, D. Oncel, S. Chen, and H. Yu. 2004. Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol. Cell. 16:387–397. [DOI] [PubMed] [Google Scholar]
- 10.Fang, G. W., H. T. Yu, and M. W. Kirschner. 1998. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 12:1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang, Z. Y., R. Bharadwaj, B. Li, and H. T. Yu. 2001. Mad2-independent inhibition of APC(Cdc20) by the mitotic checkpoint protein BubR1. Dev. Cell. 1:227–237. [DOI] [PubMed] [Google Scholar]
- 12.Sudakin, V., G. K. T. Chan, and T. J. Yen. 2001. Checkpoint inhibition of the APC/C in Hela Cells is mediated by a complex of BURB1, BUB3, CDC20, and MAD2. J. Cell Biol. 154:925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray, A. W. 2004. Recycling the cell cycle: cyclins revisited. Cell. 116:221–234. [DOI] [PubMed] [Google Scholar]
- 14.Chen, R. H., A. Shevchenko, M. Mann, and A. W. Murray. 1998. Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J. Cell Biol. 143:283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan, G. K., and T. J. Yen. 2003. The mitotic checkpoint: a signaling pathway that allows a single unattached kinetochore to inhibit mitotic exit. In Progress in Cell Cycle Research. L. Meijer, A. Jezequel, and M. Roberge, editors. Springer, New York. 431–439. [PubMed]
- 16.Reimann, J. D. R., E. Freed, J. Y. Hsu, E. R. Kramer, J. M. Peters, and P. K. Jackson. 2001. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell. 105:645–655. [DOI] [PubMed] [Google Scholar]
- 17.Hsu, J. Y., J. D. R. Reimann, C. S. Sorensen, J. Lukas, and P. K. Jackson. 2002. E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC(Cdh1). Nat. Cell Biol. 4:358–366. [DOI] [PubMed] [Google Scholar]
- 18.Yu, H. T. 2002. Regulation of APC-Cdc20 by the spindle checkpoint. Curr. Opin. Cell Biol. 14:706–714. [DOI] [PubMed] [Google Scholar]
- 19.Elson, E. L., and D. Magde. 1974. Fluorescence correlation spectroscopy. 1. Conceptual basis and theory. Biopolymers. 13:1–27. [DOI] [PubMed] [Google Scholar]
- 20.Magde, D., W. W. Webb, and E. Elson. 1972. Thermodynamic fluctuations in a reacting system—measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett. 29:705–708. [Google Scholar]
- 21.Berland, K. M., P. T. C. So, and E. Gratton. 1995. 2-Photon fluorescence correlation spectroscopy—method and application to the intracellular environment. Biophys. J. 68:694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brock, R., M. A. Hink, and T. M. Jovin. 1998. Fluorescence correlation microscopy of cells in the presence of autofluorescence. Biophys. J. 75:2547–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwille, P., U. Haupts, S. Maiti, and W. W. Webb. 1999. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys. J. 77:2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen, Y., J. D. Muller, Q. Q. Ruan, and E. Gratton. 2002. Molecular brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys. J. 82:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohler, R. H., P. Schwille, W. W. Webb, and M. R. Hanson. 2000. Active protein transport through plastid tubules: velocity quantified by fluorescence correlation spectroscopy. J. Cell Sci. 113:3921–3930. [DOI] [PubMed] [Google Scholar]
- 26.Wang, Z., J. V. Shah, Z. Chen, C. H. Sun, and M. W. Berns. 2004. Fluorescence correlation spectroscopy investigation of a GFP mutant-enhanced cyan fluorescent protein and its tubulin fusion in living cells with two-photon excitation. J. Biomed. Opt. 9:395–403. [DOI] [PubMed] [Google Scholar]
- 27.Larson, D. R., Y. M. Ma, V. M. Vogt, and W. W. Webb. 2003. Direct measurement of Gag-Gag interaction during retrovirus assembly with FRET and fluorescence correlation spectroscopy. J. Cell Biol. 162:1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah, J. V., E. Botvinick, Z. Bonday, F. Furnari, M. Berns, and D. W. Cleveland. 2004. Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr. Biol. 14:942–952. [DOI] [PubMed] [Google Scholar]
- 29.Thompson, N. L. 1991. Fluorescence correlation spectroscopy. In Topics in Fluorescence Spectroscopy. J. R. Lakowicz, editor. Plenum, New York. 337–378.
- 30.Hess, S. T., S. H. Huang, A. A. Heikal, and W. W. Webb. 2002. Biological and chemical applications of fluorescence correlation spectroscopy: a review. Biochemistry. 41:697–705. [DOI] [PubMed] [Google Scholar]
- 31.Chen, Y., J. D. Muller, K. M. Berland, and E. Gratton. 1999. Fluorescence fluctuation spectroscopy. Methods. 19:234–252. [DOI] [PubMed] [Google Scholar]
- 32.Meseth, U., T. Wohland, R. Rigler, and H. Vogel. 1999. Resolution of fluorescence correlation measurements. Biophys. J. 76:1619–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kallio, M. J., V. A. Beardmore, J. Weinstein, and G. J. Gorbsky. 2002. Rapid microtubule-independent dynamics of Cdc20 at kinetochores and centrosomes in mammalian cells. J. Cell Biol. 158:841–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howell, B. J., B. Moree, E. M. Farrar, S. Stewart, G. Fang, and E. D. Salmon. 2004. Spindle checkpoint protein dynamics at kinetochores in living cells. Curr. Biol. 14:953–964. [DOI] [PubMed] [Google Scholar]
- 35.Dong, X. Z., K. H. Zavitz, B. J. Thomas, M. Lin, S. Campbell, and S. L. Zipursky. 1997. Control of G1 in the developing Drosophila eye: rca1 regulates Cyclin A. Genes Dev. 11:94–105. [DOI] [PubMed] [Google Scholar]
- 36.Reimann, J. D. R., B. E. Gardner, F. Margottin-Goguet, and P. K. Jackson. 2001. Emi1 regulates the anaphase-promoting complex by a different mechanism than Mad2 proteins. Genes Dev. 15:3278–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfleger, C. M., and M. W. Kirschner. 2000. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 14:655–665. [PMC free article] [PubMed] [Google Scholar]
- 38.Prinz, S., E. S. Hwang, R. Visintin, and A. Amon. 1998. The regulation of Cdc20 proteolysis reveals a role for the APC components Cdc23 and Cdc27 during S phase and early mitosis. Curr. Biol. 8:750–760. [DOI] [PubMed] [Google Scholar]
- 39.Shirayama, M., W. Zachariae, R. Ciosk, and K. Nasmyth. 1998. The polo-like kinase Cdc5p and the WD-repeat protein Cdc2Op/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J. 17:1336–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang, Y. K., and E. Lees. 2001. Identification of an overlapping binding domain on Cdc20 for Mad2 and anaphase-promoting complex: model for spindle checkpoint regulation. Mol. Cell. Biol. 21:5190–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zachariae, W., and K. Nasmyth. 1999. Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev. 13:2039–2058. [DOI] [PubMed] [Google Scholar]
- 42.den Elzen, N., and J. Pines. 2001. Cyclin a is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol. 153:121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geley, S., E. Kramer, C. Gieffers, J. Gannon, J. M. Peters, and T. Hunt. 2001. Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin a starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol. 153:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dawson, I. A., S. Roth, and S. Artavanistsakonas. 1995. The Drosophila cell cycle gene fizzy is required for normal degradation of cyclins A and B during mitosis and has homology to the CDC20 gene of Saccharomyces-cerevisiae. J. Cell Biol. 129:725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hames, R. S., S. L. Wattam, H. Yamano, R. Bacchieri, and A. M. Fry. 2001. APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J. 20:7117–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]