

Abstract
A relationship between nelfinavir antiretroviral efficacy and plasma concentrations has been previously established. As physiological changes associated with pregnancy have a large impact on the pharmacokinetics of many drugs, a nelfinavir population study with women was developed, and the large intersubject variability was analyzed in order to optimize individual treatment schedules for this drug during pregnancy. A population pharmacokinetic model was developed in order to describe the concentration time course of nelfinavir and its metabolite M8 in pregnant and nonpregnant women. Individual characteristics, such as age, body weight, and weeks of gestation or delivery, which may influence nelfinavir-M8 pharmacokinetics were investigated. Data from therapeutic drug monitoring in 133 women treated with nelfinavir were retrospectively analyzed with NONMEM. Nelfinavir pharmacokinetics was described by a one-compartment model with linear absorption and elimination and M8 produced from the nelfinavir central compartment. Mean pharmacokinetic estimates and the corresponding intersubject percent variabilities for a nonpregnant woman were the following: absorption rate, 0.83 h−1; absorption lag time, 0.85 h; apparent nelfinavir elimination clearance (CL10/F), 35.5 liters/h (50%); apparent volume of distribution (V/F), 596 liters (118%); apparent formation clearance to M8 (CL1M/F), 0.65 liters/h (69%); and M8 elimination rate constant (kM0), 3.3 h−1 (59%). During pregnancy, we observed significant increases in nelfinavir (44.4 liters/h) and M8 (5 h−1) elimination but unchanged nelfinavir transformation clearance to M8, suggesting an induction of CYP3A4 but no effect on CYP2C19. Apparent nelfinavir clearance and volume showed a twofold increase on the day of delivery, suggesting a decrease in bioavailability on this day. The M8 elimination rate was increased by concomitant administration of nonnucleoside reverse transcriptase inhibitors. A trough nelfinavir plasma concentration above 1 mg/liter was previously shown to improve the antiretroviral response. The Bayesian individual pharmacokinetic estimates suggested that the dosage should not be changed in pregnant women but may be doubled on the day of delivery.
Nelfinavir is a human immunodeficiency virus (HIV) protease inhibitor used in highly active antiretroviral therapy. It has been widely used in pregnancy as therapy for the woman's own HIV infection as well as for the prevention of mother-to-child transmission because of its tolerability and potency. However, the physiological changes associated with pregnancy can lead to important variations in pharmacokinetic processes of absorption, distribution, and elimination. The oral bioavailability of nelfinavir is highly variable in adults (20 to 80%) and increases two- to threefold when administered with food (Viracept package insert, 2001; Agouron Pharmaceuticals, La Jolla, Calif.). The nelfinavir apparent volume of distribution is 2 to 7 liters/kg of body weight. Nelfinavir is metabolized into the active metabolite hydroxy-tert-butylamide (M8) via the CYP2C19 enzyme, and both drugs are metabolized via CYP3A4 (14, 25). It was reported that pregnant women have lower nelfinavir plasma concentrations than nonpregnant women. Nellen et al. (19) found that the mean nelfinavir concentration ratio was significantly lower in pregnant women. Van Heeswijk et al. (24) reported a significant reduction in nelfinavir minimum plasma concentration, supported by an Angel et al. (2) study showing a 2.5-fold increase in nelfinavir apparent oral clearance. However, these studies included a small number of pregnant women. A population pharmacokinetic study was performed in order to confirm the results in a larger number of pregnant women, study the influence of covariates (such as weight and pregnancy duration), and assess whether changes during pregnancy and delivery affect nelfinavir pharmacokinetics to such an extent that dose adjustments should be considered.
The aim of the present study was to characterize the nelfinavir and M8 pharmacokinetics in pregnant women and during delivery. This was achieved by (i) developing an integrated pharmacokinetic model to simultaneously describe the nelfinavir and M8 pharmacokinetics and (ii) using a pharmacostatistic model to identify the patient characteristics that can influence nelfinavir and M8 pharmacokinetics. Such results should be useful to optimize nelfinavir treatment, since a significant relationship has already been demonstrated between nelfinavir antiretroviral efficacy/safety and minimum plasma concentration (22, 23).
MATERIALS AND METHODS
Patients.
The population included nonpregnant women, pregnant women, and women on the day of delivery receiving oral nelfinavir for treatment of HIV infection and whose antiretroviral drug plasma concentrations were monitored on a routine basis. Nelfinavir was administered as a 750-mg thrice-daily (TID) or a 1,250-mg twice-daily (BID) regimen using 250-mg tablets. For each woman, the time elapsed between administration and sampling times, time of dosing (a.m. or p.m.), body weight (BW), age, weeks of gestation, and combined treatments, particularly other antiretroviral drugs, were carefully recorded. When repeated low compliance was suspected by the clinician or by the pharmacologist (undetectable plasma concentrations of nelfinavir and M8), the data were not included in the analysis. When the times elapsed between administration and sampling were longer than 15 h for BID and 11 h for TID regimens, samples were not included in the analysis. The four women who were administered ritonavir associated with nelfinavir were excluded from the analysis. Ethics committee approval and patient consent are not compulsory in France in order to retrospectively use therapeutic drug monitoring data, so for nonpregnant women, no informed consent was collected. All pregnant women were enrolled with informed consent in the French Perinatal Cohort, as approved by the institutional review board.
Analytical method.
Nelfinavir and M8 plasma concentrations were measured by high-pressure liquid chromatography. Briefly, the method involved the extraction of the drugs and the internal standard (clazepam SL 72469) from 200 μl of plasma with a 6-ml mixture of ethyl acetate-hexane (vol/vol) in alkaline medium (0.2 M sodium carbonate; 0.5 ml). After evaporation, the residue was dissolved in eluent consisting of acetonitrile-perchlorate tetramethyl ammonium (0.01 M) in trifluoroacetic acid (0.01%) (37:63, vol/vol). Chromatography was performed using a reverse-phase C8 analytical column (Nucleosil C8; 125 by 4.6 mm, 3 μm; Macherey-Nagel) and gradient elution with an increase of acetonitrile from 37 to 45%. UV detection at 205 nm was used. Linearity of the method was obtained in the concentration ranges of 0.2 to 20 mg/liter and 0.05 to 8 mg/liter for nelfinavir and M8, respectively. Based on standard samples, interday accuracy for the two analytes ranged from 92.9 to 97.6%, and based on quality control samples, interday precision expressed as a percent coefficient of variation was less than 10%.
Population pharmacokinetic modeling of nelfinavir and M8.
Concentrations that were beyond the limit of quantification were set to half the limit of quantification (i.e., 0.1 mg/liter for nelfinavir and 0.025 mg/liter for M8) (4). Data were analyzed using the nonlinear mixed effect modeling software program NONMEM (version V, level 1.1, double precision) with the DIGITAL FORTRAN compiler (5). The first-order conditional estimation method was used. The pharmacokinetics of nelfinavir and M8 were studied sequentially. Nelfinavir data were first analyzed according to a one-compartment open model. Nelfinavir concentrations versus time were fitted using the NONMEM subroutine ADVAN2 TRANS2. Parameters of the model were the absorption rate constant (ka), absorption lag time (Tlag), apparent distribution volume (V/F), and elimination clearance (CL/F). The pharmacokinetic parameters of the parent compound were then used to produce the input function into the metabolite compartment (Fig. 1). Parameters of the nelfinavir-M8 model were the absorption rate constant (ka), absorption lag time (Tlag), apparent distribution volume of nelfinavir (V/F), apparent nelfinavir elimination clearance (CL10/F), M8 apparent formation clearance (CL1M/F), and M8 elimination rate constant (kM0), where F is the bioavailability fraction. The M8 distribution volume was not identifiable. The equations for the metabolite pharmacokinetics, used in a $PRED section in NONMEM, are derived in the Appendix.
FIG. 1.

Pharmacokinetic compartment model for the simultaneous prediction of nelfinavir and M8 plasma concentration after nelfinavir oral dose. Nelfinavir (in compartment 1) undergoes irreversible biotransformation to produce M8 (in compartment 2). Ka denotes the absorption rate constant, V the nelfinavir distribution volume, CL10 the nelfinavir elimination clearance, CL1M the nelfinavir-to-M8 formation clearance, and KM0 the M8 elimination rate constant.
Several error models were investigated (i.e., exponential, proportional, and additive error models) to describe intersubject and residual variabilities.
The influence of each patient covariate was systematically tested via a generalized additive modeling according to the following equation using CL: for example, CL = TV(CL) × (BW/median BW)θBW, where TV(CL) is the typical value of clearance for a patient with the median covariate value and θBW is the estimated influential factor for body weight. Such covariates included age and body weight.
Categorical covariates (CC), including pregnancy (PREG), delivery (DEL), diurnal variation in nelfinavir disposition, and combined antiretroviral drugs, were tested using an inducing drug effect: for example, CL = TV(CL) × [1 + θCC × (CC = 0 or 1)], or in the case of an inhibitory drug effect, CL = TV(CL)/[1 + θCC × (CC = 0 or 1)], where θCC is the estimated influential factor for the categorical covariate.
Weeks gestation (GW) was tested in pregnant women as a combination of the two previous equations: CL = TV(CL) × [1 + θPREG × (PREG = 0 or 1) × (GW/median GW)θGW], where θGW and θPREG are the estimated influential factors for weeks gestation and pregnancy, respectively. On delivery day, the coding was 0 for pregnancy (PREG = 0) and 1 for delivery (DEL = 1).
Covariates were selected in the final population model if (i) their effect was biologically plausible, (ii) they produced a minimum reduction of 4 in the objective function value (OFV), and (iii) they produced a reduction in the variability of the pharmacokinetic parameter, assessed by the associated intersubject variability. An intermediate multivariate model including all significant covariates was then obtained. In order to keep only those covariates with the largest contribution in the final multivariate model, a change of 7 (P < 0.01, 1 degree of freedom) of the objective function was required for the retention of a single parameter during backward stepwise multiple regression analysis.
For evaluation of the goodness of fit, the following graphs were compared: observed and predicted concentrations versus time, observed concentrations versus predictions, weighted residuals versus time, and weighted residuals versus predictions, as well as the corresponding graphs issued from the POSTHOC estimation step. Diagnostic graphics and distribution statistics were obtained using the R program (10).
Bootstrap validation.
The accuracy and robustness of the final population model were assessed using a bootstrap method, as previously described in detail (20). Briefly, this includes the following steps. (i) From the original data set of n individuals, B bootstrap sets (B = 1,000) of n individuals are drawn with replacement (resampling). (ii) For each of the B bootstrap sets, the population pharmacokinetic parameters are estimated. (iii) With the B estimates of each population pharmacokinetic parameter, the corresponding mean and standard deviation are estimated. (iv) To validate the model, the parameters estimated from the bootstrap must be close to estimates obtained from the original population set. The entire procedure was performed in an automated fashion using Wings for NONMEM (http://wfn.sourceforge.net/wfninst.htm). This procedure also provided nonparametric statistics (median and 2.5th and 97.5th percentiles) of the population parameters.
Individual minimum plasma concentrations.
Individual pharmacokinetic parameters obtained from the POSTHOC option of NONMEM were used to calculate the daily dosage to obtain a minimum plasma concentration of 1 mg/liter (22). We considered twice daily (BID) and thrice daily (TID) regimens for nonpregnant women, pregnant women, and women on the day of delivery. For all of the women, the daily dose needed to obtain a minimum plasma concentration above 1 mg/liter was simulated with an administration every 8 and every 12 h. For each group and regimen, a cumulative curve was drawn to show for a given daily dose the percentage of women with a minimum plasma concentration above 1 mg/liter. Current nelfinavir doses (FDA recommendations) of 1,250 mg BID and 750 mg TID were evaluated.
RESULTS
Demographic data.
One hundred thirty-three women (259 samples) were available for pharmacokinetic evaluation: 69 women were not pregnant (129 samples), 60 were pregnant (87 samples), and 42 were at the time of delivery (43 samples). Six samples out of 265 were excluded: times elapsed between administration and sampling were up to 15 h for BID or 11 h for TID. Table 1 summarizes patient characteristics: age, weight, regimen, percentage of dosing after the morning dose, distribution of dosing time, and other concomitant medications, broken down by pregnancy group. The nine samples from four women who were coadministered ritonavir were excluded from the analysis. All plasma samples were collected at steady state.
TABLE 1.
Patient characteristics
| Characteristic | Nonpregnant women | Pregnant women | Women on the day of delivery |
|---|---|---|---|
| Age (yr) | 33.4 ± 4.9 | 32.6 ± 4.9 | 33.3 ± 4.6 |
| Weight (kg) | 64 ± 15 | 70 ± 16 | 71 ± 12 |
| BID regimen (%) | 75 | 87 | 90 |
| % Dosing after morning dose | 60 | 70 | 72 |
| Distribution of dosing time (h) | 6.1 ± 4.7 | 6.0 ± 4.4 | 6.6 ± 4.1 |
| Saquinavir (no.) | 1 | 2 | 2 |
| Nonnucleoside inhibitors (INN) (no.) | 9 | 4 | 5 |
Population pharmacokinetics. (i) Nelfinavir pharmacokinetic model building.
A one-compartment model adequately described the data. Intersubject and residual variabilities were best described by exponential and additive error models, respectively. The available data were not sufficient to estimate intersubject variability for ka and Tlag, and exclusion of these random effects had no influence on the objective function value. Among all of the covariates, delivery (DEL) had a significant effect on CL10 and V, resulting in 40 and 10 U decreases in the OFV, respectively. Adding the same delivery effect on both V and CL10 resulted in a 48 U decrease in OFV. The use of two different delivery effects for V and CL10 did not improve the OFV. Adding the covariate pregnancy (PREG) also had a significant effect on CL10, resulting in a 7 U OFV decrease. The following equations describe the final covariate model for nelfinavir: delivery effect = [1 + θDEL × (DEL = 0 or 1)]; CL = TV(CL) × delivery effect × [1 + θPREG × (PREG = 0 or 1)]; V = TV(V) × delivery effect.
(ii) M8 pharmacokinetic model building.
The M8 pharmacokinetics was modeled as a metabolite compartment connected to the central compartment (Fig. 1). The nelfinavir pharmacokinetic parameters, including the effect of bioavailability on CL10 and V and pregnancy on CL10, were fixed, and M8 parameters were estimated. The covariate submodeling was then established for M8 formation (CL1M) and elimination (kM0). We did not find that any covariates had a significant effect on CL1M. In the pregnancy group, kM0 significantly increased, resulting in a 12 U decrease in OFV. During pregnancy, kM0 was increased, so those women had a shorter nelfinavir-M8 half-life than nonpregnant women. Adding weight effect and inductor effect of nonnucleoside transcriptase inverse inhibitors (INN) led to 10 and 5 U decreases in OFV, respectively. At this step, the following equation described the final covariate model: kM0 = TV(kM0) × (BW/median BW)θBW × [1 + θPREG × (PREG = 0 or 1)] × [1 + θINN × (INN = 0 or 1)], where θPREG and θINN are the estimated influential factors for pregnancy and INN coadministration, respectively.
(iii) Nelfinavir-M8 pharmacokinetic model building.
Nelfinavir and M8 were simultaneously fitted to the parent-metabolite model, including the covariate submodelings, in order to verify and refine the parameter estimates. This step led to minor changes in the previous estimates. The addition of a significant covariance term between residual variability for nelfinavir and residual variability for M8 (an L2 item was coded in the database for NONMEM) led to a 41 U decrease in OFV. Then covariate deletion was performed to verify the nelfinavir-M8 pharmacokinetic model. Table 2 summarizes changes in OFV from the backward elimination step from the final model. At this step, the following equations described the covariate model: CL10 = 35.5 (±7%) × [1 + 0.25 (±34%) × (PREG = 0 or 1)] × [1 + 2.63 (±20%) × (DEL = 0 or 1)]; V = 596 (±23%) × [1 + 2.63 (±20%) × (DEL = 0 or 1)]; kM0 = 3.3 (±26%) × [1 + 0.51 (±33%) × (PREG = 0 or 1)] × (BW/median BW)1.18 (±26%) × [1 + 1.03 (±37%) × (INN = 0 or 1)], in which the percentages in parentheses denote the coefficients of variation (standard error of estimate/estimate × 100). Table 3 summarizes the final population pharmacokinetic estimates. The correlation coefficient between nelfinavir and M8 residual variability estimates, σnelfinavir and σM8, was 0.47 (30%). Calculating median population pharmacokinetic parameter estimates for each group, apparent nelfinavir elimination clearance was then 35.5 liters/h in nonpregnant women, 44.4 liters/h in pregnant women, and 128.9 liters/h during delivery. The nelfinavir apparent volume of distribution was 596 liters in nonpregnant and pregnant women and 2,163 liters for women on the day of delivery. For a woman weighing 64 kg, the M8 elimination rate constant was 3.3 h−1 in nonpregnant women and women on the day of delivery and 5.0 h−1 in pregnant women. The M8 elimination rate constant increased linearly with weight. These two values were 103% higher when the woman was given a nonnucleoside transcriptase inverse inhibitor.
TABLE 2.
OFV changes from the backward elimination step from the final model
| Covariate deleted | OFV increase |
|---|---|
| None | 0 |
| Delivery on CL10 and V | +79 |
| Pregnancy on CL10 | +7 |
| Weight on kM0 | +13 |
| Pregnancy on kM0 | +31 |
| INN on kM0 | +7 |
TABLE 3.
Population pharmacokinetic parameters of nelfinavir and M8 and bootstrap validationa
| Parameter | Final model with original dataset mean (CV%) | Bootstrapb
|
|
|---|---|---|---|
| Median | 2.5th-97.5th percentiles | ||
| Structural model | |||
| ka (h−1) | 0.83 (16) | 0.84 | 0.15-1.98 |
| Tlag (h−1) | 0.85 (2) | 0.86 | 0.29-1.74 |
| V (liters) | 596 (23) | 564 | 200-978 |
| CL10 (liters/h) | 35.5 (7) | 34.3 | 29.4-39.7 |
| CL10, θPREG | 0.25 (34) | 0.31 | 0.07-0.84 |
| CL10 and V, θDEL | 2.63 (20) | 2.75 | 1.83-4.16 |
| CL1M (liters/h) | 0.65 (22) | 0.73 | 0.34-2.02 |
| kM0 (h−1) | 3.3 (26) | 3.73 | 1.71-10 |
| kM0, θPREG | 0.51 (33) | 0.50 | 0.18-0.87 |
| kM0, θBW | 1.18 (26) | 1.13 | 0.50-1.76 |
| kM0, θINN | 1.03 (37) | 1.10 | 0.50-3.35 |
| Statistical model | |||
| σNELFI (ng/ml) | 1.17 (13) | 1.15 | 0.96-1.33 |
| σM8 (ng/ml) | 0.27 (24) | 0.26 | 0.20-0.33 |
| ω(V) (%) | 118 (37) | 118 | 59-185 |
| ω(CL10) (%) | 50 (20) | 49 | 36-60 |
| ω(CL1M) (%) | 62 (69) | 63 | 36-84 |
| ω(kM0) (%) | 59 (22) | 58 | 45-72 |
CV%, coefficient of variation (standard error of estimate/estimate × 100); ka, absorption rate constant; Tlag, absorption lag time; V, distribution volume of nelfinavir; CL10, nelfinavir elimation clearance; CL1M, nelfinavir-to-M8 formation clearance; kM0, M8 elimination rate constant; kM0, θINN, INN influential factor on kM0; σ, residual variability estimate (CV of residual variability, %); and ω, interindividual variability estimate (CV of intersubject variability, %).
Statistics are from 1,000 bootstrap analyses.
Finally, we tested the assumption that F from 1,250 BID is similar to 750 TID by introducing an effect from a BID regimen compared to TID: CL = TV(CL) × delivery effect × [1 + θPREG × (PREG = 0 or 1)] × (θREGIMEN)BID; V = TV(V) × delivery effect × (θREGIMEN)BID. As we found that θREGIMEN was 1.05 ± 0.17 and there was no modification of the OFV, we concluded that there is a linear bioavailability and dose-independent absorption.
Model performance.
Final model performance can be appreciated by comparing population predicted and observed plasma concentrations versus time (Fig. 2). Bayesian estimates obtained with the POSTHOC option of NONMEM showed a good correlation between individual predicted and observed concentrations: r = 0.91 for nelfinavir and r = 0.92 for M8 (not shown).
FIG. 2.

Predicted (line) and observed (○) nelfinavir (CMT = 2, top) and M8 (CMT = 3, bottom) plasma concentrations versus time after administration. Data for nonpregnant women (ANC = 0, left), pregnant women (ANC = 1, middle), and women at delivery (ANC = 2, right) are shown.
Bootstrap assessment of the final population model.
The final model obtained with the original data set was subjected to a bootstrap analysis. As shown in Table 3, the median and variability of parameter estimates obtained from the bootstrap process—1,000 runs—were reasonably close to the estimates previously obtained with the original data set.
Individual minimum plasma concentrations.
The minimum plasma concentration and the daily dose to obtain the target concentration of 1 mg/liter were calculated using the Bayesian pharmacokinetic estimates (obtained with the POSTHOC option of NONMEM).
The mean minimum plasma concentrations were 2.5 mg/liter in nonpregnant women, 1.5 mg/liter in pregnant women, and 0.6 mg/liter in women on the day of delivery. These three minimum plasma concentrations were significantly different (P < 10−4), but only women at delivery had a minimum plasma concentration lower than the target concentration of 1 mg/liter. For the 2,500-mg/day recommended dosage in adults, 90% of the nonpregnant women (n = 62/69), 85% of the pregnant women (n = 51/60), and 10% of women on the day of delivery (n = 4/42) had a minimum plasma concentration above 1 mg/liter (Fig. 3). These three percentages were significantly different (P < 10−4). Compared two by two, the percentage of women with a minimum plasma concentration above 1 mg/liter was significantly lower on the day of delivery than in pregnant (P < 10−4) and nonpregnant (P < 10−4) women, but we did not find a significant difference between the percentages of nonpregnant and pregnant women (P = 0.43).
FIG. 3.

Percentage of women with a minimum plasma concentration above 1 mg/liter as a function of daily dose and group. Dashed line, nonpregnant women; thin line, pregnant women; and thick line, women on the day of delivery. The vertical dashed line denotes the recommended 2,500-mg dose.
Then the influence of drug regimen (i.e., BID or TID) on minimum plasma concentrations was analyzed in each of the three groups. For nonpregnant women, the predicted minimum plasma concentration was above 1 mg/liter in 94% (n = 65/69) of women receiving 750 mg of drug every 8 h compared to 91% (n = 63/69) of those receiving 1,250 mg every 12 h (Fig. 4). We did not find a significant difference between the BID and TID regimens (P = 0.74). For pregnant women, the predicted minimum plasma concentration was above 1 mg/liter in 95% (n = 57/60) of women when they received 750 mg every 8 h compared to 85% (n = 51/60) when they received 1,250 mg every 12 h. No significant difference between the BID and TID regimens was found (P = 0.13) (Fig. 5). Then we divided pregnant women into two groups, those in the second trimester of pregnancy and those in the third trimester (n = 20 and 67 samples, respectively), and we obtained similar curves for the percentage of women with a minimum plasma concentration above 1 mg/liter.
FIG. 4.

Percentage of the 69 nonpregnant women with a minimum plasma concentration above 1 mg/liter as a function of daily dose and frequency of administration. Solid curve, twice-daily regimen (BID); dotted curve, thrice-daily regimen (TID). The vertical lines denote the minimal FDA-recommended doses: 1,250 mg BID (solid line) or 750 mg TID (dotted line).
FIG. 5.

Percentage of the 60 pregnant women with a minimum plasma concentration above 1 mg/liter as a function of daily dose and frequency of administration. Solid line, twice-daily regimen (BID); dotted line, thrice-daily regimen (TID). The vertical lines denote the minimal FDA-recommended doses: 1,250 mg BID (solid line) or 750 mg TID (dotted line).
At the time of delivery, the predicted minimum plasma concentration was above 1 mg/liter in 7% (n = 3/42) of women receiving 750 mg every 8 h and 10% (n = 4/42) of those receiving 1,250 mg every 12 h (Fig. 6). To obtain the target minimum plasma concentration in most of the women, nelfinavir dosage should be doubled on the day of delivery. A 5,000-mg dose should be given this day, preferably in three administrations (Fig. 6).
FIG. 6.

Percentage of the 42 women on the day of delivery with a minimum plasma concentration above 1 mg/liter as a function of daily dose and frequency of administration. Solid line, twice-daily regimen (BID); dotted line, thrice-daily regimen (TID). The vertical lines denote the minimal FDA-recommended doses: 1,250 mg BID (solid line) or 750 mg TID (dotted line).
DISCUSSION
The nelfinavir-M8 pharmacokinetics was satisfactorily described by the proposed compartmental model. The present study showed a great consistency in the final nelfinavir-M8 population model derived from sequential analyses of nelfinavir and M8, confirming the robustness of the process. The basic one-compartment model used for nelfinavir was already used in adults (22). The pharmacokinetics of the metabolite produced from the parent drug should be described by a two-exponent equation, but the sparse data set did not allow the identification of two exponential components. Only an integrated modeling of parent-metabolite pharmacokinetics can provide a reliable estimate of M8 elimination, since the information for the fast exponential decay is provided by nelfinavir data. Indeed, in this approach, data on the metabolite may add information to the observations on the parent and vice versa.
The following observations support the use of this pharmacokinetic model. (i) The nelfinavir mean plasma clearance in pregnant women (CL10/F = 44.4 liters/h) was consistent with previously reported values: 49.6 liters/h in 11 pregnant women (24) and 56 liters/h in 1 pregnant woman (2). (ii) The nelfinavir apparent plasma clearance (CL10/F) increased by 25% during pregnancy, going from 35.5 liters/h in nonpregnant women to 44.4 liters/h in pregnant women, in agreement with previous studies. Van Heeswijk et al. (24) compared nelfinavir clearance in women postpartum and during pregnancy and reported an increase of the clearance by 33% from 37.3 to 49.6 liters/h. (iii) The median M8-to-nelfinavir concentration ratio was 13% in pregnant women and 23% in nonpregnant women compared to 11.4 and 27.4% during pregnancy and postpartum reported by Van Heeswijk et al. (24). (iv) The final model could be appreciated by the goodness of fit depicted in Fig. 2.
A major aim of population pharmacokinetics is to determine which measurable pathophysiological factor can cause changes in the dose-concentration relationship and to estimate the degree to which it does so, so that an appropriate dose adjustment can be made. This is particularly relevant for nelfinavir in pregnant women because the drug exhibits an appreciable degree of intersubject variability, increased by physiological changes during pregnancy.
In pregnant women, an increase of plasma progesterone, which is believed to increase gastric and intestinal emptying time, could increase nelfinavir absorption variability (21). Moreover, an increase of plasma volume, fat storage, and total body mass could increase the apparent volume of distribution for nelfinavir (12). Nelfinavir binds extensively (98%) to both α-1-acid glycoprotein and albumin in plasma; however, during pregnancy it was shown that protein binding to albumin is decreased and binding to α-1-acid glycoprotein is equivocal (7). Finally, it is known that pregnancy may produce alterations in hepatic drug metabolism (involving the cytochrome 450 isoenzymes) as a possible result of simulated microsomal enzyme activity induced by progesterone (15).
In this study, pregnancy, delivery and coadministration of INN influenced the nelfinavir-M8 pharmacokinetics.
During pregnancy, nelfinavir and M8 elimination (elimination clearance for nelfinavir and elimination rate constant for M8) increased, whereas the M8-to-nelfinavir concentration ratio decreased. Two hypotheses were previously proposed: pregnancy induces CYP3A4 metabolism (CYP3A4 metabolizes M8 and to a lower extent nelfinavir) and/or pregnancy inhibits CYP2C19 metabolism (nelfinavir metabolism to M8 is exclusively mediated by CYP2C19). Kosel et al. (11) reported that in two women who were administered nelfinavir, the 6-β-hydroxycortisol-to-cortisol ratio increased by 40 and 78% between the third trimester and postpartum (a similar report was made for indinavir [8]). These authors suggested that CYP3A4 activity was enhanced during late pregnancy. McGready et al. (17) reported an inhibition of CYP2C19 during late pregnancy, reducing the transformation of proguanil to its active metabolite cycloguanil embonate. In our model, the effect of pregnancy could be tested separately on nelfinavir and M8 elimination and on nelfinavir-to-M8 formation clearance. No significant effect was observed on nelfinavir metabolism to M8, suggesting that pregnancy did not inhibit CYP2C19 metabolism. Significant increases of nelfinavir (24%) and M8 (52%) elimination support the induction of CYP3A4 during pregnancy; nelfinavir is metabolized to a low extent by CYP3A4 while M8 is essentially metabolized by CYP3A4. This difference in pregnancy's effect on nelfinavir and M8 elimination could explain the decrease of the M8-to-nelfinavir concentration ratio (42%).
Previous studies have already shown that exposure to other protease inhibitors, such as indinavir (8, 11), ritonavir, and saquinavir (1), was reduced in pregnant women. During delivery, nelfinavir and M8 concentrations were lower than those during pregnancy and those in nonpregnant women, and those concentrations were very homogeneous, so we don't think that doses were missed because it was the time of delivery. Moreover, a decrease in plasma drug concentrations during delivery has already been reported. Chappuy et al. (6) observed that on this day, most maternal protease inhibitor plasma concentrations were below the trough concentration that was recommended for therapeutic drug monitoring. Marzolini et al. (16) also noted that maternal concentrations of protease inhibitors and nevirapine measured at delivery were lower than those observed in a general HIV-infected population and suggested that the decrease in plasma levels resulted from the nonspecific stimulation of general metabolism and enzyme expression level. Mirochnick et al. (18) reported that women in labor given nevirapine demonstrated an increase in nevirapine half-life and a decrease in bioavailability.
In our study, low nelfinavir and M8 concentrations at delivery could be explained by a decrease in bioavailability. It is recommended to administer nelfinavir with food because oral bioavailability increases two- to threefold. However, HIV-infected women whose delivery is planned should be fasting, and this could explain a decreased nelfinavir bioavailability on this day.
The M8 elimination clearance was doubled in patients treated with INN, consistent with an induction of CYP3A4 by this drug, since M8 is metabolized via CYP3A4 (3). Furthermore, very low and variable M8 elimination was observed for all samples from four women who received ritonavir, a known CYP3A4 inhibitor (13), but these data were too scanty to reach statistical significance. We already showed these effects in a previous study on nelfinavir in children (9).
A minimum nelfinavir plasma concentration above 1 mg/liter was previously shown to improve the antiretroviral response (22). Using a Bayesian approach, we showed that with 2,500 mg daily the percentage of women who had a minimum plasma concentration above 1 mg/liter was not significantly different between nonpregnant and pregnant women but was significantly lower on the day of delivery. Thus, the dosage should not be changed during pregnancy but may be increased on the day of delivery. In nonpregnant women, the regimen (TID or BID) did not much influence the percentage of women with a minimum plasma concentration above 1 mg/liter (94 to 91%), as already shown by Pellegrin et al. (22). For pregnant women, the TID regimen produced a nonsignificantly higher percentage of women with a minimum plasma concentration above 1 mg/liter than the BID regimen (95 to 85%). Nelfinavir dosage should not be increased during pregnancy. At delivery, nelfinavir minimum plasma concentrations were low, suggesting that dosage may be doubled on the day of delivery, probably due to a decrease in bioavailability on this day. This conclusion is based on pharmacokinetic considerations; HIV RNA was not determined this day to confirm this. Also, if viral load is suppressed to <1,000 copies, then it is very unlikely that rebound will occur in a 24-h interval, and thus perinatal transmission would still be unlikely. Even if one increased the dose and maternal nelfinavir exposure, the drug would not get to the infant. There are also potentially increased gastrointestinal intolerability issues which could complicate delivery. One could make the case for more frequent administration (up to 1,250 mg every 6 h), but larger doses would need to be evaluated in a controlled environment.
During pregnancy, important physiological and pharmacokinetic changes occur, and pregnant women are at an increased risk of having subtherapeutic nelfinavir concentrations compared to nonpregnant women. Dosing strategies should be tested to circumvent this.
Acknowledgments
This work was supported by a CRES (contrat de recherche stratégie) from INSERM.
We acknowledge one of the referees for his helpful suggestions.
APPENDIX
The following differential system is connected with the model depicted in Fig. 1: dG/dt = −ka × G, where G = D from t = 0 to t = Tlag; dA/dt = ka × G − K × A, where A = 0 from t = 0 to t = Tlag; dM/dt = CL1M/V × A − kM0 × M, where M = 0 from t = 0 to t = Tlag. G and A denote the nelfinavir amounts in gut and body, M denotes the metabolite amount in the body, ka is the absorption rate, Tlag is the absorption lag time, V is the nelfinavir distribution volume, k = (CL10 + CL1M)/V is the total nelfinavir constant rate, CL10 is the nelfinavir elimination clearance, CL1M is the nelfinavir-to-M8 formation clearance, and kM0 is the M8 elimination rate constant (kM0 = CLM0/Vm, with Vm = 1).
The solution giving the profile of the metabolite compartment is
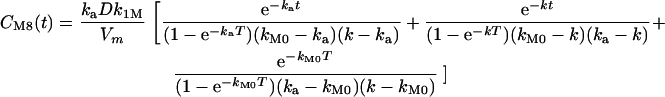 |
where t is the time elapsed between drug administration and blood sampling minus the absorption lag time and T is the time interval between two administrations.
REFERENCES
- 1.Acosta, E. P., A. Bardeguez, C. D. Zorrilla, R. Van Dyke, M. D. Hugues, S. Huang, L. Pompeo, A. M. Stek, J. Pitt, D. H. Watts, E. Smith, E. Jimenez, L. Mofenson, and the Pediatric AIDS Clinical Trials Group 386 Protocol Team. 2004. Pharmacokinetics of saquinavir plus low-dose ritonavir in human immunodeficiency virus-infected pregnant women. Antimicrob. Agents Chemother. 48:430-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angel, J. B., Y. Khaliq, M. L. Monpetit, D. W. Cameron, and K. Gallicano. 2001. An argument for therapeutic drug monitoring of HIV-1 protease inhibitors during pregnancy. AIDS 15:417-419. [DOI] [PubMed] [Google Scholar]
- 3.Baede-van Dijk, P. A., P. W. Hugen, C. P. Verweij-van Wissen, P. P. Koopmans, D. M. Burger, and Y. A. Hekster. 2001. Analysis of variation in plasma concentrations of nelfinavir and its active metabolite M8 in HIV-positive patients. AIDS 15:991-998. [DOI] [PubMed] [Google Scholar]
- 4.Beal, S. L. 2001. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 28:481-504. [DOI] [PubMed] [Google Scholar]
- 5.Beal, S. L., and L. B. Sheiner. 1998. NONMEM user's guide: NONMEM project group. University of California at San Francisco, San Francisco, Calif.
- 6.Chappuy, H., J. M. Treluyer, V. Jullien, J. Dimet, E. Rey, M. Fouché, G. Firtion, G. Pons, and L. Mandelbrot. 2004. Maternal-fetal transfer and amniotic fluid accumulation of protease inhibitors in pregnant women who are infected with immunodeficiency virus. Am. J. Obstet. Gynecol. 191:558-562. [DOI] [PubMed] [Google Scholar]
- 7.Dean, M., B. Stock, R. J. Patterson, and G. Levy. 1980. Serum protein binding of drugs during and after pregnancy in humans. Clin. Pharmacol. Ther. 28:253-260. [DOI] [PubMed] [Google Scholar]
- 8.Hayashi, S., K. P. Beckerman, M. Homma, B. Kosel, and F. Aweeka. 2000. Pharmacokinetics of indinavir in HIV-positive pregnant women. AIDS 14:1061-1063. [DOI] [PubMed] [Google Scholar]
- 9.Hirt, D., S. Urien, V. Jullien, G. Firtion, E. Rey, G. Pons, S. Blanche, and J. M. Treluyer. 2006. Age-related effects on nelfinavir and M8 pharmacokinetics: a population study in 182 children. Antimicrob. Agents Chemother. 50:910-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ihaka, R., and R. Gentleman. 1996. R: a language for data analysis and graphics. J. Comput. Graphic Stat. 5:299. [Google Scholar]
- 11.Kosel, B., K. P. Beckerman, S. Hayashi, M. Homma, and F. Aweeka. 2003. Pharmacokinetics of nelfinavir and indinavir in HIV-1 infected pregnant women. AIDS 17:1195-1199. [DOI] [PubMed] [Google Scholar]
- 12.Krauer, B., F. Krauer, and F. E. Hytten. 1980. Drug disposition and pharmacokinetics in the maternal-placental-fetal unit. Pharmacol. Ther. 10:301-328. [DOI] [PubMed] [Google Scholar]
- 13.Kurowski, M., B. Kaeser, A. Sawyer, M. Popescu, and A. Mrozikiewicz. 2002. Low-dose ritonavir moderately enhances nelfinavir exposure. Clin. Pharmacol. Ther. 72:123-132. [DOI] [PubMed] [Google Scholar]
- 14.Lillibridge, J. H., C. A. Lee, and Y. K. Pithavala. 1998. The role of CYP2C19 in the metabolism of nelfinavir mesylate. Abstr. XII Annu. Meet. Expo. Am. Assoc. Pharm. Sci., San Francisco, Calif., abstr. 3035.
- 15.Loebstein, R., A. Lalkin, and G. Koren. 1997. Pharmacokinetics changes during pregnancy and their clinical relevance. Clin. Pharmacokinet. 33:328-343. [DOI] [PubMed] [Google Scholar]
- 16.Marzolini, C., C. Rudin, L. A. Decosterd, A. Telenti, A. Schreyer, J. Biollaz, T. Buclin, and the Swiss Mother + Child HIV Cohort Study. 2002. Transplacental passage of protease inhibitors at delivery. AIDS 16:889-893. [DOI] [PubMed] [Google Scholar]
- 17.McGready, R., K. Stepniewska, E. Seaton, T. Cho, D. Cho, A. Ginsberg, M. D. Edstein, E. Ashley, S. Looareesuwan, N. J. White, and F. Nosten. 2003. Pregnancy and use of oral contraceptives reduces the biotransformation of proguanil to cycloguanil. Eur. J. Clin. Pharmacol. 59:553-557. [DOI] [PubMed] [Google Scholar]
- 18.Mirochnick, M. 2000. Antiretroviral pharmacology in pregnant women and their newborns. Ann. N. Y. Acad. Sci. 918:287-297. [DOI] [PubMed] [Google Scholar]
- 19.Nellen, J. F., I. Schillevoort, F. W. Wit, A. S. Bergshoeff, M. H. Godfried, K. Boer, J. M. Lange, D. M. Burger, and J. M. Prins. 2004. Nelfinavir plasma concentrations are low during pregnancy. Clin. Infect. Dis. 39:736-740. [DOI] [PubMed] [Google Scholar]
- 20.Parke, J., N. H. Holford, and B. Charles. 1999. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects population models. Comput. Methods Programs Biomed. 59:19. [DOI] [PubMed] [Google Scholar]
- 21.Parry, E., R. Shields, and A. Turnbull. 1970. Transit time in the small intestine in pregnancy. J. Obstet. Gynaecol. Br. Commonw. 77:900-901. [DOI] [PubMed] [Google Scholar]
- 22.Pellegrin, I., D. Breilh, F. Montestruc, A. Caumont, I. Garrigue, P. Morlat, C. Le Camus, M. C. Saux, H. J. Fleury, and J. L. Pellegrin. 2002. Virologic response to nelfinavir-based regimens: pharmacokinetics and drug resistance mutations (VIRAPHAR study). AIDS 16:1331-1340. [DOI] [PubMed] [Google Scholar]
- 23.Treluyer, J. M., J. P. Morini, J. Dimet, I. Gorin, E. Rey, J. Deleuze, P. F. Ceccaldi, J. P. Escande, G. Pons, and N. Dupin. 2002. High concentrations of nelfinavir as an independent risk factor for lipodystrophy in human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 46:4009-4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Heeswijk, R. P. G., Y. Khaliq, K. D. Gallicano, M. Bourbeau, I. Seguin, E. J. Phillips, and D. W. Cameron. 2004. The pharmacokinetics of nelfinavir and M8 during pregnancy and post partum. Clin. Pharmacol. Ther. 76:588-597. [DOI] [PubMed] [Google Scholar]
- 25.Zhang, K. E., E. Wu, A. K. Patick, B. Kerr, M. Zorbas, A. Lankford, T. Kobayashi, Y. Maeda, B. Shetty, and S. Webber. 2001. Circulating metabolites of the human immunodeficiency virus protease inhibitor nelfinavir in humans: structural identification, levels in plasma, and antiviral activities. Antimicrob. Agents Chemother. 45:1086-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]