

NEONATAL IMMUNITY TO GROUP B STREPTOCOCCUS
Streptococcus agalactiae (group B streptococcus [GBS]) became recognized about 30 years ago as the dominant pathogen of neonatal sepsis (110). GBS is a Janus-faced organism. About 20% of healthy pregnant women and 10% of all newborn infants are colonized; hence, GBS is a component of the normal mucosal flora. Despite the recent implementation of intrapartum antibiotic prophylaxis, GBS remains the single most important cause of early neonatal sepsis in newborn infants (65, 73, 134, 160). Moreover, GBS is isolated with increasing frequency from elderly and diabetic patients (120) and is the third most frequent cause of bacterial meningitis in countries with routine immunization against Haemophilus influenzae type b (73, 135). Similar to the case for newborn infants, bacteremia is the most common presentation of GBS disease in adults (60%) (19). Infections due to GBS are important causes of morbidity: the mortality rate is about 10% for infants and adults (19, 65), and nearly half of all infants who survive an episode of GBS meningitis suffer from long-term neurodevelopmental sequelae (44, 65).
The predisposition of the very young and of elderly or immunocompromised adults remains to be explained. Infant susceptibility to GBS occurs mainly in the first 3 months of life, which suggests that this predisposition may be due to immaturity of the innate immune responses. If so, this immaturity must be relatively common, since prior to the introduction of screening or prophylactic measures, GBS affected 0.6% of all infants (11). It is also tempting to speculate that this immaturity may be mimicked later in life by the onset of diabetes mellitus or other immunocompromising conditions.
The defense of the newborn infant relies in part on placental transfer of maternal GBS antibodies. Infants suffering from GBS sepsis exhibit reduced serum concentrations of immunoglobulins specific for GBS capsular polysaccharide compared to infants who are only colonized with GBS (9). Furthermore, sialic acid constituents of the GBS capsular polysaccharide mimic the human Lewis X antigen, thus making GBS capsular polysaccharides poor immunogens (78). The adaptive immune systems of newborn and, particularly, preterm infants are significantly impaired due to both decreased synthesis of immunoglobulin G and constraints in the VH gene repertoire (13). Hence, in many cases the major burden of the neonatal defense to infection lies on the more ancient arm of immunity, the innate immune system. The study of the newborn, especially preterm, infant response to GBS thus constitutes a unique human model to gain insight into the contribution of the innate immune system to the prevention of sepsis.
It has long been assumed that irrespective of the age of disease onset, GBS invasive disease likely begins with a breach of the epithelial barrier of skin or mucous membranes. For a comprehensive review of how GBS manages to translocate across this immunological barrier, see the recent publication by Doran and Nizet (38). Here we focus our review on the interplay between GBS and cells of the neonatal innate immune system. This interaction determines whether GBS will be efficiently cleared or not and whether the immune response will be polarized to systemic inflammation. A deeper understanding of this interaction will facilitate the development of novel antisepsis therapies.
RECEPTOR-BASED RECOGNITION OF GBS BY TLRs
Ten human Toll-like receptors (TLRs) have recently been recognized as key recognition molecules of the innate immune system. TLRs are ubiquitously expressed on many cell types. TLRs sense and discriminate between minute concentrations of microbial substructures, including flagellin (TLR5) and hypomethylated bacterial DNA (TLR9) (74, 85). The intracellular portion of TLRs exhibits striking homology to the interleukin-1 (IL-1) receptor and is called the Toll/interleukin-1 receptor resistance (TIR) domain (106, 154). Upon TLR activation, several intracellular proteins (MAL/TIRAP, TRIF, TRAM, and TOLLIP), some of which contain TIR domains as well, are recruited to form an adapter (24, 47, 48, 71, 155). It is currently believed that these adapter proteins and downstream kinases, such as the four interleukin-1-receptor-associated kinases (IRAKs), determine the specificity of the immune response (6, 108, 148). It is fair to state that TLR-induced immune cell activation contains several levels of control and specific activation that have been resolved only partially so far (66).
With respect to GBS, early studies showed that inactivated preparations of whole bacteria did not significantly activate epithelial cells that expressed TLR2, a receptor that was believed to recognize all gram-positive bacteria (49). Subsequent attempts to find TLRs that recognize GBS substructures were not successful, despite the absolute dependence of GBS-mediated inflammatory signaling on the common TLR adapter protein myeloid differentiation antigen 88 (MyD88) (69). Accordingly, a TLR beyond the TLRs that have been connected to stimulation by other gram-positive bacteria has been invoked in a signaling response that comprises the mitogen-activated protein (MAP) kinases p38 and c-Jun kinase and the transcription factors NF-κB and activator protein 1 (Fig. 1) (79). In contrast to the whole bacterium, a released factor of GBS engaged TLR2 (70; see below). Accordingly, a neonatal mouse model of GBS sepsis revealed that TLR2 and, to a greater extent, MyD88 mediate tumor necrosis factor alpha (TNF-α) production and lethality (95).
FIG. 1.

Signaling pathways engaged by GBS cell wall. CD11b/CD18 mediates the uptake of opsonized GBS without directly affecting the transcription of inflammatory genes. In contrast, MyD88 mediates both the formation of cytokines and the antibacterial oxygen radical attack via distinct but interrelated pathways.
Relatively little is known about TLR expression and function in newborn infants or other fetal or newborn mammals. Recent data implicate a relevant increase in lipopolysaccharide (LPS)-induced TLR4 expression and the cytokine response of the fetal lung between days 14 and 17 (term, 20 days) as well as a further increase of both parameters after birth (61). Similarly, TLR4 expression on peripheral blood monocytes increases with gestational age (51). Furthermore, MyD88 expression appears to be lower in neonatal monocytes and correlates with TNF-α production (156). The expression of TLR4 and its coreceptor, CD14, on LPS-stimulated neonatal phagocytes appears to be somewhat reduced compared to that on adult cells (28, 68, 156). However, the biological significance of age-dependent differences in TLR4 (and CD14) expression remains to be established, since low TLR4 copy numbers are sufficient for a full LPS response (41).
With respect to phagocyte function, some consistent response patterns to TLR stimulation of cells from newborn infants have been observed. Notably, in terms of cytokine induction, neonatal monocytes are stimulated by GBS at least as potently as adult cells (Table 1) (18, 90, 121, 151). More importantly, in septic newborn infants, serum concentrations of inflammatory cytokines such as IL-1β and IL-8 are extremely elevated, with a trend toward higher values in very immature infants (16, 81). In line with these laboratory observations, newborn infants infected with GBS exhibit all the cardinal clinical signs of septic shock that have been shown to be mediated by endogenous pyrogens such as TNF-α and IL-1 (95). In contrast, published data on the inflammatory response to purified bacterial TLR agonists are less consistent. Several studies found that following stimulation with purified TLR agonists (TLRs 2, 4, and 7), cord blood mononuclear cells of newborn infants exhibited inferior inflammatory cytokine responses, particularly TNF-α responses, compared to cells from healthy adults. Other studies, however, did not find these differences (15, 28, 92, 122, 151, 156). A diminished response to bacterial components may be important for some aspects of disease progression but not for others. For example, an early and powerful immune response may limit the invasion of bacteria at very early stages, when only traces of microbial components are detected by innate immune cells; such a response later in the pathogenesis of infection may be deleterious.
TABLE 1.
Comparative studies of TNF-α/IL-1 responses by neonatal and adult peripheral blood mononuclear cells or macrophages
| Investigators (reference) | Stimulus | Key findings (relative response levels)a |
|---|---|---|
| Berner et al. (18) | GBS | For TNF-α and IL-1, neonatal ≥ adult PBMC |
| LPS | For TNF-α and IL-1, neonatal ≥ adult PBMC | |
| Levy et al. (90) | GBS | For TNF-α, neonatal ≥ adult PBMC |
| Peat et al. (121) | GBS | For TNF-α, neonatal > adult PBMC |
| Williams et al. (152) | GBS | For TNF-α, neonatal > adult PBMC |
| Chelvarajan et al. (28) | LPS | For TNF-α and IL-1, neonatal < adult macrophages |
| Forster-Waldl et al. (51) | LPS | For TNF-α and IL-1β, neonatal < adult PBMC |
| Levy et al. (92) | Lipopeptides, LPS, imiquimod | For TNF-α, neonatal < adult monocytes for TLR2, -4, and -7 agonists |
| Manimtim et al. (97) | LPS | For TNF-α, neonatal < adult monocytes |
| Peters et al. (122) | LPS | For TNF-α and IL-1, neonatal < adult monocytes |
| Weatherstone and Rich (148) | LPS | For TNF-α, neonatal < adult monocytesb; IL-1 results similar between groups |
| Yan et al. (157) | LPS | For TNF-α, neonatal < adult monocytes |
PBMC, peripheral blood mononuclear cells.
Only cells from preterm infants, not term infants, exhibited an impaired response.
Plasma factors appear to be involved in the differences between newborn and adult cells, since the addition of adult plasma to neonatal monocytes significantly increases the response to some TLR agonists and neonatal plasma suppresses the response of adult cells (92, 156). IL-10 may be an attractive candidate for this activity because an exaggerated anti-inflammatory IL-10 response following stimulation with TLR agonists has been associated with the decreased ability of neonatal phagocytes to mount a proinflammatory cytokine response (139). In accord with these data, neonatal IL-10−/− mouse macrophages secrete “adult concentrations” of IL-6 and TNF-α (28).
GBS SUBSTRUCTURES AS PUTATIVE TLR AGONISTS
Inactivated whole GBS cells are powerful stimulators of macrophages and monocytes, exceeding the capacity of similarly inactivated virulent S. pneumoniae at least 100-fold. Which components of bacterial structures are actually sensed by TLRs remains to be determined, however. The polysaccharide capsule and the adjacent cell wall form obvious sites of the interface between a microbe and host cell and have thus been the most adequately studied to date.
Polysaccharide.
GBS expresses two distinct carbohydrate entities, i.e., a type-specific and a group-specific polysaccharide. The polysaccharide capsule represents a logical candidate for recognition since it forms the outermost lining of GBS. Nine antigenically distinct capsular types of GBS are associated with human infection. The capsular polysaccharides of all serotypes contain various amounts of glucose, galactose, and N-acetylneuraminic acid and have a conserved terminal sialic acid. Furthermore, all serotypes but VI and VIII contain N-acetylglucosamine, whereas rhamnose is only found in type VIII (10, 30). It has been well established that the polysaccharide contributes to GBS virulence by interfering with C3 opsonization through inhibition of the alternative complement pathway in the absence of type-specific capsule antibodies (100, 131). Furthermore, type-specific polysaccharide contributes to immune evasion by host structure mimicry via the sialic acid residue (150). A role of type-specific polysaccharide as a positive inflammatory agent of the innate immune system has repeatedly been proposed both in vitro and in vivo (96, 146). As an example, increased serotype-specific invasion and cytokine induction for GBS VIII compared to GBS III was recently reported (109). However, the TNF-α-inducing properties of type-specific polysaccharide are weak, and acapsular GBS mutants are as able to induce cytokines as the isogenic encapsulated wild-type GBS parent strain (32, 37, 151). In contrast to type-specific polysaccharide, the group-specific polysaccharide from GBS has been found to be a more potent inflammatory stimulus. In particular, the dimeric adhesion molecule CD11b/CD18 has been implicated in the recognition of this carbohydrate (31, 32, 96). However, studies on the inflammatory characteristics of both polysaccharide species must be interpreted with caution. The type- and group-specific polysaccharides are covalently linked to peptidoglycan (PGN) and, possibly, other cell wall components of GBS (34). Therefore, it is difficult to ascertain the role of the polysaccharide alone in the absence of other potentially inflammatory stimuli.
PGN.
The GBS cell wall is a network of cross-linked PGN, surface proteins, polyanionic teichoic acid, and lipoteichoic acid (LTA). The polysaccharide capsule is covalently phosphodiester linked to N-acetylglucosamine, part of the minimal disaccharide unit [N-acetylmuramic acid (MurNAc)-(β1-4)-N-acetylglucosamine (GlcNAc)], within the macromolecular PGN assembly of the cell wall. The d-lactyl moiety of each MurNAc unit is amide linked to a short peptide component of PGN. These “wall peptides” are cross-linked with other peptides that are attached to a neighboring glycan strand (114). Whereas the repeating disaccharides are conserved between bacterial species, both “wall peptides” and cross-linking peptides exhibit considerable interspecies variability. GBS shares the PGN stem pentapeptide with many other gram-positive bacteria. However, the cross-linking interpeptide that bridges the l-lysine of one stem peptide to the d-alanine at position 4 of a neighboring subunit is l-Ala-l-Ala or l-Ala-l-Ser (Fig. 2) (133). To date, it remains unknown whether the composition and potential modifications, such as (serine) phosphorylation, of this peptide affect the inflammatory properties of the molecule.
FIG. 2.
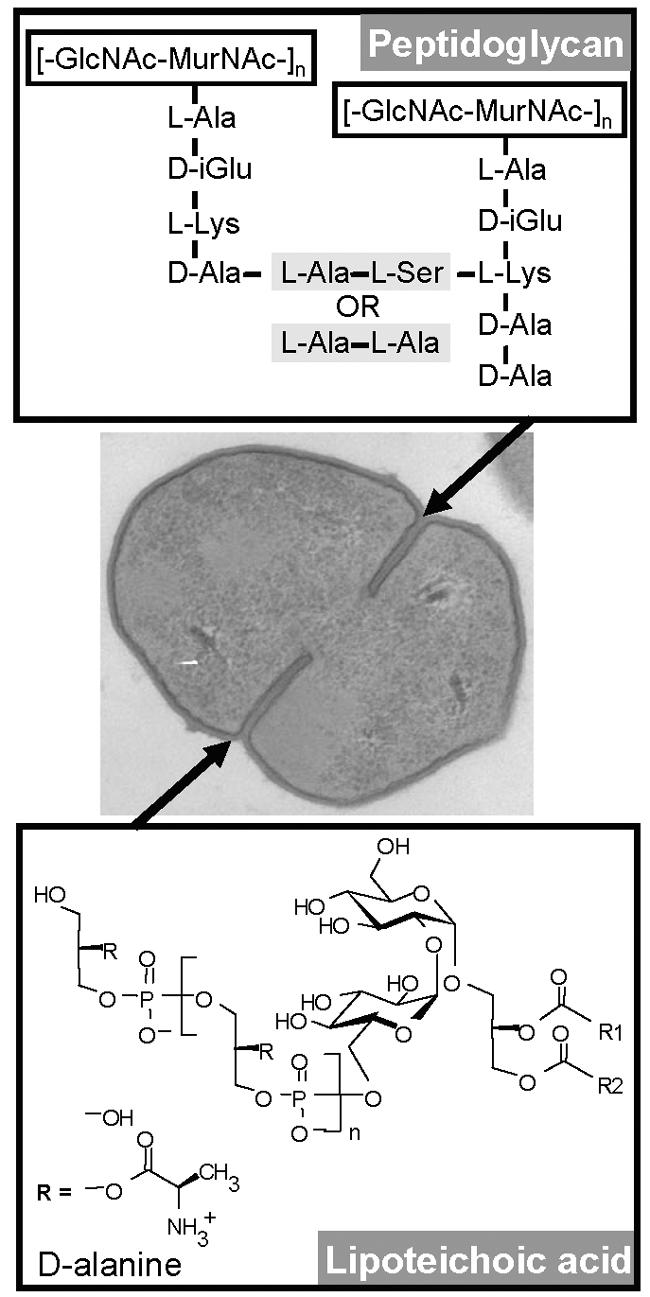
Structure of GBS cell-wall-associated patterns. The peptidoglycan macromolecule of the GBS cell wall is distinct from those of many other gram-positive bacteria by the cross-linking interpeptide (l-Ala-l-Ser or l-Ala-l-Ala). The structure of the glycolipid Glc(α1-2)Glc(α1-3)diacylglycerol in GBS LTA is different from those revealed in S. aureus and S. pneumoniae but similar to those in Enterococcus, Leuconostoc, and Lactococcus spp. (86).
PGN preparations, whether generated by cell wall digestion or by growth in the presence of β-lactam antibiotics, constitute a heterogeneous mixture of cell wall fragments. These preparations have been shown to activate immune cells via the cell-wall-anchored glycoprotein CD14 and TLR2 (136, 159). PGN isolated from GBS was shown to induce cytokine formation in cord blood monocytes, but host cell molecules that mediated the response were not investigated (145). Accordingly, it remains unknown whether PGN purified from GBS engages similar receptors as PGN from Staphylococcus aureus.
The interaction of PGN preparations with TLR2 has been mapped to amino acids 107 to 115 within the extracellular leucine-rich portion of TLR2 (52, 107). TLR2 is believed to function only in concert with other TLRs, since homodimerization of the cytoplasmic domain of TLR2 does not induce inflammatory cytokines in macrophages, whereas similar dimerization of TLR4 is believed to do so (117). TLR2 has been shown to form heterodimers with TLR1 and TLR6 that discriminate between subtle differences in acylation of bacterial proteins: diacylated lipoproteins are recognized by TLR2/TLR6, while triacylated lipoproteins are recognized by TLR1/TLR2 (118, 141, 143, 142). It seems noteworthy that so far, inflammatory lipoproteins have only been identified in gram-negative bacteria, mycobacteria, and Mycoplasmataceae, but not in gram-positive bacteria. In contrast to lipoproteins, PGN preparations appear to activate immune cells independently of TLR6 and TLR1 (142). Hence, either PGN engages a different and unique TLR2-containing heteromer or the heterogeneity of the PGN preparation is reflected by the engagement of various heteromers that share TLR2 but otherwise compensate for each other. Doubts concerning PGN as a true TLR2 ligand have increased recently due to a report by Travassos et al. (144). In their hands, further purification of PGN from various organisms to remove lipidated structures not only eliminated TLR2 activation but also abrogated the stimulation of macrophages altogether. In contrast, both highly purified PGN and synthetic minimal PGN fragments (MurNAc-l-Ala-d-IsoGln) from gram-positive and gram-negative bacteria were detected by the intracellular receptor NOD2. Other PGN fragments specific for gram-negative bacteria (GlcNAc-MurNAc-l-Ala-γ-d-Glu-meso-diaminopimelic acid) were detected by NOD1 (55, 56). Interestingly, the only highly purified streptococcal PGN preparation examined (from S. pneumoniae) did not activate NOD2 either, in striking contrast to PGN from S. aureus, Listeria, and Escherichia coli (144). Both the association of PGN with cell-wall-embedded lipoproteins and the essential role of PGN for bacterial cell wall integrity have been scientific obstacles to the analysis of PGN-induced innate immune mechanisms, since PGN-deficient bacteria are not viable. Accordingly, studies on whether TLR2 stimulation by PGN preparations is due to PGN itself or to an as yet unidentified copurified molecule remain inconclusive (42).
LTA.
LTA, the second important GBS cell wall component, resembles the potent gram-negative endotoxin (LPS) in some respects. LTA and LPS are both anchored via their glycolipids to the membrane and carry a polysaccharide chain extending into the PGN layer of the cell wall (114). In contrast to the apparent role of LTA in cell morphology, its contribution to the activation of innate immune cells by gram-positive bacteria remains less well understood. Similar to PGN, LTA has been shown to engage TLR2. According to data generated with targeted deletion mice, TLR6, but not TLR1, serves as an essential coreceptor for TLR2 in the recognition of LTA from both GBS and S. aureus (67). Other studies performed with human cells suggested that TLR1 may contribute significantly to the recognition of LTA (60, 144). The conflicting data might be due to species-related differences (mouse versus human cells). Alternatively, interference with formation of the entire TLR1/2/6 complex by TLR1-specific antibodies or heterologous expression of TLR1 in the context of endogenous TLR1 and TLR6 might have affected the specificities of these approaches (60, 69, 144). Clearly, engagement of the TLR2-TLR6 multimer by LTA corresponds to observations on other diacylated agonists (e.g., a lipoprotein from Mycoplasma fermentas) (142). Despite the engagement of a similar receptor heteromer, GBS LTA appears to have a significantly lower inflammatory potency than S. aureus LTA. Nuclear magnetic resonance analysis revealed that GBS cells contain type I LTA, similar to other streptococci and staphylococci (101, 115). In type I LTA, polyglycerophosphate is attached to C-6 of the nonreducing glucosyl of the glycolipid anchor. However, beyond their basic structure, GBS LTA and S. aureus LTA are distinct with respect to the average length of the hydrophilic glycerophosphate chain (19 units in GBS and >45 units in S. aureus) (111) and the structure of the glycolipid [Glc(α1-2)Glc(α1-3)diacylglycerol in GBS versus Glc(β1-6)Glc(β1-3)diacylglycerol in S. aureus and S. pneumoniae] (86). Additionally, in GBS LTA, the polyglycerophosphate backbone is substituted with d-alanine only, whereas in S. aureus and many other species, d-alanine substituents alternate with N-acetylglucosamine (Fig. 2). Structural analysis of native and synthetic LTA from S. aureus revealed that next to the lipid anchor itself, alanine substituents in the d-configuration are required for proper inflammatory activity of the molecule (111-113). In contrast, the individual contributions of the glycolipid configuration, N-acetylglucosamine, and the length of the polyglycerophosphate to the inflammatory potency of GBS are unknown.
However, the engagement of TLR2 by GBS LTA seems of minor importance for recognition of the GBS cell wall. First, despite the apparent dependency of LTA d-alanylation on cytokine induction, cell walls from GBS with a genetically disrupted incorporation of d-alanine into LTA (DltA−) exhibit similar inflammatory activity to that of the isogenic parental strain (33, 123). Mouse and rat models of invasive infections with DltA− gram-positive organisms remain difficult to interpret with respect to the specific role of d-alanine (and therefore LTA) in the innate immune response. The observed loss in virulence of DltA− GBS correlates with an increased susceptibility to killing by phagocytes (125), since deletion of d-alanine increases the net anionic charge of the bacterial cell wall and therefore alters binding of cationic peptides. Furthermore, in contrast to GBS LTA, the cell wall interacts with phagocytes via a receptor that is dependent on the TLR adapter protein MyD88 but largely independent of TLR2 (and TLRs 1, 4, 6, and 9). Finally, a GBS mutant lacking IagA glycosyltransferase, which mediates membrane anchoring of LTA, exhibits attenuated virulence, independent of TLR2, in a rat sepsis model (36).
Accordingly, in contrast to the important contributions of alanylated LTA (and PGN) to bacterial survival in the host and endothelial invasion, additional and as yet unidentified GBS components are the dominant inflammatory stimuli of the cell wall. Interestingly, this observation contrasts to findings for Lactococcus plantarum, where d-alanylation of LTA and the interaction of LTA with TLR2 critically influence the proinflammatory activity of the organism (58).
Membrane proteins.
In GBS, similar to other gram-positive bacteria, cell wall proteins express carboxy-terminal signaling sequences, which are putative enzyme cleavage sites (LPXTG) that, together with a hydrophobic domain and charged tail, are sufficient for PGN anchoring by an enzyme called sortase (103). As an example, the NEM316 (GBS III) strain's genome encodes 35 putative surface proteins bearing a cell wall sorting signal motif. In GBS, a deletion of the major sortase SrtA affects the colonization frequency of the rat intestine, but virulence is not affected (82).
Probably the best-studied class of surface proteins in GBS are the C proteins alpha C, beta C, and Rib. The C proteins are variably expressed among different serotypes, and active immunization with these proteins has been shown to induce protection against invasive disease. Furthermore, antigenic variation by variable expression of tandem repeats in these proteins might be an immune evasion mechanism of GBS (17, 59). On the other hand, expression of the alpha C protein on the surface of GBS mediates entry into cervical epithelial cells involving interaction of the alpha C protein N-terminal domain and the host cell glycosaminoglycan and Rho GTPase-dependent actin rearrangements (8, 12, 21). Another interesting GBS surface protein that inhibits opsonophagocytosis is CspA. CspA is a serine protease that mediates cleavage of human fibrinogen and contributes to GBS virulence. However, the exact mechanism by which CspA is involved in GBS immune evasion has not been firmly established (64).
A host of other surface proteins important for GBS virulence have recently been identified by the employment of proteomics and molecular approaches, such as phage display and signature-tagged mutagenesis (72, 76, 94). As an example, C5a peptidase was shown to have fibronectin binding properties (14). However, the precise role, if any, of these proteins in innate immune recognition of GBS remains to be elucidated.
INFLAMMATORY FACTORS RELEASED DURING GROWTH OF GBS
Extracellular TLR2/6 activity.
Starting during early logarithmic growth, GBS releases an extracellular factor that induces NF-κB and MAP kinases via CD14, TLR2, and TLR6 in phagocytes (8). Whereas the identity of the secreted factor is still unknown, it represents a potent inflammatory stimulus for phagocytes, since GBS supernatant enriched for TLR2 activity by size-exclusion chromatography induces the transcription of cytokine genes at concentrations as low as 100 ng/ml (P. Henneke and D. T. Golenbock, unpublished observations). Since the expression of TLR2 is critical for the course of neonatal GBS sepsis in mice, but not for the macrophage response to the GBS cell wall in vitro, the described extracellular factor likely contributes to phagocyte activation in vivo (31, 67, 69, 95).
Several proteins are secreted by GBS, e.g., CAMP factor, a pore-forming cohemolysin for beta-toxin of S. aureus, the protein of cell wall separation B, enolase, fructose bisphosphonate aldolase, heat shock protein 70, and surface immunogenic protein (23, 50, 84). Whether any of these proteins is involved in the activation of innate immune cells remains unclear. Furthermore, the membrane-anchored LTA can be isolated from GBS supernatant. However, LTA from GBS represents a comparatively weak stimulus for phagocytes. If GBS supernatant is purified for TLR2 activity by size-exclusion chromatography, the resulting fractions are 10,000-fold more potent than their LTA contents (P. Henneke, unpublished observations). Therefore, LTA either synergizes with other released factors or is just one among many TLR2/6 agonists in the GBS supernatant. Interesting candidates are the more than 30 putatively amino-terminally lipidated proteins encoded in the GBS genome (www.mrc-lmb.cam.ac.uk/genomes/dolop/). Nothing is known about the inflammatory potencies of these lipoproteins.
β-Hemolysin.
A zone of β-hemolysis surrounding colonies on blood-agar medium is a key phenotypic feature of group B streptococcus. Functional information on β-hemolysin derived from nonhemolytic and hyperhemolytic GBS mutants and relatively crude GBS extracts stabilized by starch suggests that β-hemolysin induces cytolytic injury to epithelial and endothelial cells and both the formation of nitric oxide and apoptosis in macrophages (128). Furthermore, the beta-hemolytic activity of GBS promotes the invasion of human lung epithelial cells and is a critical virulence factor in a neonatal rabbit model of GBS pneumonia (35, 46). Although the causative beta-hemolytic exotoxin has been known for decades, a single gene (cylE) encoding a predicted 78-kDa protein sufficient to confer a hemolytic phenotype to nonhemolytic E. coli has only recently been identified (126). However, the gene product of cylE has evaded purification to homogeneity, probably due to low stability of the protein. In addition to the hemolytic phenotype, CylE is also linked to the occurrence of a carotenoid pigment in these bacteria (93). The carotenoid itself carries the ability to shield GBS from oxidative damage. The molecular mechanism by which the cylE gene controls two apparently independent functions remains to be established. As an in vivo correlate, an increased virulence of beta-hemolytic compared to nonhemolytic GBS was demonstrated in several animal models of invasive GBS disease, such as an adult mouse model of GBS arthritis, a rabbit sepsis model, and rabbit and rat models of GBS pneumonia (38). The molecular mechanism by which GBS hemolysin activates innate immune cells remains unknown, however, since a hemolysin receptor has not yet been identified. As an alternative to direct receptor engagement, β-hemolysin might induce cellular activation via perturbation of the phagocyte membrane and subsequent transmembrane calcium flux.
Recently, two groups independently identified a functional two-component regulatory system in GBS, denominated CsrR/CsrS (75) or CovS/CovR (83), that controls the transcription of several exported virulence factors, including beta-hemolysin/cytolysin (cylE), CAMP factor (cfb), and C5a peptidase (scpB). A GBS mutant with mutation of either gene exhibited significantly reduced virulence. However, the individual contributions of the multiple virulence factors altered in CsrR/CsrS mutant GBS to the host-pathogen interaction are currently poorly understood.
PHAGOCYTE RECEPTORS FOR GBS BEYOND TLRs
In addition to TLRs, the TLR coreceptor CD14, a glycosylphosphatidylinositol-linked membrane protein without a signaling domain, has been implicated in the response to GBS. As evidenced by studies of isolated GBS substructures and by gain-of-function models, CD14 may be involved in inflammatory signaling under certain circumstances (31, 32, 105) but does not appear to be necessary for the phagocyte response to whole inactivated GBS (18, 49, 69). Similar to TLR2, CD14 clearly mediates the response to secreted factors from GBS (70).
Furthermore, the heterodimeric β2-integrins, in particular CD11b/CD18, have been repeatedly evaluated as signaling molecules for GBS (32, 57). Heterologous expression of CD11b/CD18 on fibroblasts confers some signaling properties to these cells in response to GBS, such as translocation of NF-κB (31, 105). Furthermore, incubation of whole blood from CD11b-deficient mice with GBS results in lower cytokine concentrations than those in blood from wild-type controls (90). However, macrophages from the same CD11b-deficient mice are normal with respect to a host of inflammatory signaling events, including the formation of cytokines, oxygen radicals, and signaling intermediates (69). An important caveat of experiments with CD11b−/− cells is that CD11b is a very promiscuous molecule that interacts with several extracellular matrix proteins, such as fibronectin, fibrin, and collagen (62). Fibronectin, in turn, has been shown to bind to GBS and to contribute to the formation of TNF-α, opsonin-dependent actin polymerization, phagocytosis, and oxygen radical formation (121, 157). Furthermore, CD11b deficiency results in a lack of expression of CD18, the subunit common to all β2-integrins, which results in alteration of many cellular functions, such as adhesion and costimulatory events. Hence, the phenotype of these mice remains difficult to interpret. In summary, in contrast to the important role of CD11b in clearance of opsonized GBS, its role in the inflammatory reaction against GBS is less clear. CD11b appears not to be a critical cognate receptor for the GBS cell wall on resident phagocytes; however, it might affect cellular activation in a mixed leukocyte environment.
DIRECTLY ANTIBACTERIAL PROPERTIES OF NEONATAL PHAGOCYTES
Currently, it is believed that the most common route of early-onset GBS infection in newborn infants occurs via perinatal aspiration of GBS that colonize the female genital tract. Shortly after the inoculation of bacteria into the parenchyma of the lung, GBS may reach a density as high as 109 to 1011 CFU/g lung tissue, as confirmed in the primate model of neonatal GBS pneumonia (130). Furthermore, 30 GBS organisms injected subcutaneously represented the 50% lethal dose in a neonatal mouse model of GBS sepsis, whereas the equivalent bacterial dosage was approximately 1,000,000-fold higher for adult mice of the same mouse strain (95). Hence, host factors that directly influence the elimination of GBS from tissue are likely to be insufficiently developed in newborn infants. Notably, diabetic rodents and phagocytes from patients with type 2 diabetes exhibit a reduced capacity to eliminate GBS, whereas cytokine formation appears to be preserved (43, 53, 102).
The elimination of GBS from tissues and the bloodstream constitutes a sequence of tightly controlled interdigitating events, such as recognition by resident macrophages, chemotactic approaching of GBS by additional phagocytes, and finally, phagocytosis of GBS and killing of the internalized bacteria (2). In this section, we aim to illuminate the specific capabilities of neonatal phagocytes that lead to rapid destruction of invasive GBS organisms.
Chemotaxis.
Chemotaxis, or directed cell movement to inflammatory stimuli, was shown to be impaired in both preterm neonates and those brought to term (term neonates), with neutrophils from newborn infants migrating at about half the speed of adult cells (5, 27, 80, 119, 153). Within the complicated process of chemotaxis (recognition of a signal, adherence to extracellular matrix proteins, and polarized movement by rearrangement of the actin cytoskeleton), several deficiencies in neutrophils have been defined for preterm and term infants (although the deficiencies are less obvious in the latter). First, in very preterm infants, the total neutrophil cell mass is decreased (26). Second, rolling adhesion to activated endothelium or glass and transmigration to the subendothelial tissue are decreased in cells from newborn infants compared to those from adults (4). Third, the formation of lamellipodia and directed movements towards a stimulus are impaired in neonatal cells (5). On the molecular level, the expression of L-selectin, which controls neutrophil rolling along the endothelium, and therefore chemotaxis, is impaired on neutrophils from newborn, particularly preterm, infants (4, 98). Beyond the expression of surface molecules, neonatal neutrophils exhibit a reduced ability to rapidly polymerize monomeric G-actin to form microtubules, resulting in an imperfect bipolar shape change in these cells (63, 132). Strikingly, postnatal improvement in neutrophil chemotaxis is quite rapid and reaches “normal” values at a median of 10 days, whereas the same development takes considerably more time for very preterm infants (for a review, see reference 25). With particular respect to GBS, it has been established that GBS induces chemokines, such as IL-8 and leukotriene B4, and complement factors, in particular C3b and C5a, that in turn indirectly induce chemotaxis by neutrophils (129, 138). On the other hand, the conserved GBS surface protein C5a peptidase contributes to immune evasion by abolishing the chemoattractant activity of C5a (140). Furthermore, the GBS β-protein increases binding of the complement factor H that inhibits the alternative pathway (7). In contrast, a GBS substructure and a phagocyte receptor that directly induce chemotaxis to GBS have not been described.
Phagocytosis of GBS.
Phagocytosis, the binding and ingestion of particles such as GBS, comprises recognition by cell surface receptors, the subsequent formation of actin-rich membrane extensions around the particle, phagosome formation, and maturation into a phagolysosome. Resident macrophages are the first to encounter GBS that enter the lung via aspiration. In a rat model of GBS pneumonia, Martin et al. found that clearance of GBS that were inoculated directly into the trachea was compromised in neonatal rats. Moreover, alveolar macrophages from newborn rats phagocytosed GBS less efficiently than adult cells (100). On the molecular level, the recognition of GBS that precedes phagocytosis can be indirect, via host factors such as surfactant proteins A and D, antibodies, or complement components, i.e., via opsonization, or direct, via interactions of GBS substructures with host cell receptors (2, 88). Accordingly, low serum concentrations of complement factors in newborn infants might negatively affect both chemotaxis and phagocytosis in newborn infants (40).
The following two types of host cell receptors are critical for GBS phagocytosis: (i) the β2-integrin CD11b/CD18 that has already been described and (ii) two classes of Fcγ receptors (FcγRII and -III). In various experimental models, phagocytosis of GBS is heavily dependent on CD11b/CD18 and opsonization with complement, whereas specific serum does not seem to be required (3, 69, 137, 158). Accordingly, mice deficient in the CD11b/CD18 ligand C3 were significantly more susceptible to GBS than wild-type controls (149). In the absence of specific anti-GBS serum, the capsular polysaccharide appears to promote an interaction with CD11b/CD18, probably through the lectin binding site of the receptor (3, 45). Interestingly, the expression of CD11b/CD18 is decreased on both resting and stimulated phagocytes of preterm infants but not on those of term infants (1, 68, 104, 116). Whether the deficiency of the crucial phagocyte receptor CD11b/CD18 contributes to the particular susceptibility of preterm infants to GBS sepsis needs to be further studied. Among FcγRs, only FcγRIII, a phosphoinositol-linked structure involved in neutrophil granule exocytosis, is significantly less expressed on preterm neonatal cells than on term neonatal or adult cells (25). However, FcR-mediated phagocytosis is likely to be further impaired in preterm infants, as it relies on the synthesis of specific immunoglobulins that are lacking in these patients.
In contrast to FcγR and β2-integrins, TLRs appear not to be important for the phagocytosis of GBS, since macrophages deficient in MyD88 or individual TLRs and wild-type macrophages internalize GBS at similar speeds (69). In contrast, both phagocytosis of S. aureus and subsequent phagosome maturation appear to be dependent on the expression of TLR2 (20).
Intracellular killing of GBS.
On the global level, cord blood granulocytes and monocytes were found to be similar with respect to intracellular killing of E. coli and S. pyogenes compared to cells from adults. In contrast, killing of GBS was almost completely abrogated in cord blood phagocytes (99). Antibacterial killing mechanisms are best categorized as (i) oxygen dependent and (ii) oxygen independent, which can be further divided into enzymatic mechanisms (involving, e.g., lysozyme, cathepsin G, elastase, and 14-kDa phospholipases A2 and 2b) and nonenzymatic, mostly membrane-active mechanisms (such as defensins, cathelicidins, lactoferrin, and bactericidal/permeability-increasing proteins) (89). Unfortunately, little specific data are available on anti-GBS killing properties by both oxygen-dependent and -independent means inside neonatal phagocytes (127).
In piglet and mouse models of GBS pneumonia and sepsis, oxygen radical species are critical for killing of GBS (22). GBS possess a single Mn-cofactored superoxide dismutase (SodA) that serves as a virulence factor by counteracting intracellular killing in macrophages (124). Clearly, the engagement of Fc receptors on phagocytes by antibody-opsonized GBS mediates intracellular killing of GBS, which is in part dependent on oxygen radicals (29). In neutrophils from very preterm infants, the intracellular oxidative burst was found to be markedly reduced compared to that in cells from adults in response to opsonized GBS (77).
On the molecular level, the GBS cell wall activates the formation of oxygen radicals via a distinct MyD88-dependent but NF-κB-independent pathway. Again, this response is independent of TLR2 (69). Furthermore, p38, but not Jun N-terminal kinase, appears to be crucial in this context (Fig. 1) (79). Notably, costimulation of alveolar macrophages by granulocyte-macrophage colony-stimulating factor (GM-CSF) is a prerequisite for proper antibacterial superoxide and hydrogen peroxide formation by mouse alveolar macrophages and for GBS clearance from the lung, as evidenced in GM-CSF−/− mice. Moreover, these mice exhibited an exaggerated cytokine response by alveolar macrophages, again highlighting the counteracting effects of GBS clearance and the cytokine response (87). Importantly, GBS is not a typical isolate from patients with defects in the NADPH oxidase system, i.e., those with chronic granulomatous disease. Surprisingly, however, GBS are less susceptible to H2O2 and more susceptible to hydroxyl radicals than S. aureus. The molecular basis for these findings has not been established, but species-specific expression of scavenger proteins appears not to be critical in this context (152). Furthermore, oxygen-independent mechanisms might be particularly relevant in the killing of GBS (and other catalase-deficient bacteria). In preterm infants, but not term infants, elastase formation in whole blood upon endotoxin stimulation is reduced (68). Lysozyme is found in low concentrations in neonatal polymorphonuclear granulocytes (PMN); however, it is increased to reach concentrations similar to adult PMN upon stimulation with zymosan (54, 91). The complex nonoxidative, nonenzymatic mechanisms that contribute to bacterial killing by phagocytes have been covered by a recent comprehensive review (89). With respect to GBS, cathelicidin and β-defensin-2 exhibit synergistic antimicrobial activity against GBS. Moreover, the expression of cathelicidin is increased in neonatal mouse skin (39). Whereas defensins are found in normal concentrations in neonatal PMN, other nonenzymatic proteins (lactoferrin and bactericidal/permeability-increasing protein) are decreased in both resting and activated PMN (54, 91). In light of the partially redundant, partially interdigitating nature of the various oxidative and nonoxidative killing mechanisms, their individual contributions to the decreased antibacterial activity toward GBS in phagocytes from newborn infants remain to be fully elucidated (127).
CONCLUSION
GBS is a common colonizer of the normal human mucosa. In contrast, invasive GBS disease constitutes a rare event, for which newborn infants and diabetics are clearly at increased risk. As model residents, macrophages sense spurious amounts of TLR2/6 ligands released from GBS, such as lipoteichoic acid. Activation of TLRs will activate both the cytoskeleton, via phosphatidylinositol 3-kinase, and the release of cytokines and chemokines, via MyD88, which results in the recruitment and activation of other phagocytes. At the same time, β2-integrins and FcγR mediate the engulfment of particulate GBS material in dependence on complement and specific antiserum, although GBS has developed strategies for evading phagocytosis. TLRs sample the contents of the phagolysosome and instruct the radical attack via the MAP kinase p38. In accordance with this model, the TLR system is essential for the inflammatory cytokine response and killing—but not phagocytosis—of GBS both in vivo and in vitro. In contrast, the β2-integrin CD11b/CD18 is essential for GBS phagocytosis, but its direct contribution to the phagocytic cytokine response may be less important. It is reasonable to hypothesize that immunocompetent humans eliminate GBS that translocate across the mucosa without eliciting a systemic inflammatory response. In accordance with this, normal mice clear a small local GBS inoculum without mounting measurable cytokine formation in the blood, whereas immunodeficient mice frequently succumb to septic shock. Hence, a tight balance between a sufficient local proinflammatory response and a nonphlogistic, antibacterial removal of invading bacteria may be the key to efficient clearance of invasive GBS.
Monocytes and macrophages from newborn infants mount a powerful inflammatory cytokine response to GBS. In contrast, several components of the clearance apparatus, such as directed movement to the microorganism, expression of adhesion molecules, and the intracellular killing of GBS, are deficient in newborn, particularly preterm, infants. The neonatal innate immune system appears to be less capable of concentrating and compartmentalizing inflammation at the site of local microbial invasion. Notably, a similar imbalance of low antibacterial and powerful inflammatory responses to GBS can be observed in animal models of diabetes. Thus, a preponderance of proinflammatory factors is likely to account for the particular susceptibility of both newborn infants and diabetic patients to hyperinflammation in invasive GBS disease. Accordingly, the identification of GBS substructures and host factors that trigger potentially detrimental systemic inflammation seems critical for the development of adjunctive sepsis therapy.
Acknowledgments
This work was supported in part by Deutsche Forschungsgemeinschaft (He 3127/2-1) and by the National Institutes of Health (AI52455).
We are grateful to Richard Malley (Boston, MA) for many insightful comments and careful review of the manuscript. We acknowledge Sybille Kenzel's (Freiburg, Germany) valuable help with this work. The electron microscopy image of GBS was a kind gift from Patrick Trieu-Cuot (Paris, France).
Editor: J. B. Kaper
REFERENCES
- 1.Abughali, N., M. Berger, and M. F. Tosi. 1994. Deficient total cell content of CR3 (CD11b) in neonatal neutrophils. Blood 83:1086-1092. [PubMed] [Google Scholar]
- 2.Aderem, A. 2003. Phagocytosis and the inflammatory response. J. Infect. Dis. 187(Suppl. 2):S340-S345. [DOI] [PubMed] [Google Scholar]
- 3.Albanyan, E. A., J. G. Vallejo, C. W. Smith, and M. S. Edwards. 2000. Nonopsonic binding of type III group B streptococci to human neutrophils induces interleukin-8 release mediated by the p38 mitogen-activated protein kinase pathway. Infect. Immun. 68:2053-2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson, D. C., O. Abbassi, T. K. Kishimoto, J. M. Koenig, L. V. McIntire, and C. W. Smith. 1991. Diminished lectin-, epidermal growth factor-, complement binding domain-cell adhesion molecule-1 on neonatal neutrophils underlies their impaired CD18-independent adhesion to endothelial cells in vitro. J. Immunol. 146:3372-3379. [PubMed] [Google Scholar]
- 5.Anderson, D. C., B. J. Hughes, and C. W. Smith. 1981. Abnormal mobility of neonatal polymorphonuclear leukocytes. Relationship to impaired redistribution of surface adhesion sites by chemotactic factor or colchicine. J. Clin. Investig. 68:863-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arbibe, L., J. P. Mira, N. Teusch, L. Kline, M. Guha, N. Mackman, P. J. Godowski, R. J. Ulevitch, and U. G. Knaus. 2000. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat. Immunol. 1:533-540. [DOI] [PubMed] [Google Scholar]
- 7.Areschoug, T., M. Stalhammar-Carlemalm, I. Karlsson, and G. Lindahl. 2002. Streptococcal beta protein has separate binding sites for human factor H and IgA-Fc. J. Biol. Chem. 277:12642-12648. [DOI] [PubMed] [Google Scholar]
- 8.Auperin, T. C., G. R. Bolduc, M. J. Baron, A. Heroux, D. J. Filman, L. C. Madoff, and J. M. Hogle. 2005. Crystal structure of the N-terminal domain of the group B streptococcus alpha C protein. J. Biol. Chem. 280:18245-18252. [DOI] [PubMed] [Google Scholar]
- 9.Baker, C. J., and D. L. Kasper. 1976. Correlation of maternal antibody deficiency with susceptibility to neonatal group B streptococcal infection. N. Engl. J. Med. 294:753-756. [DOI] [PubMed] [Google Scholar]
- 10.Baker, C. J., D. L. Kasper, and C. E. Davis. 1976. Immunochemical characterization of the “native” type III polysaccharide of group B streptococcus. J. Exp. Med. 143:258-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baltimore, R. S., S. M. Huie, J. I. Meek, A. Schuchat, and K. L. O'Brien. 2001. Early-onset neonatal sepsis in the era of group B streptococcal prevention. Pediatrics 108:1094-1098. [DOI] [PubMed] [Google Scholar]
- 12.Baron, M. J., G. R. Bolduc, M. B. Goldberg, T. C. Auperin, and L. C. Madoff. 2004. Alpha C protein of group B streptococcus binds host cell surface glycosaminoglycan and enters cells by an actin-dependent mechanism. J. Biol. Chem. 279:24714-24723. [DOI] [PubMed] [Google Scholar]
- 13.Bauer, K., M. Zemlin, M. Hummel, S. Pfeiffer, J. Karstaedt, G. Steinhauser, X. Xiao, H. Versmold, and C. Berek. 2002. Diversification of Ig heavy chain genes in human preterm neonates prematurely exposed to environmental antigens. J. Immunol. 169:1349-1356. [DOI] [PubMed] [Google Scholar]
- 14.Beckmann, C., J. D. Waggoner, T. O. Harris, G. S. Tamura, and C. E. Rubens. 2002. Identification of novel adhesins from group B streptococci by use of phage display reveals that C5a peptidase mediates fibronectin binding. Infect. Immun. 70:2869-2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berner, R., J. Csorba, and M. Brandis. 2001. Different cytokine expression in cord blood mononuclear cells after stimulation with neonatal sepsis or colonizing strains of Streptococcus agalactiae. Pediatr. Res. 49:691-697. [DOI] [PubMed] [Google Scholar]
- 16.Berner, R., C. M. Niemeyer, J. U. Leititis, A. Funke, C. Schwab, U. Rau, K. Richter, M. S. Tawfeek, A. Clad, and M. Brandis. 1998. Plasma levels and gene expression of granulocyte colony-stimulating factor, tumor necrosis factor-alpha, interleukin (IL)-1beta, IL-6, IL-8, and soluble intercellular adhesion molecule-1 in neonatal early onset sepsis. Pediatr. Res. 44:469-477. [DOI] [PubMed] [Google Scholar]
- 17.Berner, R., M. Ruess, S. Bereswill, and M. Brandis. 2002. Polymorphisms in the cell wall-spanning domain of the C protein beta-antigen in clinical Streptococcus agalactiae isolates are caused by genetic instability of repeating DNA sequences. Pediatr. Res. 51:106-111. [DOI] [PubMed] [Google Scholar]
- 18.Berner, R., P. Welter, and M. Brandis. 2002. Cytokine expression of cord and adult blood mononuclear cells in response to Streptococcus agalactiae. Pediatr. Res. 51:304-309. [DOI] [PubMed] [Google Scholar]
- 19.Blancas, D., M. Santin, M. Olmo, F. Alcaide, J. Carratala, and F. Gudiol. 2004. Group B streptococcal disease in nonpregnant adults: incidence, clinical characteristics, and outcome. Eur. J. Clin. Microbiol. Infect. Dis. 23:168-173. [DOI] [PubMed] [Google Scholar]
- 20.Blander, J. M., and R. Medzhitov. 2004. Regulation of phagosome maturation by signals from Toll-like receptors. Science 304:1014-1018. [DOI] [PubMed] [Google Scholar]
- 21.Bolduc, G. R., M. J. Baron, C. Gravekamp, C. S. Lachenauer, and L. C. Madoff. 2002. The alpha C protein mediates internalization of group B streptococcus within human cervical epithelial cells. Cell. Microbiol. 4:751-758. [DOI] [PubMed] [Google Scholar]
- 22.Bowdy, B. D., S. L. Marple, T. H. Pauly, J. D. Coonrod, and M. N. Gillespie. 1990. Oxygen radical-dependent bacterial killing and pulmonary hypertension in piglets infected with group B streptococci. Am. Rev. Respir. Dis. 141:648-653. [DOI] [PubMed] [Google Scholar]
- 23.Brodeur, B. R., M. Boyer, I. Charlebois, J. Hamel, F. Couture, C. R. Rioux, and D. Martin. 2000. Identification of group B streptococcal Sip protein, which elicits cross-protective immunity. Infect. Immun. 68:5610-5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burns, K., J. Clatworthy, L. Martin, F. Martinon, C. Plumpton, B. Maschera, A. Lewis, K. Ray, J. Tschopp, and F. Volpe. 2000. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat. Cell Biol. 2:346-351. [DOI] [PubMed] [Google Scholar]
- 25.Carr, R. 2000. Neutrophil production and function in newborn infants. Br. J. Haematol. 110:18-28. [DOI] [PubMed] [Google Scholar]
- 26.Carr, R., and T. W. Huizinga. 2000. Low soluble FcRIII receptor demonstrates reduced neutrophil reserves in preterm neonates. Arch. Dis. Child. 83:F160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carr, R., D. Pumford, and J. M. Davies. 1992. Neutrophil chemotaxis and adhesion in preterm babies. Arch. Dis. Child. 67:813-817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chelvarajan, R. L., S. M. Collins, I. E. Doubinskaia, S. Goes, J. Van Willigen, D. Flanagan, W. J. De Villiers, J. S. Bryson, and S. Bondada. 2004. Defective macrophage function in neonates and its impact on unresponsiveness of neonates to polysaccharide antigens. J. Leukoc. Biol. 75:982-994. [DOI] [PubMed] [Google Scholar]
- 29.Cheng, Q., B. Carlson, S. Pillai, R. Eby, L. Edwards, S. B. Olmsted, and P. Cleary. 2001. Antibody against surface-bound C5a peptidase is opsonic and initiates macrophage killing of group B streptococci. Infect. Immun. 69:2302-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cieslewicz, M. J., D. Chaffin, G. Glusman, D. Kasper, A. Madan, S. Rodrigues, J. Fahey, M. R. Wessels, and C. E. Rubens. 2005. Structural and genetic diversity of group B streptococcus capsular polysaccharides. Infect. Immun. 73:3096-3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuzzola, M., G. Mancuso, C. Beninati, C. Biondo, F. Genovese, F. Tomasello, T. H. Flo, T. Espevik, and G. Teti. 2000. Beta 2 integrins are involved in cytokine responses to whole gram-positive bacteria. J. Immunol. 164:5871-5876. [DOI] [PubMed] [Google Scholar]
- 32.Cuzzola, M., G. Mancuso, C. Beninati, C. Biondo, C. von Hunolstein, G. Orefici, T. Espevik, T. H. Flo, and G. Teti. 2000. Human monocyte receptors involved in tumor necrosis factor responses to group B streptococcal products. Infect. Immun. 68:994-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Debabov, D. V., M. Y. Kiriukhin, and F. C. Neuhaus. 2000. Biosynthesis of lipoteichoic acid in Lactobacillus rhamnosus: role of DltD in d-alanylation. J. Bacteriol. 182:2855-2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng, L., D. L. Kasper, T. P. Krick, and M. R. Wessels. 2000. Characterization of the linkage between the type III capsular polysaccharide and the bacterial cell wall of group B streptococcus. J. Biol. Chem. 275:7497-7504. [DOI] [PubMed] [Google Scholar]
- 35.Doran, K. S., J. C. Chang, V. M. Benoit, L. Eckmann, and V. Nizet. 2002. Group B streptococcal beta-hemolysin/cytolysin promotes invasion of human lung epithelial cells and the release of interleukin-8. J. Infect. Dis. 185:196-203. [DOI] [PubMed] [Google Scholar]
- 36.Doran, K. S., E. J. Engelson, A. Khosravi, H. C. Maisey, I. Fedtke, O. Equils, K. S. Michelsen, M. Arditi, A. Peschel, and V. Nizet. 2005. Blood-brain barrier invasion by group B streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. J. Clin. Investig. 115:2499-2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doran, K. S., G. Y. Liu, and V. Nizet. 2003. Group B streptococcal beta-hemolysin/cytolysin activates neutrophil signaling pathways in brain endothelium and contributes to development of meningitis. J. Clin. Investig. 112:736-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doran, K. S., and V. Nizet. 2004. Molecular pathogenesis of neonatal group B streptococcal infection: no longer in its infancy. Mol. Microbiol. 54:23-31. [DOI] [PubMed] [Google Scholar]
- 39.Dorschner, R. A., K. H. Lin, M. Murakami, and R. L. Gallo. 2003. Neonatal skin in mice and humans expresses increased levels of antimicrobial peptides: innate immunity during development of the adaptive response. Pediatr. Res. 53:566-572. [DOI] [PubMed] [Google Scholar]
- 40.Drossou, V., F. Kanakoudi, E. Diamanti, V. Tzimouli, T. Konstantinidis, A. Germenis, G. Kremenopoulos, and V. Katsougiannopoulos. 1995. Concentrations of main serum opsonins in early infancy. Arch. Dis. Child. 72:F172-F175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du, X., A. Poltorak, M. Silva, and B. Beutler. 1999. Analysis of Tlr4-mediated LPS signal transduction in macrophages by mutational modification of the receptor. Blood Cells Mol. Dis. 25:328-338. [DOI] [PubMed] [Google Scholar]
- 42.Dziarski, R., and D. Gupta. 2005. Staphylococcus aureus peptidoglycan is a Toll-like receptor 2 activator: a reevaluation. Infect. Immun. 73:5212-5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edwards, M. S., and P. A. Fuselier. 1983. Enhanced susceptibility of mice with streptozotocin-induced diabetes to type II group B streptococcal infection. Infect. Immun. 39:580-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edwards, M. S., M. A. Rench, A. A. Haffar, M. A. Murphy, M. M. Desmond, and C. J. Baker. 1985. Long-term sequelae of group B streptococcal meningitis in infants. J. Pediatr. 106:717-722. [DOI] [PubMed] [Google Scholar]
- 45.Edwards, M. S., M. R. Wessels, and C. J. Baker. 1993. Capsular polysaccharide regulates neutrophil complement receptor interactions with type III group B streptococci. Infect. Immun. 61:2866-2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fettucciari, K., E. Rosati, L. Scaringi, P. Cornacchione, G. Migliorati, R. Sabatini, I. Fetriconi, R. Rossi, and P. Marconi. 2000. Group B streptococcus induces apoptosis in macrophages. J. Immunol. 165:3923-3933. [DOI] [PubMed] [Google Scholar]
- 47.Fitzgerald, K. A., E. M. Palsson-McDermott, A. G. Bowie, C. A. Jefferies, A. S. Mansell, G. Brady, E. Brint, A. Dunne, P. Gray, M. T. Harte, D. McMurray, D. E. Smith, J. E. Sims, T. A. Bird, and L. A. O'Neill. 2001. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413:78-83. [DOI] [PubMed] [Google Scholar]
- 48.Fitzgerald, K. A., D. C. Rowe, B. J. Barnes, D. R. Caffrey, A. Visintin, E. Latz, B. Monks, P. M. Pitha, and D. T. Golenbock. 2003. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the Toll adapters TRAM and TRIF. J. Exp. Med. 198:1043-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flo, T. H., O. Halaas, E. Lien, L. Ryan, G. Teti, D. T. Golenbock, A. Sundan, and T. Espevik. 2000. Human Toll-like receptor 2 mediates monocyte activation by Listeria monocytogenes, but not by group B streptococci or lipopolysaccharide. J. Immunol. 164:2064-2069. [DOI] [PubMed] [Google Scholar]
- 50.Fluegge, K., O. Schweier, E. Schiltz, S. Batsford, and R. Berner. 2004. Identification and immunoreactivity of proteins released from Streptococcus agalactiae. Eur. J. Clin. Microbiol. Infect. Dis. 23:818-824. [DOI] [PubMed] [Google Scholar]
- 51.Forster-Waldl, E., K. Sadeghi, D. Tamandl, B. Gerhold, U. Hallwirth, K. Rohrmeister, M. Hayde, A. R. Prusa, K. Herkner, G. Boltz-Nitulescu, A. Pollak, and A. Spittler. 2005. Monocyte Toll-like receptor 4 expression and LPS-induced cytokine production increase during gestational aging. Pediatr. Res. 58:121-124. [DOI] [PubMed] [Google Scholar]
- 52.Fujita, M., T. Into, M. Yasuda, T. Okusawa, S. Hamahira, Y. Kuroki, A. Eto, T. Nisizawa, M. Morita, and K. Shibata. 2003. Involvement of leucine residues at positions 107, 112, and 115 in a leucine-rich repeat motif of human Toll-like receptor 2 in the recognition of diacylated lipoproteins and lipopeptides and Staphylococcus aureus peptidoglycans. J. Immunol. 171:3675-3683. [DOI] [PubMed] [Google Scholar]
- 53.Furudoi, S., T. Yoshii, and T. Komori. 2003. Balance of tumor necrosis factor alpha and interleukin-10 in a buccal infection in a streptozotocin-induced diabetic rat model. Cytokine 24:143-149. [DOI] [PubMed] [Google Scholar]
- 54.Gahr, M., M. Schulze, D. Scheffczyk, C. P. Speer, and J. H. Peters. 1987. Diminished release of lactoferrin from polymorphonuclear leukocytes of human neonates. Acta Haematol. 77:90-94. [DOI] [PubMed] [Google Scholar]
- 55.Girardin, S. E., I. G. Boneca, L. A. Carneiro, A. Antignac, M. Jehanno, J. Viala, K. Tedin, M. K. Taha, A. Labigne, U. Zahringer, A. J. Coyle, P. S. DiStefano, J. Bertin, P. J. Sansonetti, and D. J. Philpott. 2003. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300:1584-1587. [DOI] [PubMed] [Google Scholar]
- 56.Girardin, S. E., R. Tournebize, M. Mavris, A. L. Page, X. Li, G. R. Stark, J. Bertin, P. S. DiStefano, M. Yaniv, P. J. Sansonetti, and D. J. Philpott. 2001. CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2:736-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goodrum, K. J., J. Dierksheide, and B. J. Yoder. 1995. Tumor necrosis factor alpha acts as an autocrine second signal with gamma interferon to induce nitric oxide in group B streptococcus-treated macrophages. Infect. Immun. 63:3715-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grangette, C., S. Nutten, E. Palumbo, S. Morath, C. Hermann, J. Dewulf, B. Pot, T. Hartung, P. Hols, and A. Mercenier. 2005. Enhanced antiinflammatory capacity of a Lactobacillus plantarum mutant synthesizing modified teichoic acids. Proc. Natl. Acad. Sci. USA 102:10321-10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gravekamp, C., D. L. Kasper, J. L. Michel, D. E. Kling, V. Carey, and L. C. Madoff. 1997. Immunogenicity and protective efficacy of the alpha C protein of group B streptococci are inversely related to the number of repeats. Infect. Immun. 65:5216-5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han, S. H., J. H. Kim, M. Martin, S. M. Michalek, and M. H. Nahm. 2003. Pneumococcal lipoteichoic acid (LTA) is not as potent as staphylococcal LTA in stimulating Toll-like receptor 2. Infect. Immun. 71:5541-5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harju, K., M. Ojaniemi, S. Rounioja, V. Glumoff, R. Paananen, R. Vuolteenaho, and M. Hallman. 2005. Expression of Toll-like receptor 4 and endotoxin responsiveness in mice during perinatal period. Pediatr. Res. 57:644-648. [DOI] [PubMed] [Google Scholar]
- 62.Harris, E. S., T. M. McIntyre, S. M. Prescott, and G. A. Zimmerman. 2000. The leukocyte integrins. J. Biol. Chem. 275:23409-23412. [DOI] [PubMed] [Google Scholar]
- 63.Harris, M. C., M. Shalit, and F. S. Southwick. 1993. Diminished actin polymerization by neutrophils from newborn infants. Pediatr. Res. 33:27-31. [DOI] [PubMed] [Google Scholar]
- 64.Harris, T. O., D. W. Shelver, J. F. Bohnsack, and C. E. Rubens. 2003. A novel streptococcal surface protease promotes virulence, resistance to opsonophagocytosis, and cleavage of human fibrinogen. J. Clin. Investig. 111:61-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heath, P. T., G. Balfour, A. M. Weisner, A. Efstratiou, T. L. Lamagni, H. Tighe, L. A. O'Connell, M. Cafferkey, N. Q. Verlander, A. Nicoll, and A. C. McCartney. 2004. Group B streptococcal disease in UK and Irish infants younger than 90 days. Lancet 363:292-294. [DOI] [PubMed] [Google Scholar]
- 66.Henneke, P., and D. T. Golenbock. 2002. Innate immune recognition of lipopolysaccharide by endothelial cells. Crit. Care Med. 30:S207-S213. [DOI] [PubMed] [Google Scholar]
- 67.Henneke, P., S. Morath, S. Uematsu, S. Weichert, M. Pfitzenmaier, O. Takeuchi, A. Muller, C. Poyart, S. Akira, R. Berner, G. Teti, A. Geyer, T. Hartung, P. Trieu-Cuot, D. L. Kasper, and D. T. Golenbock. 2005. Role of lipotechoic acid in the phagocyte response to group B streptococcus. J. Immunol. 174:6449-6455. [DOI] [PubMed] [Google Scholar]
- 68.Henneke, P., I. Osmers, K. Bauer, N. Lamping, H. T. Versmold, and R. R. Schumann. 2003. Impaired CD14-dependent and independent response of polymorphonuclear leukocytes in preterm infants. J. Perinat. Med. 31:176-183. [DOI] [PubMed] [Google Scholar]
- 69.Henneke, P., O. Takeuchi, R. Malley, E. Lien, R. R. Ingalls, M. W. Freeman, T. Mayadas, V. Nizet, S. Akira, D. L. Kasper, and D. T. Golenbock. 2002. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J. Immunol. 169:3970-3977. [DOI] [PubMed] [Google Scholar]
- 70.Henneke, P., O. Takeuchi, J. A. van Strijp, H. K. Guttormsen, J. A. Smith, A. B. Schromm, T. A. Espevik, S. Akira, V. Nizet, D. L. Kasper, and D. T. Golenbock. 2001. Novel engagement of CD14 and multiple Toll-like receptors by group B streptococci. J. Immunol. 167:7069-7076. [DOI] [PubMed] [Google Scholar]
- 71.Horng, T., G. M. Barton, and R. Medzhitov. 2001. TIRAP: an adapter molecule in the Toll signaling pathway. Nat. Immunol. 2:835-841. [DOI] [PubMed] [Google Scholar]
- 72.Hughes, M. J., J. C. Moore, J. D. Lane, R. Wilson, P. K. Pribul, Z. N. Younes, R. J. Dobson, P. Everest, A. J. Reason, J. M. Redfern, F. M. Greer, T. Paxton, M. Panico, H. R. Morris, R. G. Feldman, and J. D. Santangelo. 2002. Identification of major outer surface proteins of Streptococcus agalactiae. Infect. Immun. 70:1254-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hyde, T. B., T. M. Hilger, A. Reingold, M. M. Farley, K. L. O'Brien, and A. Schuchat. 2002. Trends in incidence and antimicrobial resistance of early-onset sepsis: population-based surveillance in San Francisco and Atlanta. Pediatrics 110:690-695. [DOI] [PubMed] [Google Scholar]
- 74.Imler, J. L., and J. A. Hoffmann. 2001. Toll receptors in innate immunity. Trends Cell Biol. 11:304-311. [DOI] [PubMed] [Google Scholar]
- 75.Jiang, S. M., M. J. Cieslewicz, D. L. Kasper, and M. R. Wessels. 2005. Regulation of virulence by a two-component system in group B streptococcus. J. Bacteriol. 187:1105-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jones, A. L., K. M. Knoll, and C. E. Rubens. 2000. Identification of Streptococcus agalactiae virulence genes in the neonatal rat sepsis model using signature-tagged mutagenesis. Mol. Microbiol. 37:1444-1455. [DOI] [PubMed] [Google Scholar]
- 77.Kallman, J., J. Schollin, C. Schalen, A. Erlandsson, and E. Kihlstrom. 1998. Impaired phagocytosis and opsonisation towards group B streptococci in preterm neonates. Arch. Dis. Child. 78:F46-F50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kasper, D. L., C. J. Baker, R. S. Baltimore, J. H. Crabb, G. Schiffman, and H. J. Jennings. 1979. Immunodeterminant specificity of human immunity to type III group B streptococcus. J. Exp. Med. 149:327-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kenzel, S., G. Mancuso, R. Malley, G. Teti, D. T. Golenbock, and P. Henneke. 2006. c-Jun kinase is a critical signaling molecule in a neonatal model of group B streptococcal sepsis. J. Immunol. 176:3181-3188. [DOI] [PubMed] [Google Scholar]
- 80.Klein, R. B., T. J. Fischer, S. E. Gard, M. Biberstein, K. C. Rich, and E. R. Stiehm. 1977. Decreased mononuclear and polymorphonuclear chemotaxis in human newborns, infants, and young children. Pediatrics 60:467-472. [PubMed] [Google Scholar]
- 81.Krueger, M., M. S. Nauck, S. Sang, R. Hentschel, H. Wieland, and R. Berner. 2001. Cord blood levels of interleukin-6 and interleukin-8 for the immediate diagnosis of early-onset infection in premature infants. Biol. Neonate 80:118-123. [DOI] [PubMed] [Google Scholar]
- 82.Lalioui, L., E. Pellegrini, S. Dramsi, M. Baptista, N. Bourgeois, F. Doucet-Populaire, C. Rusniok, M. Zouine, P. Glaser, F. Kunst, C. Poyart, and P. Trieu-Cuot. 2005. The SrtA sortase of Streptococcus agalactiae is required for cell wall anchoring of proteins containing the LPXTG motif, for adhesion to epithelial cells, and for colonization of the mouse intestine. Infect. Immun. 73:3342-3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lamy, M. C., M. Zouine, J. Fert, M. Vergassola, E. Couve, E. Pellegrini, P. Glaser, F. Kunst, T. Msadek, P. Trieu-Cuot, and C. Poyart. 2004. CovS/CovR of group B streptococcus: a two-component global regulatory system involved in virulence. Mol. Microbiol. 54:1250-1268. [DOI] [PubMed] [Google Scholar]
- 84.Lang, S., and M. Palmer. 2003. Characterization of Streptococcus agalactiae CAMP factor as a pore-forming toxin. J. Biol. Chem. 278:38167-38173. [DOI] [PubMed] [Google Scholar]
- 85.Le-Barillec, K., P. Henneke, and D. T. Golenbock. 2000. Toll receptors: guardians of the immune system and effectors of septic shock. Adv. Sepsis 1:23-30. [Google Scholar]
- 86.Leopold, K., and W. Fischer. 1991. Separation of the poly(glycerophosphate) lipoteichoic acids of Enterococcus faecalis Kiel 27738, Enterococcus hirae ATCC 9790 and Leuconostoc mesenteroides DSM 20343 into molecular species by affinity chromatography on concanavalin A. Eur. J. Biochem. 196:475-482. [DOI] [PubMed] [Google Scholar]
- 87.LeVine, A. M., J. A. Reed, K. E. Kurak, E. Cianciolo, and J. A. Whitsett. 1999. GM-CSF-deficient mice are susceptible to pulmonary group B streptococcal infection. J. Clin. Investig. 103:563-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.LeVine, A. M., J. A. Whitsett, J. A. Gwozdz, T. R. Richardson, J. H. Fisher, M. S. Burhans, and T. R. Korfhagen. 2000. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J. Immunol. 165:3934-3940. [DOI] [PubMed] [Google Scholar]
- 89.Levy, O. 2004. Antimicrobial proteins and peptides: anti-infective molecules of mammalian leukocytes. J. Leukoc. Biol. 76:909-925. [DOI] [PubMed] [Google Scholar]
- 90.Levy, O., R. M. Jean-Jacques, C. Cywes, R. B. Sisson, K. A. Zarember, P. J. Godowski, J. L. Christianson, H. K. Guttormsen, M. C. Carroll, A. Nicholson-Weller, and M. R. Wessels. 2003. Critical role of the complement system in group B streptococcus-induced tumor necrosis factor alpha release. Infect. Immun. 71:6344-6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Levy, O., S. Martin, E. Eichenwald, T. Ganz, E. Valore, S. F. Carroll, K. Lee, D. Goldmann, and G. M. Thorne. 1999. Impaired innate immunity in the newborn: newborn neutrophils are deficient in bactericidal/permeability-increasing protein. Pediatrics 104:1327-1333. [DOI] [PubMed] [Google Scholar]
- 92.Levy, O., K. A. Zarember, R. M. Roy, C. Cywes, P. J. Godowski, and M. R. Wessels. 2004. Selective impairment of TLR-mediated innate immunity in human newborns: neonatal blood plasma reduces monocyte TNF-alpha induction by bacterial lipopeptides, lipopolysaccharide, and imiquimod, but preserves the response to R-848. J. Immunol. 173:4627-4634. [DOI] [PubMed] [Google Scholar]
- 93.Liu, G. Y., K. S. Doran, T. Lawrence, N. Turkson, M. Puliti, L. Tissi, and V. Nizet. 2004. Sword and shield: linked group B streptococcal beta-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proc. Natl. Acad. Sci. USA 101:14491-14496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maione, D., I. Margarit, C. D. Rinaudo, V. Masignani, M. Mora, M. Scarselli, H. Tettelin, C. Brettoni, E. T. Iacobini, R. Rosini, N. D'Agostino, L. Miorin, S. Buccato, M. Mariani, G. Galli, R. Nogarotto, V. Nardi Dei, F. Vegni, C. Fraser, G. Mancuso, G. Teti, L. C. Madoff, L. C. Paoletti, R. Rappuoli, D. L. Kasper, J. L. Telford, and G. Grandi. 2005. Identification of a universal group B streptococcus vaccine by multiple genome screen. Science 309:148-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mancuso, G., A. Midiri, C. Beninati, C. Biondo, R. Galbo, S. Akira, P. Henneke, D. Golenbock, and G. Teti. 2004. Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J. Immunol. 172:6324-6329. [DOI] [PubMed] [Google Scholar]
- 96.Mancuso, G., F. Tomasello, C. von Hunolstein, G. Orefici, and G. Teti. 1994. Induction of tumor necrosis factor alpha by the group- and type-specific polysaccharides from type III group B streptococci. Infect. Immun. 62:2748-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Manimtim, W. M., J. D. Hasday, L. Hester, K. D. Fairchild, J. C. Lovchik, and R. M. Viscardi. 2001. Ureaplasma urealyticum modulates endotoxin-induced cytokine release by human monocytes derived from preterm and term newborns and adults. Infect. Immun. 69:3906-3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mariscalco, M. M., M. H. Tcharmtchi, and C. W. Smith. 1998. P-selectin support of neonatal neutrophil adherence under flow: contribution of L-selectin, LFA-1, and ligand(s) for P-selectin. Blood 91:4776-4785. [PubMed] [Google Scholar]
- 99.Marodi, L., P. C. Leijh, and R. van Furth. 1984. Characteristics and functional capacities of human cord blood granulocytes and monocytes. Pediatr. Res. 18:1127-1131. [DOI] [PubMed] [Google Scholar]
- 100.Martin, T. R., J. T. Ruzinski, C. E. Rubens, E. Y. Chi, and C. B. Wilson. 1992. The effect of type-specific polysaccharide capsule on the clearance of group B streptococci from the lungs of infant and adult rats. J. Infect. Dis. 165:306-314. [DOI] [PubMed] [Google Scholar]
- 101.Maurer, J. J., and S. J. Mattingly. 1991. Molecular analysis of lipoteichoic acid from Streptococcus agalactiae. J. Bacteriol. 173:487-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mazade, M. A., and M. S. Edwards. 2001. Impairment of type III group B streptococcus-stimulated superoxide production and opsonophagocytosis by neutrophils in diabetes. Mol. Genet. Metab. 73:259-267. [DOI] [PubMed] [Google Scholar]
- 103.Mazmanian, S. K., G. Liu, H. Ton-That, and O. Schneewind. 1999. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 285:760-763. [DOI] [PubMed] [Google Scholar]
- 104.McEvoy, L. T., H. Zakem-Cloud, and M. F. Tosi. 1996. Total cell content of CR3 (CD11b/CD18) and LFA-1 (CD11a/CD18) in neonatal neutrophils: relationship to gestational age. Blood 87:3929-3933. [PubMed] [Google Scholar]
- 105.Medvedev, A. E., T. Flo, R. R. Ingalls, D. T. Golenbock, G. Teti, S. N. Vogel, and T. Espevik. 1998. Involvement of CD14 and complement receptors CR3 and CR4 in nuclear factor-kappaB activation and TNF production induced by lipopolysaccharide and group B streptococcal cell walls. J. Immunol. 160:4535-4542. [PubMed] [Google Scholar]
- 106.Medzhitov, R., and C. Janeway, Jr. 2000. Innate immunity. N. Engl. J. Med. 343:338-344. [DOI] [PubMed] [Google Scholar]
- 107.Medzhitov, R., P. Preston-Hurlburt, and C. A. Janeway, Jr. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394-397. [DOI] [PubMed] [Google Scholar]
- 108.Medzhitov, R., P. Preston-Hurlburt, E. Kopp, A. Stadlen, C. Chen, S. Ghosh, and C. A. Janeway, Jr. 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 2:253-258. [DOI] [PubMed] [Google Scholar]
- 109.Mikamo, H., A. K. Johri, L. C. Paoletti, L. C. Madoff, and A. B. Onderdonk. 2004. Adherence to, invasion by, and cytokine production in response to serotype VIII group B streptococci. Infect. Immun. 72:4716-4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moore, M. R., S. J. Schrag, and A. Schuchat. 2003. Effects of intrapartum antimicrobial prophylaxis for prevention of group-B-streptococcal disease on the incidence and ecology of early-onset neonatal sepsis. Lancet Infect. Dis. 3:201-213. [DOI] [PubMed] [Google Scholar]
- 111.Morath, S., A. Geyer, and T. Hartung. 2001. Structure-function relationship of cytokine induction by lipoteichoic acid from Staphylococcus aureus. J. Exp. Med. 193:393-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Morath, S., A. Geyer, I. Spreitzer, C. Hermann, and T. Hartung. 2002. Structural decomposition and heterogeneity of commercial lipoteichoic acid preparations. Infect. Immun. 70:938-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Morath, S., A. Stadelmaier, A. Geyer, R. R. Schmidt, and T. Hartung. 2002. Synthetic lipoteichoic acid from Staphylococcus aureus is a potent stimulus of cytokine release. J. Exp. Med. 195:1635-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Navarre, W. W., and O. Schneewind. 1999. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 63:174-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Neuhaus, F. C., and J. Baddiley. 2003. A continuum of anionic charge: structures and functions of d-alanyl-teichoic acids in gram-positive bacteria. Microbiol. Mol. Biol. Rev. 67:686-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nupponen, I., E. Pesonen, S. Andersson, A. Makela, R. Turunen, H. Kautiainen, and H. Repo. 2002. Neutrophil activation in preterm infants who have respiratory distress syndrome. Pediatrics 110:36-41. [DOI] [PubMed] [Google Scholar]
- 117.Ozinsky, A., K. D. Smith, D. Hume, and D. M. Underhill. 2000. Co-operative induction of pro-inflammatory signaling by Toll-like receptors. J. Endotoxin Res. 6:393-396. [PubMed] [Google Scholar]
- 118.Ozinsky, A., D. M. Underhill, J. D. Fontenot, A. M. Hajjar, K. D. Smith, C. B. Wilson, L. Schroeder, and A. Aderem. 2000. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc. Natl. Acad. Sci. USA 97:13766-13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pahwa, S. G., R. Pahwa, E. Grimes, and E. Smithwick. 1977. Cellular and humoral components of monocyte and neutrophil chemotaxis in cord blood. Pediatr. Res. 11:677-680. [DOI] [PubMed] [Google Scholar]
- 120.Palazzi, D. L., M. A. Rench, M. S. Edwards, and C. J. Baker. 2004. Use of type V group B streptococcal conjugate vaccine in adults 65-85 years old. J. Infect. Dis. 190:558-564. [DOI] [PubMed] [Google Scholar]
- 121.Peat, E. B., N. H. Augustine, W. K. Drummond, J. F. Bohnsack, and H. R. Hill. 1995. Effects of fibronectin and group B streptococci on tumour necrosis factor-alpha production by human culture-derived macrophages. Immunology 84:440-445. [PMC free article] [PubMed] [Google Scholar]
- 122.Peters, A. M., P. Bertram, M. Gahr, and C. P. Speer. 1993. Reduced secretion of interleukin-1 and tumor necrosis factor-alpha by neonatal monocytes. Biol. Neonate 63:157-162. [DOI] [PubMed] [Google Scholar]
- 123.Poyart, C., M. C. Lamy, C. Boumaila, F. Fiedler, and P. Trieu-Cuot. 2001. Regulation of d-alanyl-lipoteichoic acid biosynthesis in Streptococcus agalactiae involves a novel two-component regulatory system. J. Bacteriol. 183:6324-6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Poyart, C., E. Pellegrini, O. Gaillot, C. Boumaila, M. Baptista, and P. Trieu-Cuot. 2001. Contribution of Mn-cofactored superoxide dismutase (SodA) to the virulence of Streptococcus agalactiae. Infect. Immun. 69:5098-5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Poyart, C., E. Pellegrini, M. Marceau, M. Baptista, F. Jaubert, M. C. Lamy, and P. Trieu-Cuot. 2003. Attenuated virulence of Streptococcus agalactiae deficient in d-alanyl-lipoteichoic acid is due to an increased susceptibility to defensins and phagocytic cells. Mol. Microbiol. 49:1615-1625. [DOI] [PubMed] [Google Scholar]
- 126.Pritzlaff, C. A., J. C. Chang, S. P. Kuo, G. S. Tamura, C. E. Rubens, and V. Nizet. 2001. Genetic basis for the beta-haemolytic/cytolytic activity of group B streptococcus. Mol. Microbiol. 39:236-247. [DOI] [PubMed] [Google Scholar]
- 127.Reeves, E. P., H. Lu, H. L. Jacobs, C. G. Messina, S. Bolsover, G. Gabella, E. O. Potma, A. Warley, J. Roes, and A. W. Segal. 2002. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416:291-297. [DOI] [PubMed] [Google Scholar]
- 128.Ring, A., J. S. Braun, V. Nizet, W. Stremmel, and J. L. Shenep. 2000. Group B streptococcal beta-hemolysin induces nitric oxide production in murine macrophages. J. Infect. Dis. 182:150-157. [DOI] [PubMed] [Google Scholar]
- 129.Rowen, J. L., C. W. Smith, and M. S. Edwards. 1995. Group B streptococci elicit leukotriene B4 and interleukin-8 from human monocytes: neonates exhibit a diminished response. J. Infect. Dis. 172:420-426. [DOI] [PubMed] [Google Scholar]
- 130.Rubens, C. E., H. V. Raff, J. C. Jackson, E. Y. Chi, J. T. Bielitzki, and S. L. Hillier. 1991. Pathophysiology and histopathology of group B streptococcal sepsis in Macaca nemestrina primates induced after intraamniotic inoculation: evidence for bacterial cellular invasion. J. Infect. Dis. 164:320-330. [DOI] [PubMed] [Google Scholar]
- 131.Rubens, C. E., M. R. Wessels, L. M. Heggen, and D. L. Kasper. 1987. Transposon mutagenesis of type III group B streptococcus: correlation of capsule expression with virulence. Proc. Natl. Acad. Sci. USA 84:7208-7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sacchi, F., N. H. Augustine, M. M. Coello, E. Z. Morris, and H. R. Hill. 1987. Abnormality in actin polymerization associated with defective chemotaxis in neutrophils from neonates. Int. Arch. Allergy Appl. Immunol. 84:32-39. [DOI] [PubMed] [Google Scholar]
- 133.Schleifer, K. H., and O. Kandler. 1972. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36:407-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schuchat, A., T. Hilger, E. Zell, M. M. Farley, A. Reingold, L. Harrison, L. Lefkowitz, R. Danila, K. Stefonek, N. Barrett, D. Morse, and R. Pinner. 2001. Active bacterial core surveillance of the emerging infections program network. Emerg. Infect. Dis. 7:92-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schuchat, A., K. Robinson, J. D. Wenger, L. H. Harrison, M. Farley, A. L. Reingold, L. Lefkowitz, and B. A. Perkins. 1997. Bacterial meningitis in the United States in 1995. N. Engl. J. Med. 337:970-976. [DOI] [PubMed] [Google Scholar]
- 136.Schwandner, R., R. Dziarski, H. Wesche, M. Rothe, and C. J. Kirschning. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. J. Biol. Chem. 274:17406-17409. [DOI] [PubMed] [Google Scholar]
- 137.Shappell, S. B., C. Toman, D. C. Anderson, A. A. Taylor, M. L. Entman, and C. W. Smith. 1990. Mac-1 (CD11b/CD18) mediates adherence-dependent hydrogen peroxide production by human and canine neutrophils. J. Immunol. 144:2702-2711. [PubMed] [Google Scholar]
- 138.Shigeoka, A. O., R. J. Gobel, J. Janatova, and H. R. Hill. 1988. Neutrophil mobilization induced by complement fragments during experimental group B streptococcal (GBS) infection. Am. J. Pathol. 133:623-629. [PMC free article] [PubMed] [Google Scholar]
- 139.Sun, C. M., E. Deriaud, C. Leclerc, and R. Lo-Man. 2005. Upon TLR9 signaling, CD5+ B cells control the IL-12-dependent Th1-priming capacity of neonatal DCs. Immunity 22:467-477. [DOI] [PubMed] [Google Scholar]
- 140.Takahashi, S., Y. Nagano, N. Nagano, O. Hayashi, F. Taguchi, and Y. Okuwaki. 1995. Role of C5a-ase in group B streptococcal resistance to opsonophagocytic killing. Infect. Immun. 63:4764-4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Takeuchi, O., A. Kaufmann, K. Grote, T. Kawai, K. Hoshino, M. Morr, P. F. Muhlradt, and S. Akira. 2000. Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a Toll-like receptor 2- and MyD88-dependent signaling pathway. J. Immunol. 164:554-557. [DOI] [PubMed] [Google Scholar]
- 142.Takeuchi, O., T. Kawai, P. F. Muhlradt, M. Morr, J. D. Radolf, A. Zychlinsky, K. Takeda, and S. Akira. 2001. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 13:933-940. [DOI] [PubMed] [Google Scholar]
- 143.Takeuchi, O., S. Sato, T. Horiuchi, K. Hoshino, K. Takeda, Z. Dong, R. L. Modlin, and S. Akira. 2002. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 169:10-14. [DOI] [PubMed] [Google Scholar]
- 144.Travassos, L. H., S. E. Girardin, D. J. Philpott, D. Blanot, M. A. Nahori, C. Werts, and I. G. Boneca. 2004. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 5:1000-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vallejo, J. G., C. J. Baker, and M. S. Edwards. 1996. Roles of the bacterial cell wall and capsule in induction of tumor necrosis factor alpha by type III group B streptococci. Infect. Immun. 64:5042-5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.von Hunolstein, C., A. Totolian, G. Alfarone, G. Mancuso, V. Cusumano, G. Teti, and G. Orefici. 1997. Soluble antigens from group B streptococci induce cytokine production in human blood cultures. Infect. Immun. 65:4017-4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Weatherstone, K. B., and E. A. Rich. 1989. Tumor necrosis factor/cachectin and interleukin-1 secretion by cord blood monocytes from premature and term neonates. Pediatr. Res. 25:342-346. [DOI] [PubMed] [Google Scholar]
- 148.Wesche, H., X. Gao, X. Li, C. J. Kirschning, G. R. Stark, and Z. Cao. 1999. IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J. Biol. Chem. 274:19403-19410. [DOI] [PubMed] [Google Scholar]
- 149.Wessels, M. R., P. Butko, M. Ma, H. B. Warren, A. L. Lage, and M. C. Carroll. 1995. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc. Natl. Acad. Sci. USA 92:11490-11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wessels, M. R., C. E. Rubens, V. J. Benedi, and D. L. Kasper. 1989. Definition of a bacterial virulence factor: sialylation of the group B streptococcal capsule. Proc. Natl. Acad. Sci. USA 86:8983-8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Williams, P. A., J. F. Bohnsack, N. H. Augustine, W. K. Drummond, C. E. Rubens, and H. R. Hill. 1993. Production of tumor necrosis factor by human cells in vitro and in vivo, induced by group B streptococci. J. Pediatr. 123:292-300. [DOI] [PubMed] [Google Scholar]
- 152.Wilson, C. B., and W. M. Weaver. 1985. Comparative susceptibility of group B streptococci and Staphylococcus aureus to killing by oxygen metabolites. J. Infect. Dis. 152:323-329. [DOI] [PubMed] [Google Scholar]
- 153.Wolach, B., D. Sonnenschein, R. Gavrieli, O. Chomsky, A. Pomeranz, and I. Yuli. 1998. Neonatal neutrophil inflammatory responses: parallel studies of light scattering, cell polarization, chemotaxis, superoxide release, and bactericidal activity. Am. J. Hematol. 58:8-15. [DOI] [PubMed] [Google Scholar]
- 154.Xu, Y., X. Tao, B. Shen, T. Horng, R. Medzhitov, J. L. Manley, and L. Tong. 2000. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature 408:111-115. [DOI] [PubMed] [Google Scholar]
- 155.Yamamoto, M., S. Sato, H. Hemmi, S. Uematsu, K. Hoshino, T. Kaisho, O. Takeuchi, K. Takeda, and S. Akira. 2003. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat. Immunol. 4:1144-1150. [DOI] [PubMed] [Google Scholar]
- 156.Yan, S. R., G. Qing, D. M. Byers, A. W. Stadnyk, W. Al-Hertani, and R. Bortolussi. 2004. Role of MyD88 in diminished tumor necrosis factor alpha production by newborn mononuclear cells in response to lipopolysaccharide. Infect. Immun. 72:1223-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yang, K. D., N. H. Augustine, M. F. Shaio, J. F. Bohnsack, and H. R. Hill. 1994. Effects of fibronectin on actin organization and respiratory burst activity in neutrophils, monocytes, and macrophages. J. Cell Physiol. 158:347-353. [DOI] [PubMed] [Google Scholar]
- 158.Yang, K. D., J. M. Bathras, A. O. Shigeoka, J. James, S. H. Pincus, and H. R. Hill. 1989. Mechanisms of bacterial opsonization by immune globulin intravenous: correlation of complement consumption with opsonic activity and protective efficacy. J. Infect. Dis. 159:701-707. [DOI] [PubMed] [Google Scholar]
- 159.Yoshimura, A., E. Lien, R. R. Ingalls, E. Tuomanen, R. Dziarski, and D. Golenbock. 1999. Cutting edge: recognition of gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J. Immunol. 163:1-5. [PubMed] [Google Scholar]
- 160.Zaleznik, D. F., M. A. Rench, S. Hillier, M. A. Krohn, R. Platt, M. L. Lee, A. E. Flores, P. Ferrieri, and C. J. Baker. 2000. Invasive disease due to group B streptococcus in pregnant women and neonates from diverse population groups. Clin. Infect. Dis. 30:276-281. [DOI] [PubMed] [Google Scholar]