

Abstract
Campylobacter jejuni is a leading cause of bacterial food-borne diarrheal disease throughout the world and the most frequent antecedent of autoimmune neuropathy Guillain-Barré syndrome. While infection is associated with immune memory, little is known regarding the role of the epithelium in targeting dendritic cells (DC) for initiating the appropriate adaptive immune response to C. jejuni. The objective of this study was to define the role for the intestinal epithelium in the induction of the adaptive immune response in C. jejuni infection by assessing the production of DC and T-cell chemoattractants. Human T84 epithelial cells were used as model intestinal epithelia. Infection of T84 cells with C. jejuni dose- and time-dependently up-regulated DC and T-cell chemokine gene transcription and secretion. Induction required live bacteria and was in the physiologically relevant direction for attraction of mucosal immunocytes. C. jejuni-activated NF-κB signaling was shown to be essential for proinflammatory chemokine secretion. Notably, C. jejuni secretion occurred independently of flagellin identification by Toll-like receptor 5. Secretion of a DC chemoattractant by differing clinical C. jejuni isolates suggested adherence/invasion were key virulence determinants of epithelial chemokine secretion. The regulated epithelial expression of DC and T-cell chemoattractants suggests a mechanism for the directed trafficking of immune cells required for the initiation of adaptive immunity in campylobacteriosis. Chemokine secretion occurs despite Campylobacter evasion of the flagellin pattern recognition receptor, suggesting that alternate host defense strategies limit disease pathogenesis.
Campylobacter jejuni is a leading cause of bacterial food-borne diarrheal disease throughout the world and is the most frequent antecedent of the debilitating neuropathy Guillain-Barré syndrome (1, 2, 87). C. jejuni and the related Campylobacter coli are as prevalent as the well-characterized Salmonella and Shigella spp. as a cause of diarrhea, with an estimated 2.1 to 2.4 million cases per annum in the United States alone (2). In developing countries, Campylobacter infections are endemic and are a significant cause of childhood morbidity due to diarrheal illness. In addition to diarrheal disease and Guillain-Barré syndrome, C. jejuni infection has also been implicated in longer-term sequelae, such as Reiter's syndrome (reactive arthritis) (62). Despite the prevalence of disease related to infection with this pathogen, the host immune response and the mechanism(s) by which C. jejuni causes disease remain poorly defined. Lack of a reliable animal model that faithfully recapitulates human campylobacteriosis, combined with marked variation among clinical isolates, has contributed to the paucity of information on C. jejuni virulence and pathogenesis (58, 86).
Prior studies of C. jejuni enteritis have shown that this pathogen resides primarily in the intestinal mucosa, colonizing both the small and large intestines. Histological examination of humans and animals has shown pathology primarily in the colon (5, 10, 67), with C. jejuni being observed within intestinal crypts, both close to the cell surface and inside intestinal epithelial cells (4, 5, 67). Disruption of intracellular epithelial architecture and inflammation of intestinal mucosa are thus hallmark features of C. jejuni pathogenesis (10, 67). Generation of specific antibody titers (70, 72) and association with autoimmune disease (1) suggest that C. jejuni bacteria, despite rarely penetrating past the epithelial layer, are potent activators of the human adaptive immune response. The intestinal epithelium has been shown to play a critical role in orchestrating the linkage between the innate and adaptive immune responses by producing a defined set of chemoattractants. Thus, the intestinal epithelium has been shown not only to form a crucial physical barrier between the body and the luminal environment but also to actively participate in innate and adaptive immune surveillance, forming the primary defense against many mucosal enteropathogens.
A critical feature in the maintenance of the mucosal barrier is the presence of dendritic cells (DCs), specialized antigen-presenting cells that play a vital role at the intestinal epithelium interface, aiding in both immunotolerance to commensal bacteria and food antigens and also the initiation of the immune response to encountered pathogens and other noxious stimuli (8, 35, 52). In the absence of inflammatory stimuli, mucosal DCs are localized in close proximity with the intestinal epithelium in an immature form (iDCs) able to internalize and process antigens but unable to stimulate naive T cells (6, 7). Such iDCs have been shown to work closely with the intestinal epithelial barrier, actively sampling bacteria and other antigens from the external luminal environment, while preserving the integrity of the epithelial barrier by expressing tight junction proteins (66). Upon ingestion of antigens, iDCs initiate a program of functional maturation, which involves their trafficking from peripheral tissues to secondary lymphoid tissue, where they present antigens to and activate T cells, stimulating the adaptive immune system (6, 7). Activated effector T cells then migrate in a directed manner to the site of antigen or pathogen entry through chemokines produced by endothelial and epithelial cells. The complex movement of DCs and in turn effector T cells depends on the coordinate expression and regulation of chemokines, produced both at the intestinal epithelium and secondary lymphoid tissue.
The chemokine CCL20, also known as macrophage inflammatory protein 3α and the sole cognate ligand for the chemokine receptor CCR6 (15, 36, 37), is essential for the directional trafficking of iDCs to the epithelial surface and the migration of mucosal effector/memory T cells and B cells (14, 37, 47, 78, 84). CCL20's expression has characteristically been described as both constitutive and inducible and thus, at the intestinal epithelium, plays a crucial role both in mucosal homeostasis and in the host response to bacterial infection and other inflammatory stimuli, such as proinflammatory cytokines (25, 37). Studies with the enteropathogen Salmonella have shown that flagellin, a component of the bacterial flagella, recognition by the pattern recognition receptor toll-like receptor 5 (TLR5) (28), rather than invasion or lipopolysaccharide, is the key stimulating factor for CCL20 production from epithelial cells (71). Additionally, CCL20 has been shown to be important in the control of Salmonella dissemination in vivo (23). Work investigating the sole cognate receptor of CCL20 has shown that CCR6 knockout mice display an absence of subepithelial myeloid-derived DCs in Peyer's patches and an impaired ability to mount a mucosal immune response (12), further highlighting the importance of the iDC-dependent CCL20-CCR6 signaling axis in the development of the immune response at the gastrointestinal epithelium. In coordination with CCL20 expression, human intestinal epithelial cells also produce a defined set of interferon-responsive chemokines, CXCL9 (Mig), CXCL10 (IP-10), and CXCL11 (I-TAC), that bind the chemokine receptor CXCR3, expressed on activated Th1 cytokine-producing effector T cells (17). Importantly, those cells secrete gamma interferon, thereby establishing a positive-feedback regulatory loop to maintain the residence of the effector T-cell population at the site of microbial infection. Thus, the human intestinal epithelium, upon bacterial infection or proinflammatory cytokine stimulation, produces several chemokines necessary for linking the innate host defense with the appropriate adaptive immune response.
While the production of DC and T-cell chemoattractants at the intestinal epithelial surface has been shown to play a role in Escherichia coli and Salmonella spp. infection (17, 37, 65, 71), their role in C. jejuni infection has not been established. Thus, we sought to define the mechanisms regulating intestinal epithelial expression of those chemokines known to link innate and adaptive immune responses following C. jejuni infection and to investigate the virulence mechanisms whereby the bacteria induce chemokine production. Our results showed that the expression of CCL20 and CXCL10, like that of neutrophil chemoattractants, is dose-dependently up-regulated following C. jejuni infection. Moreover, unlike the case with Salmonella spp., induction of CCL20 was found to be flagellin independent, suggesting a novel mechanism of proinflammatory chemokine production whereby C. jejuni avoids signaling through TLR5. Chemokine secretion requires live bacteria and activation of the transcription factor NF-κB and was highly correlated with the efficiency of bacterial invasion. The regulated epithelial production of DC and T-cell chemoattractants by intestinal epithelial cells during C. jejuni infection suggests that regulated chemokine responses are an important component of the host defense to this clinically significant enteric pathogen.
MATERIALS AND METHODS
Cell culture.
The human colonic epithelial cell line T84 (no. CCL-248) was obtained from the American Type Culture Collection (ATCC) (Rockville, MD) (13) and cultured as described previously (18). T84 cells either were cultured on plastic or were grown as polarized monolayers on 0.45-μm-pore-size mixed cellulose ester inserts (Millipore, Bedford, MA). Transepithelial resistance (TER) was measured using a volt-ohmmeter. T84 monolayers were defined as polarized when they possessed a TER of ≥1,000 Ω · cm2.
Bacterial strains and culture conditions.
C. jejuni strain 81-176, previously shown to cause infectious diarrhea in humans (10), was kindly provided by Victor DiRita (University of Michigan Medical School, Ann Arbor, Michigan). The 81-176 flaA flaB mutant (27) was a kind gift of Patricia Guerry (Enteric Diseases Department, Naval Medical Research Center, Silver Spring, Maryland). The genome-sequenced strain NCTC11168 (61), Salmonella enterica serovar Typhimurium strain 14028s, and Escherichia coli DH5α were purchased from the ATCC. Minimally passaged C. jejuni clinical isolates were provided by David Warshauer (Wisconsin State Laboratory of Hygiene). The C. jejuni strains were cultured on Mueller-Hinton (MH) agar under microaerophilic conditions. Strain 81-176 was cultured on MH supplemented with tetracycline (Tc) (10 μg/ml). Experiments were completed using C. jejuni strains grown at 37°C that had not been subcultured more than three times. Wild-type Salmonella 14028s and E. coli DH5α were cultured overnight at 37°C on LB agar.
Infection or stimulation of epithelial monolayers.
Campylobacter infection of model epithelia was as described previously (30) with the exception that bacteria were not centrifuged onto the monolayer in order to more faithfully replicate interactions within the human gut. Briefly, confluent T84 cell monolayers were incubated for 2 h with C. jejuni at various multiplicities of infection (MOI), and the cells were then washed and incubated with fresh media containing gentamicin (100 μg/ml) to kill remaining extracellular bacteria. For chemokine analysis, cells were seeded to either 6-well or 12-well plates and allowed to grow to 90 to 100% confluence prior to inoculation. For flagellin experiments, parallel cultures of T84 cells were exposed to purified flagellin from C. jejuni strain 81-176 or Salmonella strain 14028s for the duration of the experiment.
RNA extraction and reverse transcriptase (RT)-PCR.
Total cellular RNA was isolated from epithelial cells using TRIzol reagent (Invitrogen, Carlsbad, CA) and cDNA synthesized as previously described (73). The absence of contaminating DNA was confirmed by PCR analysis using primers specific for the promoter region of human mannose binding lectin; sense, 5′-GAA GCT TAG ACC TAT GGG GCT AG-3′, and antisense, 5′-AAC TGC AGG GAA GGT TAA TCG CAG TT-3′, and the following cycling conditions: 94°C for 2 min, followed by 35 cycles of 94°C for 30 s, 60°C for 1 min, and 72°C for 2 min, followed by a final extension of 72°C for 7 min. PCRs were performed in a 50-μl reaction volume containing 5 μl cDNA. Primer pairs and cycling conditions for CCL20 have been described previously with the exception that 28 cycles were performed herein (37). Primer pairs and cycling conditions for CXCL8 (41), CXCL1 and CXCL5 (85), and CXCL9, CXCL10, and CXCL11 (17) were as described previously. Primer pairs for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were the following: sense, 5′-ACC ACA GTC CAT GCC ATC AC-3′, and antisense, 5′-TCC ACC ACC CTG TTG CTG TA-3′; PCR amplification included a program of 30 cycles of 94°C for 45 s, 60°C for 45 s, and 72°C for 2 min, followed by a final extension of 72°C for 7 min.
Real-time PCR.
Comparative levels of T84 cellular chemokine mRNA expression were determined using real-time PCR. Sequences of the oligonucleotide primer pairs used and the expected product size are outlined in Table 1. RNAs were reverse transcribed using random hexamers and Superscript II reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Real-time PCR was performed using 2× iQSYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer's instructions. Briefly, real-time PCRs were performed in a final volume of 25 μl containing 0.5 μM of each oligonucleotide. Reactions were performed using an iCycler (Bio-Rad) using the following cycling conditions: 5 min denaturation at 95°C and then 40 cycles of amplification at 95°C for 1 min, annealing and extension at 62°C for 1 min, for all genes apart from CXCL10, where annealing and extension were performed at 60°C. Each RNA sample was assayed in triplicate using primers specific for the various chemokine mRNAs. Each gene was normalized based on levels of the housekeeping gene RPL23. Results were calculated using the comparative cycle threshold (CT) 2−ΔΔCT method (51), in which the amount of target mRNA is normalized to a reference (uninfected control) relative to the internal RPL23 control.
TABLE 1.
Oligonucleotide primers used for real-time RT-PCR analysis
| mRNA | Sense primer (5′ to 3′) | Antisense primer (5′ to 3′) | Product size (bp) |
|---|---|---|---|
| CCL20 | CTGCTTTGATGTCAGTGCTGCTAC | CTGCCGTGTGAAGCCCACAATAAA | 128 |
| CXCL8 | CAGAGCACACAAGCTTCTAGGACA | GTGTGGTCCACTCTCAATCACTCT | 252 |
| CXCL1 | CCAAACCGAAGTCATAGCCACACT | GCAGCTGTGTCTCTCTTTCCTCTT | 204 |
| CXCL5 | CGTTTGTTTACAGACCACGCAAGG | ATTTCCTTCCCGTTCTTCAGGGAG | 123 |
| CXCL10 | TCACCTTTCCCATCTTCCAAGGGT | GGTAGCCACTGAAAGAATTTGGGC | 180 |
| RPL23 | CCACAGTCAAGAAAGGCAAACCAG | AGTCTGCACACTCCTTTGCTACTG | 195 |
ELISA.
The level of CCL20, CXCL8, or CXCL10 protein secreted by infected or control T84 epithelial cells was measured using enzyme-linked immunosorbent assay (ELISA). CCL20 and CXCL10 were assessed using matched antibody pairs from R&D Systems (Minneapolis, MN) as previously defined (17, 37). CXCL8 was assessed as defined previously (20).
Immunoblot analysis.
To analyze the phosphorylation of IκBα following bacterial infection, T84 cells were serum starved overnight and inoculated with C. jejuni at an MOI of 100:1 for the indicated times. As a positive control, separate cell monolayers were stimulated for 2, 5, or 30 min with 20 ng/ml tumor necrosis factor alpha (TNF-α). Following incubation, cells were solubilized in ice-cold lysis buffer (50 mM Tris HCl, pH 7.4; 150 mM NaCl; 1 mM EDTA; 0.25% [vol/vol] sodium deoxycholate; 1% [vol/vol] IGEPAL; 0.1% [vol/vol] sodium dodecyl sulfate [SDS]) supplemented with Protease Inhibitor Cocktail Set III (EMD Biosciences, La Jolla, CA) and phosphatase inhibitors (10 mM sodium orthovanadate, 40 mM β-glycerophosphate, 20 mM sodium fluoride) for 1 h on ice (73). Lysates were transferred to prechilled tubes and clarified by centrifugation at 12,000 rpm for 15 min at 4°C. Total protein concentrations were determined using the bicinchoninic acid assay (BCA). Cell lysates containing 10 μg of protein were size separated by reducing SDS-polyacrylamide gel electrophoresis (PAGE) and analyzed by Western blotting using human monoclonal antibodies to phospho-IκBα or total IκBα (Cell Signaling Technology, Beverly, MA) followed by incubation with horseradish peroxidase-conjugated donkey antirabbit antibody (Amersham Biosciences, Uppsala, Sweden). Immunoblots were visualized using the Supersignal West Pico chemiluminescent substrate kit (Pierce, Rockford, IL) and exposure to X-ray film (Eastman Kodak, Rochester, NY) according to the manufacturer's instructions.
EMSA.
To functionally assess NF-κB activity in C. jejuni-infected monolayers, electrophoretic mobility shift assays (EMSA) were performed as detailed previously (21, 38). Briefly, T84 cells were seeded to 60-mm dishes and allowed to grow to ∼90 to 100% confluence. Cells were then incubated overnight in serum-free Dulbecco's modified Eagle medium/F12 and infected with C. jejuni 81-176 in serum-free medium at an MOI of 100:1. At defined times of bacterial infection, nuclear and cytoplasmic fractions were prepared (38) and proteins quantified using the BCA assay. NF-κB which had been activated and translocated to the nucleus was detected in the nuclear protein extracts by EMSA. Complementary strands of the oligonucleotide (5′-AGTTGAGGGGACTTTCCCAGGC-3′) containing an NF-κB consensus binding motif were synthesized (Invitrogen), annealed, and then end labeled with [γ-32P]ATP (Amersham) and T4 polynucleotide kinase (Invitrogen). Unincorporated nucleotides were removed by column elution (QIAGEN, Valencia, CA).
Binding reactions were carried out in a total volume of 20 μl, containing 20 μg of nuclear protein and a reaction buffer containing final concentrations of 20 mM KCl, 5% (vol/vol) glycerol, 10 ng of salmon sperm DNA per ml, 25 mM Tris-HCl, pH 8.0, 6 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol, 4.0 μg of poly(dI-dC), and 1 μl of 32P-labeled oligonucleotide probe. Reactions were incubated at room temperature for 30 min and then immediately loaded onto a preelectrophoresed 4% (vol/vol) native 0.25 × TBE (22.3 mM Tris, 22.3 mM boric acid, 0.5 mM EDTA, pH 8.0) polyacrylamide gel, alongside a control lane containing gel loading buffer with bromophenol blue. Samples were separated from unbound probe at 130 V at 4°C. Following electrophoresis the gel was dried for 1 h at 80°C before exposure to either film (Biomax MR, Kodak) or a storage phosphor screen (Molecular Dynamics/Amersham), which was subsequently analyzed using a STORM PhosphorImager system and ImageQuant image analysis software (Molecular Dynamics).
Adenovirus constructs and adenovirus infection.
Recombinant replication-deficient adenovirus encoding either the IκBα superrepressor (Ad5IκBα-A32/A36) or, as a control, green fluorescent protein (GFP) was a kind gift from Christian Jobin (Department of Medicine, University of North Carolina). The IκBα superrepressor (Ad5IκBα-A32/36) is a mutant form of the IκBα protein with serine-to-alanine substitutions at positions 32 and 36, thereby abrogating NF-κB activation by preventing signal-induced IκBα phosphorylation (39). T84 adenovirus infections were performed as described previously (37, 39). Briefly, T84 cells grown to confluence in 12-well tissue culture plates were infected with Ad5κBα-A32/36 or Ad5GFP in serum-free medium at an MOI of 100:1 for 16 h. After viral infection, adenovirus was removed by washing, fresh medium containing serum was added, and the T84 cells were incubated for an additional 12 h before bacterial infection or TNF-α stimulation.
Genotyping Campylobacter strains.
DNA was prepared from C. jejuni isolates or, as a negative control, E. coli DH5α, using a boiling method in which bacterial colonies were resuspended in 50 μl of water, boiled for 10 min, placed on ice, and then centrifuged at room temperature for 10 min at 12,000 rpm. Whole-cell DNA extract (5 μl) was subsequently used as a template in reactions using the primer pairs and cycling conditions for either the flagellin subunit gene, flaA (81), the cytolethal distending toxin subunit B gene, cdtB (64), or the fibronectin binding protein gene, cadF (44).
Motility assay.
The motility phenotype of each of the C. jejuni isolates was determined based on a prior report (29). Briefly, C. jejuni grown for 24 h on MH agar was resuspended in MH broth to an optical density at 600 nm of 0.6 to 0.7. Each isolate was then stab inoculated using a straight wire into 0.4% (wt/vol) MH agar, and the motility phenotype was determined following 24 h of incubation at 37°C.
Invasion assays.
To assess bacterial invasion, confluent T84 cell monolayers cultured on 12-well plates were infected with approximately 1 × 108 bacteria to give an MOI of about 100:1. Following an initial 2-h infection, cells were washed three times and incubated with gentamicin (100 μg/ml) for another 4 h and then washed and lysed in 0.1% (vol/vol) Triton X-100-phosphate-buffered saline (PBS). Bacteria that had successfully infected cells were enumerated by colony count following 48 h of incubation on MH agar. Invasion is expressed as a percentage of inoculum internalized.
Heat or formalin inactivation of C. jejuni.
For heat inactivation, C. jejuni (final MOI, 100:1) was incubated at 65°C for 1 h, allowed to cool to room temperature, and then added to confluent T84 epithelial cells. Alternatively, C. jejuni was inactivated by suspension in PBS containing 5% (vol/vol) formalin and incubated for 6 h at room temperature. Following inactivation, bacteria were washed three times in PBS before addition to T84 epithelial cells. Inactivation of C. jejuni following either heat or formalin treatment was confirmed by determining growth on MH agar.
C. jejuni-conditioned supernatant.
To determine whether C. jejuni-secreted soluble factors in the presence of intestinal epithelial cells were responsible for initiation of the chemokine response, the conditioned supernatant from T84 cells inoculated with C. jejuni for 3 h was removed. This was subsequently filtered (0.22-μm pore; Millipore Corp.) to remove residual bacteria and then added to fresh epithelial cells.
Flagellin purification.
Surface flagella were isolated from C. jejuni or Salmonella according to the method of Song and colleagues (74). Ten plates of C. jejuni or Salmonella grown on MH Tc or LB agar for 3 days or 1 day, respectively, were resuspended in 5 ml of 10 mM Tris-HCl (pH 7.4) and vortexed for 5 min. Whole bacteria and bacterial cell debris were removed by centrifugation at 8,000 rpm for 30 min at 4°C. The supernatant was then ultracentrifuged in a Beckman SW50.2 Ti rotor at 25,000 rpm at 4°C for 1 h. The pellet was resuspended in 5 ml of 1% (vol/vol) SDS and ultracentrifuged as before. The resulting pellet containing flagella was resuspended in 100 μl of 10 mM Tris-HCl and stored at −20°C. The protein concentration was determined by BCA assay according to the manufacturer's instructions.
Statistical analysis.
Differences between uninfected control and experimental samples were analyzed by unpaired Student's t test using SigmaPlot (Jandel Scientific Software, San Rafael, CA). Statistical significance was defined as a P value of <0.05.
RESULTS
Infection of T84 cells with C. jejuni induces chemokine transcription in a dose- and time-dependent manner.
A reliable animal model that reproducibly mimics clinical diarrheal disease associated with acute C. jejuni infection is lacking (86). Thus, we chose the T84 cell line as a model human intestinal epithelium to define the host innate immune response to C. jejuni infection. For these studies we chose the well-characterized virulent C. jejuni isolate 81-176. Originally isolated from a diarrheal outbreak associated with raw-milk consumption (46), 81-176 was subsequently shown to cause inflammatory diarrhea in human feeding studies (10). Since C. jejuni elicits immune memory (70, 72) and since iDCs are essential immune components in linking the early innate and late adaptive immune responses, we first assessed the gene expression of CCL20. Transcriptional analyses were performed on RNA from T84 cells that had been infected with C. jejuni strain 81-176 at MOIs of 100:1, 50:1, and 25:1 and indicated that CCL20 mRNA expression increased by 2 h with maximal levels between 4 and 6 h after infection. As shown in Fig. 1, infection of T84 model epithelia led to the dose-dependent increase in CCL20 mRNA expression 6 h after C. jejuni infection with decreasing transcript expression after 12 and 24 h.
FIG. 1.

Regulated expression of CCL20 by Campylobacter jejuni-infected T84 cell monolayers. A. RT-PCR analysis of CCL20 from total RNA isolated from T84 epithelial cells inoculated with C. jejuni strain 81-176 at multiplicities of infection of 25:1, 50:1, and 100:1. RNA from uninfected cells (Ctrl) or cells treated with 20 ng/ml TNF-α were used as negative and positive controls, respectively. Reactions performed in the absence of RNA (no RNA) or with RNase-free water (H2O) served as negative controls. GAPDH verified equal loading between samples. Results are representative of three independent experiments. B. Kinetics of chemokine mRNA expression in T84 cells in response to C. jejuni infection. Epithelial cells were infected with C. jejuni strain 81-176 at an MOI of 25:1 (solid circle), 50:1 (solid triangle), or 100:1 (solid square). Total RNA was isolated 6, 12, and 24 h after C. jejuni inoculation of T84 epithelial cells and CCL20 mRNA expression assessed using real-time PCR. Samples were compared to uninfected control (empty circle), which was designated baseline. The 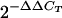 comparative CT method was used to define relative gene expression. Values are the means for triplicate samples and are representative of two separate experiments.
comparative CT method was used to define relative gene expression. Values are the means for triplicate samples and are representative of two separate experiments.
Since our data suggested that T84 model epithelia up-regulated the production of DC chemoattractants that might link to the adaptive immune response, we next sought to assess the regulated production of T-lymphocyte-specific chemoattractants by those same cells. Since we had previously shown that human intestinal epithelial cells up-regulate production of the interferon-inducible T-cell chemokines CXCL9 (Mig), CXCL10 (IP-10), and CXCL11 (I-TAC) (17), we focused on the expression of those genes in campylobacteriosis. As shown in Fig. 2A, we found that 6 h of infection with C. jejuni 81-176 up-regulated the mRNA expression of CXCL10 and to a lesser extent CXCL11 but not CXCL9. Real-time PCR analyses to assess the time course for CXCL10 gene expression indicated that CXCL10 was up-regulated at 6 h postinfection and was diminishing by 12 and 24 h (Table 2).
FIG. 2.

Regulated expression of T-cell and neutrophil chemoattractants in T84 epithelial cells infected with Campylobacter jejuni. RT-PCR analysis of the T-cell chemoattractants CXCL9, CXCL10, and CXCL11 (A), or the neutrophil chemokines CXCL1, CXCL5, and CXCL8 (B) from total RNA isolated from T84 epithelial cells inoculated with C. jejuni strain 81-176 at an MOI of 25:1, 50:1, or 100:1. RNA from uninfected cells (Ctrl) or cells treated with 20 ng/ml TNF-α were used as negative and positive controls, respectively. Reactions performed in the absence of RNA (No RNA) as a negative control. GAPDH verified equal loading between samples. Data are representative of three independent experiments.
TABLE 2.
Kinetics of T-cell and neutrophil chemokine mRNA expression in T84 cells infected with Campylobacter jejuni
| Gene | MOI | Fold change in mRNA expressiona
|
||
|---|---|---|---|---|
| 6 h | 12 h | 24 h | ||
| CXCL10 | 25:1 | 8.7 | 5.0 | 2.5 |
| 50:1 | 37.6 | 4.5 | 9.5 | |
| 100:1 | 40.3 | 10.5 | 5.5 | |
| CXCL8 | 25:1 | 12.7 | 7 | 3.2 |
| 50:1 | 25.4 | 9.5 | 6.4 | |
| 100:1 | 37.6 | 22.3 | 5.1 | |
| CXCL1 | 25:1 | 5 | 1.8 | 1.6 |
| 50:1 | 8.8 | 2.9 | 1.6 | |
| 100:1 | 15.3 | 4 | 1.8 | |
| CXCL5 | 25:1 | 84 | 105 | 15.9 |
| 50:1 | 147 | 156 | 21 | |
| 100:1 | 223 | 265 | 41 | |
Values are mean n-fold changes for triplicate samples. Data are representative of two independently completed real-time RT-PCR analyses.
In conjunction with DCs and T cells, intestinal epithelial recruitment of polymorphonuclear leukocytes plays an important role in the innate mucosal immune response to enteric pathogens. The production of CXCL8 (interleukin 8) and CXCL1 (GROα) in response to C. jejuni infection from intestinal epithelial cells has previously been characterized (30, 33, 82). Thus, as a control, we sought to define the expression of CXCL8 and CXCL1 as well as CXCL5 (ENA-78) following T84 infection with C. jejuni. In concordance with our findings for CCL20 and CXCL10, infection of T84 cells led to a dose-dependent increase in CXCL8, CXCL1, and CXCL5 mRNA expression (Fig. 2B). CXCL8 and CXCL1 were elevated at 6 h, with a similar decrease as shown for CCL20 and CXCL10 after 12 and 24 h of infection (Table 2). In contrast to those chemokines, expression of CXCL5 was sustained from 6 to 12 h following C. jejuni infection (Table 2), as previously noted for Salmonella-infected epithelial cells (85). Taken together, these findings indicate that the human intestinal epithelium coordinately up-regulates a defined battery of DC, T-cell, and neutrophil chemoattractants in response to C. jejuni infection.
Chemokine secretion from intestinal epithelial cells infected with C. jejuni.
To determine whether the increased transcription of CCL20 and CXCL10 was paralleled with an increase in chemokine secretion, we next used ELISA to define protein production from C. jejuni-infected model epithelia. T84 cell monolayers were infected with C. jejuni at various MOIs, and supernatants were removed 6, 12, and 24 h postinfection. In agreement with increased chemokine transcription, C. jejuni induced a dose- and time-dependent increase in CCL20 secretion from 6 to 24 h (Fig. 3A). Regulated secretion of the effector-T-cell chemoattractant CXCL10 (Fig. 3B) and the neutrophil chemokine CXCL8 (Fig. 3C), examined as a control, were similarly dose- and time-dependently up-regulated in C. jejuni-infected T84 cells.
FIG. 3.

Regulated secretion of chemokines from T84 intestinal epithelial cells infected with Campylobacter jejuni. T84 epithelial cell monolayers were inoculated with C. jejuni strain 81-176 at an MOI of 25:1 (solid circle), 50:1 (solid triangle), or 100:1 (solid square). CCL20 (A), CXCL10 (B), and CXCL8 (C) secretion was assessed using ELISA, as described in Materials and Methods, 6, 12, and 24 h after bacterial inoculation. Values are means ± standard errors of the means for 3 to 11 independent experiments. An asterisk (*) indicates the significant difference between all bacterial concentrations and the uninfected control (P < 0.05). A double asterisk (**) indicates a significant difference between MOIs of 100:1 and 50:1 and the uninfected control (P < 0.05).
For CCL20 produced by epithelial cells to create a chemotactic gradient for the attraction of iDCs, T cells, and B cells, it must be secreted in the basolateral direction rather than apically into the intestinal lumen. To determine whether this was the case with C. jejuni infection, T84 cells were grown as polarized epithelial monolayers (TER ≥ 1,000 Ω · cm2) and infected apically with C. jejuni strain 81-176 to mimic human intestinal infection. Apical addition of C. jejuni induced significant secretion of CCL20 in the physiologically relevant basolateral direction. Thus, CCL20 secretion in C. jejuni-infected epithelium was found to be 1.02 ± 0.16 ng/ml in the basal chamber compared with 0.09 ± 0.04 ng/ml in the apical chamber. This contrasts with uninfected controls, which showed 0.55 ± 0.28 ng/ml and 0.0 5 ± 0.02 ng/ml in the basal and apical chambers, respectively.
C. jejuni regulates epithelial CCL20 expression through the activation of NF-κB.
The transcription factor NF-κB is a central regulator in the transcriptional activation of several genes involved in the intestinal epithelial proinflammatory response (21, 40). Prior studies have shown that CCL20 is an NF-κB target gene (37, 48, 76). Therefore, we next assessed whether C. jejuni-induced CCL20 secretion was also mediated by an NF-κB-dependent mechanism. We initially determined whether C. jejuni infection of T84 cells was capable of inducing IκBα phosphorylation, a necessary step in activation of NF-κB. As shown in Fig. 4A, phosphorylation of IκBα was observed within 15 min and was sustained over 3 h of C. jejuni infection.
FIG. 4.

CCL20 gene expression is regulated by Campylobacter jejuni activation of the NF-κB signaling pathway. A. Time course of IκBα activation in C. jejuni-infected T84 cells. Immunoblot analysis of whole-cell lysates from T84 cells infected with C. jejuni strain 81-176 (MOI of 100:1). Blots were probed with antibody to phosphorylated (p-IκBα) or total (tot-IκBα) IκBα protein. Cells were left unstimulated or were stimulated with 20 ng/ml TNF-α for 2, 5, or 30 min as negative and positive controls, respectively. Data are representative of three independent experiments. B. Nuclear extracts from C. jejuni induce the nuclear translocation of NF-κB in T84 cells. Electrophoretic mobility gel shift assays were performed using nuclear extracts from C. jejuni strain 81-176-infected (MOI of 100:1) T84 cells, obtained 30 min or 1, 2, or 3 h postinfection, and a radiolabeled probe containing an NF-κB binding site. Extracts from uninoculated cells (Ctrl) or cells stimulated with 20 ng/ml TNF-α (30 min) served as negative and positive controls, respectively. Specificity of the labeled complex was shown by incubation with unlabeled competitor probe (comp.). Data are representative of three separate experiments. C. The IκBα superrepressor inhibited CCL20 secretion. T84 cells were infected (MOI of 100:1) with recombinant adenovirus expressing either the IκBα superrepressor (Ad5IκB-A32/36) or green fluorescent protein (Ad5GFP) as indicated. Mock, uninfected cells (No Virus) were a separate internal control for Campylobacter-induced chemokine secretion. Ad5-infected cells were either inoculated with C. jejuni strain 81-176 or remained uninfected as a negative control. CCL20 secretion was assayed by ELISA as described in Materials and Methods. Values are means ± standard errors of the means for three independently completed experiments. An asterisk (*) indicates the significant difference from Campylobacter-Ad5GFP-infected cells (P < 0.05).
To determine whether Campylobacter-induced IκBα phosphorylation correlated with activation of NF-κB, we next used electrophoretic mobility shift assays on nuclear protein extracts from C. jejuni-infected T84 cells. As shown in Fig. 4B, C. jejuni infection increased DNA binding over time, with maximal binding observed 2 and 3 h after infection. These results differ slightly from those for other enteropathogens, which show maximal binding 30 to 45 min postinfection (21).
Having confirmed that C. jejuni infection of T84 cells activated NF-κB, we next sought to determine whether this transcription factor regulated CCL20 secretion in C. jejuni-infected epithelia. As shown in Fig. 4C, inhibition of NF-κB activation in T84 cells by recombinant adenovirus expressing an IκBα superrepressor (IκBα-A32/A36) markedly decreased C. jejuni-induced CCL20 secretion, an effect that was not observed in control, Ad5GFP-expressing cells. Taken together, these data indicate that C. jejuni infection of T84 model intestinal epithelia activates and in turn up-regulates CCL20 expression through the transcription factor NF-κB.
Intestinal epithelial CCL20 secretion requires live Campylobacter.
There are many bacterial virulence factors known to stimulate the NF-κB host proinflammatory chemokine response at the intestinal epithelial surface (68). To determine whether C. jejuni-induced CCL20 secretion required live bacteria, T84 cells were stimulated with either heat- or formalin-inactivated 81-176 at the same dose required to give an MOI of 100:1. As shown in Fig. 5, heat- or formalin-inactivated bacteria were incapable of inducing a CCL20 response like that observed with live bacteria, with CCL20 secretion levels being similar to those of uninfected T84 cells. Analysis of CXCL8 secretion, used as a control, showed similar results (not shown).
FIG. 5.

Live Campylobacter jejuni is required to activate CCL20 secretion from T84 epithelial cells. T84 cell monolayers were inoculated with live C. jejuni (MOI of 100:1) or C. jejuni that had been either heat killed or formalin inactivated as outlined in Materials and Methods. T84 cells were also stimulated with C. jejuni conditioned supernatants. Data are means ± standard errors of the means for three different experiments. An asterisk (*) indicates a significant difference between infected (81-176) and uninfected control (Ctrl) cells (P < 0.05). A double asterisk (**) indicates a significant difference from Campylobacter-infected cells (P < 0.05).
In addition to inactivated bacteria, we also investigated whether C. jejuni secreted any factors that were capable of inducing an epithelial CCL20 response. Initial assays of bacterium-free MH culture medium added to T84 cells failed to detect any increase in CCL20 secretion (data not shown). However, since previous reports indicate that C. jejuni virulence modalities are regulated upon environment sensing, such as interaction with the host (34), supernatants were taken from 81-176 conditioned tissue culture media as outlined in Materials and Methods. As shown in Fig. 5, T84 cells treated with filtered supernatants showed results similar to that observed for inactivated bacteria and were not different from uninfected controls.
C. jejuni-induced CCL20 secretion is flagellin independent.
Numerous studies have demonstrated that bacterial flagellin is a major stimulus of the proinflammatory response in human intestinal epithelial cells (19, 26, 75, 77), where it is recognized by TLR5 (28). Recent work has shown that Salmonella flagellin is the major component for stimulating the CCL20 response in infected epithelial cells (71). The ability of C. jejuni flagellin to induce CCL20 secretion from T84 cells was assayed in parallel with Salmonella flagellin. Flagella were purified directly from bacterial cells as described in Materials and Methods. The purity of the flagellin preparation, FlaA from C. jejuni and FliC from Salmonella, was verified by SDS-PAGE and Coomassie brilliant blue staining (Fig. 6A). Next, T84 cells were treated with various concentrations (0.1 to 1,000 ng/ml) of flagellin purified from either C. jejuni or Salmonella. As shown in Fig. 6, Campylobacter flagellin was incapable of inducing CCL20 (Fig. 6B) or CXCL8 (Fig. 6C) chemokine secretion in treated T84 epithelial cell monolayers. As expected from those data, purified Salmonella but not Campylobacter flagellin stimulated CXCL10 secretion (data not shown). TLR5 expression by T84 cells was verified by RT-PCR (not shown), indicating that the inability of Campylobacter flagellin to stimulate chemokine production did not reflect aberrant expression of the flagellin pattern recognition receptor. These data with Campylobacter flagellin are in marked contrast to those with Salmonella flagellin, used as a control, which potently up-regulated chemokine production in stimulated T84 cell monolayers (Fig. 6), and most likely reflect the inability of Campylobacter flagellin to bind and activate the TLR5 pattern recognition receptor (3, 82).
FIG. 6.

Campylobacter jejuni flagellin does not stimulate chemokine secretion by T84 epithelial cells. A. C. jejuni and S. enterica serovar Typhimurium flagellins were purified and quantified as outlined in Materials and Methods. Equal amounts of flagellin protein were size separated by SDS-PAGE and stained using Coomassie brilliant blue to verify size and purity. Flagellins of C. jejuni, FlaA (59 kDa), and S. enterica serovar Typhimurium, FliC (50 kDa), are shown. Markers (M) are as indicated. T84 cell monolayers were stimulated with increasing concentrations of flagellin (1, 10, 100, or 1,000 ng/ml) from C. jejuni or S. enterica serovar Typhimurium. Separate monolayers were left untreated or were treated with TNF-α (20 ng/ml) as negative or positive controls, respectively. CCL20 (B) and CXCL8 (C) secretion was measured 12 h after addition of flagellin. Values are means ± standard deviations from a representative experiment (n = 3).
We next tested the ability of a C. jejuni flagellar mutant to stimulate chemokine secretion from intestinal epithelial cells. Infection of T84 cell monolayers with an 81-176 flaA flaB mutant (27) produced significantly less CCL20 secretion than that of wild-type 81-176 (1.04 ± 0.29 ng/ml and 4.05 ± 0.43 ng/ml, respectively). The inability of the flagellar mutant to stimulate chemokine secretion corresponds with its lack of motility (27) and its greatly reduced ability (>700-fold less than that of the wild type) to invade intestinal epithelial cells. Together with its inability to bind TLR5, our results show that while C. jejuni flagella are not sufficient to trigger chemokine secretion from intestinal epithelial cells, they are necessary for invasion and subsequent induction of the chemokine response.
CCL20 secretion from T84 cells is differentially induced by clinical isolates.
Having shown that C. jejuni strain 81-176, a well-characterized virulent human isolate of Campylobacter, increased CCL20 expression in an NF-κB-dependent, flagellin-independent manner, we next wished to determine whether other isolates of C. jejuni were similarly capable of inducing CCL20 protein secretion from intestinal epithelial cells. Thus, we next assessed chemokine secretion from T84 cells inoculated with a number of clinical, disease-associated isolates from the Wisconsin State Laboratory of Hygiene (Madison, WI) or the genome-sequenced strain NCTC11168. Each isolate was genotyped for the virulence genes encoding flagella, using the flagellin subunit gene flaA, the cytolethal distending toxin (CDT) using the CDT B subunit gene, cdtB, and the fibronectin binding protein (CadF) gene, cadF. The virulence-associated phenotypes of motility and invasion were also assessed.
As shown in Table 3, T84 cells inoculated with differing clinical C. jejuni isolates, each of which possessed the genes encoding the flaA, cdtB, and cadF virulence determinants, consistently stimulated CCL20 secretion to various levels, from 0.87 ± 0.1 ng/ml to 3.22 ± 0.4 ng/ml. Clinical isolate 46-47 was as effective as 81-176 in stimulating the highest levels of chemokine secretion with all but two isolates secreting statistically significant levels of CCL20. Similar findings were noted for CXCL8 secretion (Table 3). In agreement with prior reports (9, 22, 24, 60), the differing clinical C. jejuni isolates as well as the strains 81-176 and NCTC11168 were found to invade cultured intestinal epithelial cells with various efficiencies. C. jejuni invasion levels correlated with chemokine secretion (Table 3), with those strains eliciting the highest level of CCL20 or CXCL8 secretion being the most invasive. Similarly, the levels of CCL20 secreted by the clinical isolates were more robust than that noted for the neutrophil chemoattractant CXCL8. The consistent increase in DC chemokine secretion by C. jejuni-infected epithelium suggests that NF-κB-regulated chemokine expression may be a common feature of clinical disease associated with campylobacteriosis.
TABLE 3.
CCL20 and CXCL8 secretion and invasion in T84 cells infected with clinical isolates of Campylobacter jejuni
| C. jejuni strain | Genotypea
|
Motilityb | % Invasionc | Chemokine secretion (ng/ml)d
|
|||
|---|---|---|---|---|---|---|---|
| flaA | cadF | cdtB | CCL20 | CXCL8 | |||
| 81-176 | + | + | + | + | 3.10 ± 0.53 | 3.36 ± 0.9* | 0.57 ± 0.15* |
| 11168 | + | + | + | + | 0.05 ± 0.04 | 1.36 ± 0.3* | 0.28 ± 0.08* |
| 45-05 | + | + | + | + | 0.02 ± 0.01 | 1.16 ± 0.2* | 0.26 ± 0.05* |
| 38-73 | + | + | + | + | 0.05 ± 0.03 | 1.39 ± 0.1* | 0.23 ± 0.06 |
| 46-47 | + | + | + | + | 0.37 ± 0.09 | 3.22 ± 0.4* | 0.54 ± 0.17* |
| 46-49 | + | + | + | + | 0.12 ± 0.01 | 0.95 ± 0.3 | 0.24 ± 0.06* |
| 39-02 | + | + | + | + | 0.27 ± 0.06 | 1.22 ± 0.3* | 0.26 ± 0.09* |
| 41-70 | + | + | + | + | 0.04 ± 0.01 | 0.87 ± 0.1 | 0.27 ± 0.06* |
E. coli DH5α and Salmonella 14028s assessed as negative controls did not possess the flaA, cadF, and cdtB genes. Data for genotype analysis are representative of two independently completed experiments.
Each of the Campylobacter isolates and Salmonella 14028s assessed as a positive control, were motile. E. coli DH5α assessed as a negative control was nonmotile. Data for motility assays are representative of two independently completed assays.
Invasion was assessed as a percentage of the initial inoculum as defined in Materials and Methods. Values are the means ± SD from two to six independent experiments.
Secretion of CCL20 or CXCL8 was assessed by ELISA as defined in the Material and Methods section. Values for chemokine secretion are means ± SEM from two to six independently completed experiments. Background levels of CCL20 and CXCL8 in uninfected T84 cells were 0.79 ± 0.26 and 0.13 ± 0.07, respectively. An asterisk denotes a statistically significant difference in chemokine secretion between C. jejuni isolates and uninfected T84 cells.
DISCUSSION
Unlike the case with many other enteroinvasive pathogens, very little is known about the mechanisms whereby C. jejuni colonizes the intestinal epithelium and induces diarrhea. However, given the similarities between C. jejuni disease pathogenesis and that of the more well-characterized enteropathogens, the ability to stimulate the secretion of proinflammatory chemokines and cytokines is likely an important factor in disease pathology. Since evidence of immune memory (70, 72) and autoimmune sequelae (1) has been found in campylobacteriosis, the adaptive immune response also likely plays a significant role in C. jejuni pathogenesis. In this work we report that C. jejuni stimulates the activation of the transcription factor NF-κB, leading to the up-regulated transcription and secretion of the DC chemokine CCL20 as well as the T-cell chemoattractant CXCL10 from intestinal epithelial cells. The up-regulation of these chemoattractants, whose major known function is to attract cells important for antigen presentation and the development of the host adaptive immune response, implicates the epithelium as a key regulator of mucosal immunity in C. jejuni infection. Further, our data suggest that DCs may play a key role in antigen processing and in directing the development of the adaptive immune response in campylobacteriosis. Moreover, we determined that chemokine expression in C. jejuni-infected epithelium occurs despite evasion of the TLR5 pattern recognition receptor, suggesting that alternate host defense responses are activated and limit morbidity with this clinically important pathogen.
In developed countries, C. jejuni infection is characteristically associated with an acute inflammatory, sometimes bloody, diarrhea and the presence of fecal leukocytes (10, 72), symptoms common to infection by many enteric pathogens. Chemokines, along with the induction of other proinflammatory mediators, are crucial in the development of the protective mucosal immune response. In particular, the chemokine CXCL8 is a potent neutrophil chemoattractant essential in early inflammatory responses and critical for both the induction of diarrhea and the clearance of bacterial infection (54, 59, 68). In agreement with the multifactoral nature of bacterium-host interactions, the production of neutrophil chemokines in C. jejuni infection has been shown to involve both C. jejuni attachment/invasion (30) and the CDT toxin (31), which elicits G2 cell cycle arrest (49, 83). Our results extend those prior findings and showed that C. jejuni infection of T84 intestinal epithelial cells also up-regulated the expression of CXCL1 and CXCL5, chemokines equally important in the attraction of neutrophils. Similar to the kinetics of expression observed in intestinal epithelial cells infected with Salmonella (85), C. jejuni showed a rapid transient upregulation of expression of CXCL8 and CXCL1 but induced a sustained response for CXCL5. This suggests that C. jejuni infection of intestinal epithelial cells induces temporal and spatial chemokine gradients, which may be important in both the early and late phases of the mucosal inflammatory response to this pathogen. Importantly, the levels of CXCL8 secretion observed in our T84 cell model were in agreement with those previously observed for C. jejuni and other enteropathogens (30, 41).
The activation of the NF-κB signaling pathway is essential for the coordinate expression of a large number of proinflammatory genes, which are critical components of the mucosal immune response (21, 40, 55). Thus, NF-κB has been shown to mediate the transcription of many of the cytokines and chemokines, such as CCL20 (37, 48, 76) and CXCL8 (21, 32), as well as many other proinflammatory mediators (40). NF-κB in nonstimulated cells is normally sequestered in the cytoplasm by its association with inhibitor of NF-κB (IκB) proteins. Activation and translocation of NF-κB to the nucleus require phosphorylation and degradation of these inhibitory molecules (43, 50). In this work we determined that C. jejuni infection of model human intestinal epithelia induced the time-dependent phosphorylation of IκBα, without subsequent degradation of total IκBα levels. In turn we noted an increase in NF-κB activity in C. jejuni-infected epithelial cells, which led to an increase in chemokine secretion. Of note, the CXCL9 and CXCL11 T-cell chemokines lacking or possessing weak NF-κB binding sites were only minimally, if at all, increased by Campylobacter infection.
While our data for C. jejuni activation of the NF-κB proinflammatory response is similar to that noted for other enteric microbial pathogens, we observed several unique features in the activation of this pathway. First, the sustained IκBα phosphorylation observed upon C. jejuni infection is in sharp contrast to other enteric microbial pathogens, such enteropathogenic E. coli, which shows rapid but transient IκBα phosphorylation upon infection of T84 cells (69). Further, in comparison to the proinflammatory cytokine TNF-α, little to incomplete degradation of total IκBα levels was observed in C. jejuni-infected epithelial cells. These data, however, are in agreement with the incomplete degradation of total IκBα observed in T84 cells infected with other enteroinvasive pathogens, such Salmonella dublin, Yersinia enterolitica, or E. coli O29:NM (21). In contrast to the incomplete IκBα degradation observed in T84 epithelia infected with live C. jejuni, a prior report showed that an undefined boiled-cell extract of C. jejuni strain NCTC11168 leads to the complete degradation of total IκBα (53), similar to that observed for enteropathogenic E. coli-infected (69) or S. enterica serovar Typhimurium-infected (57) epithelial cells. The latter difference may represent differences in the cell lines or C. jejuni isolates. Since we noted that several Campylobacter isolates reproducibly invade intestinal epithelial cells, much like enteroinvasive Salmonella, it is likely that incomplete degradation of total IκBα more closely approximates the pathophysiologic interactions at the human mucosal surface in campylobacteriosis. Taken together, we have defined for the first time a role for C. jejuni-activated NF-κB in regulation of the host epithelial chemokine response to that organism.
Intestinal epithelial cells express an array of pattern recognition receptors, Toll-like receptors, and Nods, which play a key role in the identification of pathogens and the stimulation of the NF-κB proinflammatory immune response. Each receptor is involved in the recognition of conserved microbial components, such as lipoproteins, lipopolysaccharide, and peptidoglycan (11, 63). Several studies have shown that flagellins from various gram-negative and gram-positive bacteria are capable of stimulating the proinflammatory NF-κB signaling response in intestinal epithelial cells (19, 26, 28, 77, 88), where it is recognized by TLR5 (28). Moreover, flagellin from Salmonella has specifically been shown to stimulate CCL20 gene expression and secretion from intestinal epithelial cells, resulting in the migration of iDCs (71). In this study we show that C. jejuni flagellin, in distinct contrast to Salmonella flagellin, is incapable of stimulating CCL20 secretion in human intestinal epithelial cells. This work correlates with a recent report (82), which showed a lack of CXCL8 secretion in Campylobacter flagellin-treated epithelial cells. Our data likely reflect unique amino acid differences in the flagellin protein common to several members of the α- and ɛ-Proteobacteria, including C. jejuni and C. coli (3), that prevent its binding and recognition by the human TLR5 pattern recognition receptor. This function perhaps provides a selective advantage for C. jejuni over other mucosal pathogens by avoidance of TLR5 recognition. Of note, we found that a C. jejuni flagellar mutant was unable to stimulate chemokine secretion from intestinal epithelial cells. This result was not surprising, given that Campylobacter lacking flagella are nonmotile and display decreased invasion, functions critical for C. jejuni pathogenesis. Overall, our studies indicate that while C. jejuni flagellin does not play a role in the stimulation of the proinflammatory chemokine response, flagella are still in fact necessary for successful invasion and induction of the proinflammatory response at the intestinal mucosa.
The cells of the intestinal epithelium respond to microbial threats by the activation of specific cellular signaling cascades that ultimately result in the activation of the proinflammatory gene program, subsequently leading to clearance of the offending pathogen (35, 42, 56). Our results suggest that C. jejuni activates the proinflammatory response in intestinal epithelial cells by mechanisms different from those described for other enteropathogens and may escape signaling through host innate defense recognition molecules, such as TLRs. Previous studies have shown that the ability of C. jejuni to cause disease is dependent on multiple factors, such as motility, chemotaxis, host cell adherence, toxin production, and host cell invasion (45). The major drawback in understanding pertinent virulence factors is the considerable diversity between C. jejuni strains observed both in clinical presentation and in the genotypic and phenotypic characteristics (16, 72, 80). Despite the diverse nature of C. jejuni isolates, our studies showed that the ability to induce CCL20 and CXCL8 secretion from intestinal epithelial cells, although varied among the clinical C. jejuni isolates examined, was a consistent host defense response to disease-associated Campylobacter.
Epithelial chemokine secretion induced by clinical isolates of C. jejuni was correlated with the invasion efficiency of those bacteria. Thus, those C. jejuni strains capable of robust invasion of model human intestinal epithelia induced the highest levels of CCL20 and CXCL8 secretion. Our data are in agreement with a prior report that showed that CXCL8 secretion by INT407 cells was similarly linked to C. jejuni invasion (30). This correlation of chemokine secretion and invasion was further strengthened by results using heat- or formalin-inactivated bacteria, which showed background levels of chemokine secretion compared to those for live bacteria. Thus, we have shown that to induce epithelial responses, C. jejuni must be live and in contact with epithelial cells. Moreover, these data suggest that adherence, invasion, or potentially the translocation of microbial effectors into the host cell via a secretion system may trigger the epithelial host defense response. Given the high degree of correlation between adherence and/or invasion, coupled with evasion of TLR5-flagellin recognition in the T84 model epithelium, it is tempting to speculate that intracellular pattern recognition receptors, such as Nod proteins (79), may be involved in C. jejuni signaling in the host.
In summary, our data show that C. jejuni stimulates CCL20, an important chemokine in the activation of immature dendritic cells and thus the development of the host adaptive immune response. Our work also characterizes several T-lymphocyte and neutrophil chemokines, whose coordinate expression and secretion may be vital not only for the initial acute inflammatory response but also to a later sustained innate and adaptive immune response to C. jejuni infection. Notably, C. jejuni was found to activate NF-κB and in turn the proinflammatory gene program independently of flagellin recognition by TLR5. Together, our data suggest that a redundant mechanism functionally activates epithelial host defense responses to the various virulence modalities of the food- and waterborne pathogen C. jejuni.
Acknowledgments
We thank Tara Reimer, Colleen Devaney, and Irina Ionova for technical assistance. The kind assistance of David Warshauer (Wisconsin State Laboratory of Hygiene), Victor DiRita (University of Michigan), and Patricia Guerry for providing various strains of Campylobacter jejuni used, as well as the help of Christian Jobin (University of North Carolina) in providing the adenovirus superrepressor, are gratefully acknowledged.
This work was supported by National Institutes of Health grants DK002808, AI055966, and DK062066 and, in part, by the Crohn's and Colitis Foundation of America.
Editor: V. J. DiRita
REFERENCES
- 1.Allos, B. M. 2001. Campylobacter jejuni infections: update on emerging issues and trends. Clin. Infect. Dis. 32:1201-1206. [DOI] [PubMed] [Google Scholar]
- 2.Altekruse, S. F., N. J. Stern, P. I. Fields, and D. L. Swerdlow. 1999. Campylobacter jejuni--an emerging foodborne pathogen. Emerg. Infect. Dis. 5:28-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersen-Nissen, E., K. D. Smith, K. L. Strobe, S. L. Rassoulian Barrett, B. T. Cookson, S. M. Logan, and A. Aderem. 2005. Evasion of Toll-like receptor 5 by flagellated bacteria. Proc. Natl. Acad. Sci. USA 102:9247-9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Babakhani, F. K., G. A. Bradley, and L. A. Joens. 1993. Newborn piglet model for campylobacteriosis. Infect. Immun. 61:3466-3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babakhani, F. K., and L. A. Joens. 1993. Primary swine intestinal cells as a model for studying Campylobacter jejuni invasiveness. Infect. Immun. 61:2723-2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banchereau, J., F. Briere, C. Caux, J. Davoust, S. Lebecque, Y. J. Liu, B. Pulendran, and K. Palucka. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767-811. [DOI] [PubMed] [Google Scholar]
- 7.Banchereau, J., and R. M. Steinman. 1998. Dendritic cells and the control of immunity. Nature 392:245-252. [DOI] [PubMed] [Google Scholar]
- 8.Bilsborough, J., and J. L. Viney. 2004. Gastrointestinal dendritic cells play a role in immunity, tolerance, and disease. Gastroenterology 127:300-309. [DOI] [PubMed] [Google Scholar]
- 9.Biswas, D., K. Itoh, and C. Sasakawa. 2000. Uptake pathways of clinical and healthy animal isolates of Campylobacter jejuni into INT-407 cells. FEMS Immunol. Med. Microbiol. 29:203-211. [DOI] [PubMed] [Google Scholar]
- 10.Black, R. E., M. M. Levine, M. L. Clements, T. P. Hughes, and M. J. Blaser. 1988. Experimental Campylobacter jejuni infection in humans. J. Infect. Dis. 157:472-479. [DOI] [PubMed] [Google Scholar]
- 11.Cario, E. 2005. Bacterial interactions with cells of the intestinal mucosa: Toll-like receptors and NOD2. Gut 54:1182-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cook, D. N., D. M. Prosser, R. Forster, J. Zhang, N. A. Kuklin, S. J. Abbondanzo, X. D. Niu, S. C. Chen, D. J. Manfra, M. T. Wiekowski, L. M. Sullivan, S. R. Smith, H. B. Greenberg, S. K. Narula, M. Lipp, and S. A. Lira. 2000. CCR6 mediates dendritic cell localization, lymphocyte homeostasis, and immune responses in mucosal tissue. Immunity 12:495-503. [DOI] [PubMed] [Google Scholar]
- 13.Dharmsathaphorn, K., J. A. McRoberts, K. G. Mandel, L. D. Tisdale, and H. Masui. 1984. A human colonic tumor cell line that maintains vectorial electrolyte transport. Am. J. Physiol. 246:G204-G208. [DOI] [PubMed] [Google Scholar]
- 14.Dieu, M. C., B. Vanbervliet, A. Vicari, J. M. Bridon, E. Oldham, S. Ait-Yahia, F. Briere, A. Zlotnik, S. Lebecque, and C. Caux. 1998. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 188:373-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dieu-Nosjean, M. C., C. Massacrier, B. Homey, B. Vanbervliet, J. J. Pin, A. Vicari, S. Lebecque, C. Dezutter-Dambuyant, D. Schmitt, A. Zlotnik, and C. Caux. 2000. Macrophage inflammatory protein 3alpha is expressed at inflamed epithelial surfaces and is the most potent chemokine known in attracting Langerhans cell precursors. J. Exp. Med. 192:705-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorrell, N., J. A. Mangan, K. G. Laing, J. Hinds, D. Linton, H. Al-Ghusein, B. G. Barrell, J. Parkhill, N. G. Stoker, A. V. Karlyshev, P. D. Butcher, and B. W. Wren. 2001. Whole genome comparison of Campylobacter jejuni human isolates using a low-cost microarray reveals extensive genetic diversity. Genome Res. 11:1706-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dwinell, M. B., N. Lugering, L. Eckmann, and M. F. Kagnoff. 2001. Regulated production of interferon-inducible T-cell chemoattractants by human intestinal epithelial cells. Gastroenterology 120:49-59. [DOI] [PubMed] [Google Scholar]
- 18.Dwinell, M. B., H. Ogawa, K. E. Barrett, and M. F. Kagnoff. 2004. SDF-1/CXCL12 regulates cAMP production and ion transport in intestinal epithelial cells via CXCR4. Am. J. Physiol. Gastrointest. Liver Physiol. 286:G844-G850. [DOI] [PubMed] [Google Scholar]
- 19.Eaves-Pyles, T., K. Murthy, L. Liaudet, L. Virag, G. Ross, F. G. Soriano, C. Szabo, and A. L. Salzman. 2001. Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: I kappa B alpha degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. J. Immunol. 166:1248-1260. [DOI] [PubMed] [Google Scholar]
- 20.Eckmann, L., M. F. Kagnoff, and J. Fierer. 1993. Epithelial cells secrete the chemokine interleukin-8 in response to bacterial entry. Infect. Immun. 61:4569-4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elewaut, D., J. A. DiDonato, J. M. Kim, F. Truong, L. Eckmann, and M. F. Kagnoff. 1999. NF-kappa B is a central regulator of the intestinal epithelial cell innate immune response induced by infection with enteroinvasive bacteria. J. Immunol. 163:1457-1466. [PubMed] [Google Scholar]
- 22.Everest, P. H., H. Goossens, J. P. Butzler, D. Lloyd, S. Knutton, J. M. Ketley, and P. H. Williams. 1992. Differentiated Caco-2 cells as a model for enteric invasion by Campylobacter jejuni and C. coli. J. Med. Microbiol. 37:319-325. [DOI] [PubMed] [Google Scholar]
- 23.Fahy, O. L., S. L. Townley, N. J. Coates, I. Clark-Lewis, and S. R. McColl. 2004. Control of Salmonella dissemination in vivo by macrophage inflammatory protein (MIP)-3alpha/CCL20. Lab. Investig. 84:1501-1511. [DOI] [PubMed] [Google Scholar]
- 24.Fauchere, J. L., A. Rosenau, M. Veron, E. N. Moyen, S. Richard, and A. Pfister. 1986. Association with HeLa cells of Campylobacter jejuni and Campylobacter coli isolated from human feces. Infect. Immun. 54:283-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujiie, S., K. Hieshima, D. Izawa, T. Nakayama, R. Fujisawa, H. Ohyanagi, and O. Yoshie. 2001. Proinflammatory cytokines induce liver and activation-regulated chemokine/macrophage inflammatory protein-3alpha/CCL20 in mucosal epithelial cells through NF-kappaB. Int. Immunol. 13:1255-1263. [DOI] [PubMed] [Google Scholar]
- 26.Gewirtz, A. T., T. A. Navas, S. Lyons, P. J. Godowski, and J. L. Madara. 2001. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J. Immunol. 167:1882-1885. [DOI] [PubMed] [Google Scholar]
- 27.Goon, S., C. P. Ewing, M. Lorenzo, D. Pattarini, G. Majam, and P. Guerry. 2006. A σ28-regulated nonflagella gene contributes to virulence of Campylobacter jejuni 81-176. Infect. Immun. 74:769-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi, F., K. D. Smith, A. Ozinsky, T. R. Hawn, E. C. Yi, D. R. Goodlett, J. K. Eng, S. Akira, D. M. Underhill, and A. Aderem. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099-1103. [DOI] [PubMed] [Google Scholar]
- 29.Hendrixson, D. R., B. J. Akerley, and V. J. DiRita. 2001. Transposon mutagenesis of Campylobacter jejuni identifies a bipartite energy taxis system required for motility. Mol. Microbiol. 40:214-224. [DOI] [PubMed] [Google Scholar]
- 30.Hickey, T. E., S. Baqar, A. L. Bourgeois, C. P. Ewing, and P. Guerry. 1999. Campylobacter jejuni-stimulated secretion of interleukin-8 by INT407 cells. Infect. Immun. 67:88-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hickey, T. E., A. L. McVeigh, D. A. Scott, R. E. Michielutti, A. Bixby, S. A. Carroll, A. L. Bourgeois, and P. Guerry. 2000. Campylobacter jejuni cytolethal distending toxin mediates release of interleukin-8 from intestinal epithelial cells. Infect. Immun. 68:6535-6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hobbie, S., L. M. Chen, R. J. Davis, and J. E. Galan. 1997. Involvement of mitogen-activated protein kinase pathways in the nuclear responses and cytokine production induced by Salmonella typhimurium in cultured intestinal epithelial cells. J. Immunol. 159:5550-5559. [PubMed] [Google Scholar]
- 33.Hu, L., and T. E. Hickey. 2005. Campylobacter jejuni induces secretion of proinflammatory chemokines from human intestinal epithelial cells. Infect. Immun. 73:4437-4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu, L., and D. J. Kopecko. 2000. Interactions of Campylobacter with eukaryotic cells: gut luminal colonization and mucosal invasion mechanisms, p. 191-217. In I. Nachamkin and M. J. Blaser (ed.), Campylobacter, 2nd ed. ASM Press, Washington, D.C.
- 35.Hurley, B. P., and B. A. McCormick. 2004. Intestinal epithelial defense systems protect against bacterial threats. Curr. Gastroenterol. Rep. 6:355-361. [DOI] [PubMed] [Google Scholar]
- 36.Iwasaki, A., and B. L. Kelsall. 2000. Localization of distinct Peyer's patch dendritic cell subsets and their recruitment by chemokines macrophage inflammatory protein (MIP)-3alpha, MIP-3beta, and secondary lymphoid organ chemokine. J. Exp. Med. 191:1381-1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Izadpanah, A., M. B. Dwinell, L. Eckmann, N. M. Varki, and M. F. Kagnoff. 2001. Regulated MIP-3alpha/CCL20 production by human intestinal epithelium: mechanism for modulating mucosal immunity. Am. J. Physiol. Gastrointest. Liver Physiol. 280:G710-G719. [DOI] [PubMed] [Google Scholar]
- 38.Jobin, C., S. Haskill, L. Mayer, A. Panja, and R. B. Sartor. 1997. Evidence for altered regulation of I kappa B alpha degradation in human colonic epithelial cells. J. Immunol. 158:226-234. [PubMed] [Google Scholar]
- 39.Jobin, C., A. Panja, C. Hellerbrand, Y. Iimuro, J. Didonato, D. A. Brenner, and R. B. Sartor. 1998. Inhibition of proinflammatory molecule production by adenovirus-mediated expression of a nuclear factor kappaB super-repressor in human intestinal epithelial cells. J. Immunol. 160:410-418. [PubMed] [Google Scholar]
- 40.Jobin, C., and R. B. Sartor. 2000. The I kappa B/NF-kappa B system: a key determinant of mucosalinflammation and protection. Am. J. Physiol. Cell Physiol. 278:C451-C462. [DOI] [PubMed] [Google Scholar]
- 41.Jung, H. C., L. Eckmann, S. K. Yang, A. Panja, J. Fierer, E. Morzycka-Wroblewska, and M. F. Kagnoff. 1995. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J. Clin. Investig. 95:55-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kagnoff, M. F., and L. Eckmann. 1997. Epithelial cells as sensors for microbial infection. J. Clin. Investig. 100:6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karin, M., and Y. Ben-Neriah. 2000. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 18:621-663. [DOI] [PubMed] [Google Scholar]
- 44.Konkel, M. E., S. A. Gray, B. J. Kim, S. G. Garvis, and J. Yoon. 1999. Identification of the enteropathogens Campylobacter jejuni and Campylobacter coli based on the cadF virulence gene and its product. J. Clin. Microbiol. 37:510-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Konkel, M. E., M. R. Monteville, V. Rivera-Amill, and L. A. Joens. 2001. The pathogenesis of Campylobacter jejuni-mediated enteritis. Curr. Issues Intest. Microbiol. 2:55-71. [PubMed] [Google Scholar]
- 46.Korlath, J. A., M. T. Osterholm, L. A. Judy, J. C. Forfang, and R. A. Robinson. 1985. A point-source outbreak of campylobacteriosis associated with consumption of raw milk. J. Infect. Dis. 152:592-596. [DOI] [PubMed] [Google Scholar]
- 47.Krzysiek, R., E. A. Lefevre, J. Bernard, A. Foussat, P. Galanaud, F. Louache, and Y. Richard. 2000. Regulation of CCR6 chemokine receptor expression and responsiveness to macrophage inflammatory protein-3alpha/CCL20 in human B cells. Blood 96:2338-2345. [PubMed] [Google Scholar]
- 48.Kwon, J. H., S. Keates, S. Simeonidis, F. Grall, T. A. Libermann, and A. C. Keates. 2003. ESE-1, an enterocyte-specific Ets transcription factor, regulates MIP-3alpha gene expression in Caco-2 human colonic epithelial cells. J. Biol. Chem. 278:875-884. [DOI] [PubMed] [Google Scholar]
- 49.Lara-Tejero, M., and J. E. Galan. 2000. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 290:354-357. [DOI] [PubMed] [Google Scholar]
- 50.Li, Q., and I. M. Verma. 2002. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2:725-734. [DOI] [PubMed] [Google Scholar]
- 51.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 52.Macpherson, A. J., M. B. Geuking, and K. D. McCoy. 2005. Immune responses that adapt the intestinal mucosa to commensal intestinal bacteria. Immunology 115:153-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mellits, K. H., J. Mullen, M. Wand, G. Armbruster, A. Patel, P. L. Connerton, M. Skelly, and I. F. Connerton. 2002. Activation of the transcription factor NF-kappaB by Campylobacter jejuni. Microbiology 148:2753-2763. [DOI] [PubMed] [Google Scholar]
- 54.Mumy, K. L., and B. A. McCormick. 2005. Events at the host-microbial interface of the gastrointestinal tract. II. Role of the intestinal epithelium in pathogen-induced inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 288:G854-G859. [DOI] [PubMed] [Google Scholar]
- 55.Naumann, M. 2000. Nuclear factor-kappa B activation and innate immune response in microbial pathogen infection. Biochem. Pharmacol. 60:1109-1114. [DOI] [PubMed] [Google Scholar]
- 56.Neish, A. S. 2004. Molecular aspects of intestinal epithelial cell-bacterial interactions that determine the development of intestinal inflammation. Inflamm. Bowel Dis. 10:159-168. [DOI] [PubMed] [Google Scholar]
- 57.Neish, A. S., A. T. Gewirtz, H. Zeng, A. N. Young, M. E. Hobert, V. Karmali, A. S. Rao, and J. L. Madara. 2000. Prokaryotic regulation of epithelial responses by inhibition of IkappaB-alpha ubiquitination. Science 289:1560-1563. [DOI] [PubMed] [Google Scholar]
- 58.Newell, D. G. 2001. Animal models of Campylobacter jejuni colonization and disease and the lessons to be learned from similar Helicobacter pylori models. Symp. Ser. Soc. Appl. Microbiol. 2001:57S-67S. [DOI] [PubMed] [Google Scholar]
- 59.Nusrat, A., S. V. Sitaraman, and A. Neish. 2001. Interaction of bacteria and bacterial toxins with intestinal epithelial cells. Curr. Gastroenterol. Rep. 3:392-398. [DOI] [PubMed] [Google Scholar]
- 60.Oelschlaeger, T. A., P. Guerry, and D. J. Kopecko. 1993. Unusual microtubule-dependent endocytosis mechanisms triggered by Campylobacter jejuni and Citrobacter freundii. Proc. Natl. Acad. Sci. USA 90:6884-6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parkhill, J., B. W. Wren, K. Mungall, J. M. Ketley, C. Churcher, D. Basham, T. Chillingworth, R. M. Davies, T. Feltwell, S. Holroyd, K. Jagels, A. V. Karlyshev, S. Moule, M. J. Pallen, C. W. Penn, M. A. Quail, M. A. Rajandream, K. M. Rutherford, A. H. van Vliet, S. Whitehead, and B. G. Barrell. 2000. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403:665-668. [DOI] [PubMed] [Google Scholar]
- 62.Peterson, M. C. 1994. Rheumatic manifestations of Campylobacter jejuni and C. fetus infections in adults. Scand. J. Rheumatol. 23:167-170. [DOI] [PubMed] [Google Scholar]
- 63.Philpott, D. J., and S. E. Girardin. 2004. The role of Toll-like receptors and Nod proteins in bacterial infection. Mol. Immunol. 41:1099-1108. [DOI] [PubMed] [Google Scholar]
- 64.Pickett, C. L., E. C. Pesci, D. L. Cottle, G. Russell, A. N. Erdem, and H. Zeytin. 1996. Prevalence of cytolethal distending toxin production in Campylobacter jejuni and relatedness of Campylobacter sp. cdtB genes. Infect. Immun. 64:2070-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rescigno, M., G. Rotta, B. Valzasina, and P. Ricciardi-Castagnoli. 2001. Dendritic cells shuttle microbes across gut epithelial monolayers. Immunobiology 204:572-581. [DOI] [PubMed] [Google Scholar]
- 66.Rescigno, M., M. Urbano, B. Valzasina, M. Francolini, G. Rotta, R. Bonasio, F. Granucci, J. P. Kraehenbuhl, and P. Ricciardi-Castagnoli. 2001. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2:361-367. [DOI] [PubMed] [Google Scholar]
- 67.Russell, R. G., M. O'Donnoghue, D. C. Blake, Jr., J. Zulty, and L. J. DeTolla. 1993. Early colonic damage and invasion of Campylobacter jejuni in experimentally challenged infant Macaca mulatta. J. Infect. Dis. 168:210-215. [DOI] [PubMed] [Google Scholar]
- 68.Sansonetti, P. J. 2004. War and peace at mucosal surfaces. Nat. Rev. Immunol. 4:953-964. [DOI] [PubMed] [Google Scholar]
- 69.Savkovic, S. D., A. Ramaswamy, A. Koutsouris, and G. Hecht. 2001. EPEC-activated ERK1/2 participate in inflammatory response but not tight junction barrier disruption. Am. J. Physiol. Gastrointest. Liver Physiol. 281:G890-G898. [DOI] [PubMed] [Google Scholar]
- 70.Scott, D. A., and T., D. R. 2000. Protection against Campylobacter infection and vaccine development, p. 303-319. In I. Nachamkin and M. J. Blaser (ed.), Campylobacter, 2nd ed. ASM Press, Washington, D.C.
- 71.Sierro, F., B. Dubois, A. Coste, D. Kaiserlian, J. P. Kraehenbuhl, and J. C. Sirard. 2001. Flagellin stimulation of intestinal epithelial cells triggers CCL20-mediated migration of dendritic cells. Proc. Natl. Acad. Sci. USA 98:13722-13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Skirrow, M. B., and M. J. Blaser. 2000. Clinical aspects of Campylobacter infection, p. 69-88. In I. Nachamkin and M. J. Blaser (ed.), Campylobacter. ASM Press, Washington, D.C.
- 73.Smith, J. M., P. A. Johanesen, M. K. Wendt, D. G. Binion, and M. B. Dwinell. 2005. CXCL12 activation of CXCR4 regulates mucosal host defense through stimulation of epithelial cell migration and promotion of intestinal barrier integrity. Am. J. Physiol. Gastrointest. Liver Physiol. 288:G316-G326. [DOI] [PubMed] [Google Scholar]
- 74.Song, Y. C., S. Jin, H. Louie, D. Ng, R. Lau, Y. Zhang, R. Weerasekera, S. Al Rashid, L. A. Ward, S. D. Der, and V. L. Chan. 2004. FlaC, a protein of Campylobacter jejuni TGH9011 (ATCC43431) secreted through the flagellar apparatus, binds epithelial cells and influences cell invasion. Mol. Microbiol. 53:541-553. [DOI] [PubMed] [Google Scholar]
- 75.Steiner, T. S., J. P. Nataro, C. E. Poteet-Smith, J. A. Smith, and R. L. Guerrant. 2000. Enteroaggregative Escherichia coli expresses a novel flagellin that causes IL-8 release from intestinal epithelial cells. J. Clin. Investig. 105:1769-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sugita, S., T. Kohno, K. Yamamoto, Y. Imaizumi, H. Nakajima, T. Ishimaru, and T. Matsuyama. 2002. Induction of macrophage-inflammatory protein-3alpha gene expression by TNF-dependent NF-kappaB activation. J. Immunol. 168:5621-5628. [DOI] [PubMed] [Google Scholar]
- 77.Tallant, T., A. Deb, N. Kar, J. Lupica, M. J. de Veer, and J. A. DiDonato. 2004. Flagellin acting via TLR5 is the major activator of key signaling pathways leading to NF-kappa B and proinflammatory gene program activation in intestinal epithelial cells. BMC Microbiol. 4:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tanaka, Y., T. Imai, M. Baba, I. Ishikawa, M. Uehira, H. Nomiyama, and O. Yoshie. 1999. Selective expression of liver and activation-regulated chemokine (LARC) in intestinal epithelium in mice and humans. Eur. J. Immunol. 29:633-642. [DOI] [PubMed] [Google Scholar]
- 79.Viala, J., P. Sansonetti, and D. J. Philpott. 2004. Nods and ‘intracellular’ innate immunity. C. R. Biol. 327:551-555. [DOI] [PubMed] [Google Scholar]
- 80.Wassenaar, T. M., and M. J. Blaser. 1999. Pathophysiology of Campylobacter jejuni infections of humans. Microbes Infect. 1:1023-1033. [DOI] [PubMed] [Google Scholar]
- 81.Wassenaar, T. M., and D. G. Newell. 2000. Genotyping of Campylobacter spp. Appl. Environ Microbiol. 66:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Watson, R. O., and J. E. Galan. 2005. Signal transduction in Campylobacter jejuni-induced cytokine production. Cell Microbiol. 7:655-665. [DOI] [PubMed] [Google Scholar]
- 83.Whitehouse, C. A., P. B. Balbo, E. C. Pesci, D. L. Cottle, P. M. Mirabito, and C. L. Pickett. 1998. Campylobacter jejuni cytolethal distending toxin causes a G2-phase cell cycle block. Infect. Immun. 66:1934-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang, D., O. M. Howard, Q. Chen, and J. J. Oppenheim. 1999. Cutting edge: immature dendritic cells generated from monocytes in the presence of TGF-beta 1 express functional C-C chemokine receptor 6. J. Immunol. 163:1737-1741. [PubMed] [Google Scholar]
- 85.Yang, S. K., L. Eckmann, A. Panja, and M. F. Kagnoff. 1997. Differential and regulated expression of C-X-C, C-C, and C-chemokines by human colon epithelial cells. Gastroenterology 113:1214-1223. [DOI] [PubMed] [Google Scholar]
- 86.Young, V. B., D. B. Schauer, and J. G. Fox. 2000. Animal models of Campylobacter infection, p. 287-301. In I. Nachamkin nd M. J. Blaser (ed.), Campylobacter, 2nd ed. ASM Press, Washington, D.C.
- 87.Yuki, N., K. Susuki, M. Koga, Y. Nishimoto, M. Odaka, K. Hirata, K. Taguchi, T. Miyatake, K. Furukawa, T. Kobata, and M. Yamada. 2004. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barre syndrome. Proc. Natl. Acad. Sci. USA 101:11404-11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhou, X., J. A. Giron, A. G. Torres, J. A. Crawford, E. Negrete, S. N. Vogel, and J. B. Kaper. 2003. Flagellin of enteropathogenic Escherichia coli stimulates interleukin-8 production in T84 cells. Infect. Immun. 71:2120-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]