

Abstract
Background
Urokinase-type plasminogen activator (uPA) plays a major role in extracellular proteolytic events associated with tumor cell growth, migration and angiogenesis. Consequently, uPA is an attractive target for the development of small molecule active site inhibitors. Most of the recent drug development programs aimed at nonpeptidic inhibitors targeted at uPA have focused on arginino mimetics containing amidine or guanidine functional groups attached to aromatic or heterocyclic scaffolds. There is a general problem of limited bioavailability of these charged inhibitors. In the present study, uPA inhibitors were designed on an isocoumarin scaffold containing uncharged substituents.
Results
4-Chloro-3-alkoxyisocoumarins were synthesized in which the 3-alkoxy group contained a terminal bromine; these were compared with similar inhibitors that contained a charged terminal functional group. Additional variations included functional groups attached to the seven position of the isocoumarin scaffold. N- [3-(3-Bromopropoxy)-4-chloro-1-oxo-1H-isochromen-7-yl]benzamide was identified as an uncharged lead inhibitor of uPA, Ki = 0.034 μM. Molecular modeling of human uPA with these uncharged inhibitors suggests that the bromine occupies the same position as positively charged arginino mimetic groups.
Conclusion
This study demonstrates that potent uncharged inhibitors of uPA can be developed based upon the isocoumarin scaffold. A tethered bromine in the three position and an aromatic group in the seven position are important contributors to binding. Although the aim was to develop compounds that act as mechanism-based inactivators, these inhibitors are competitive reversible inhibitors.
Background
Multiple proteases, including matrix metalloproteases (MMP-2, MMP-9 and MMP-14), cysteine proteases (cathepsin B and cathepsin L), aspartyl protease (cathepsin D) and serine proteases (plasmin, matriptase and urokinase) participate in cancer cell growth, metastasis and angiogenesis [1-4]. High expression of proteases often correlates with a poor prognosis [5,6]. Urokinase (uPA) plays an especially important role in extracellular proteolysis that contributes to cancer cell metastasis. Many cancer cells secrete pro-uPA and its receptor uPAR; binding of pro-uPA to uPAR leads to its activation, with subsequent generation of plasmin by the uPA-catalyzed hydrolysis of extracellular plasminogen [7,8]. The increased production of plasmin leads to degradation of extracellular matrix both by plasmin itself and by other proteases that are activated by plasmin. The surface location of bound uPA provides directionality to the degradation of matrix, thereby assisting the directional migration of cancer cells. uPA in complex with uPAR also affects other biological processes including signaling pathways that influence cell proliferation [9]. uPA has become a major target for development of non-peptidic small molecule inhibitors as potential anti-cancer drugs [10,11].
Most of the efforts to develop potent and selective inhibitors of uPA have focused on arginino mimetics based upon the trypsin-like specificity of uPA. The development of selective inhibitors of uPA is a challenge due to the large number of serine proteases with trypsin-like specificity, including factor VII, factor X and tissue-type plasminogen activator. Extensive structure-based drug development has provided potent and selective inhibitors of uPA; these generally are arginino mimetics with amidine or guanidine functional groups built onto aromatic or heterocyclic scaffolds (Figure 4) [12-16]. A major limitation to the use of these inhibitors is their poor bioavailability, which is at least partly owing to the presence of the positively charged amidine or guanidine group. This has limited clinical studies of these uPA inhibitors.
Figure 4.

Scaffolds that have been utilized to develop arginino mimetic uPA inhibitors in the amidine (I-VI) and guanidine (VII-IX) series [12–16].
In the present study, we have focused on the synthesis and testing of uncharged compounds as leads for the development of uPA inhibitors with improved bioavailability. 4-Chloroisocoumarin was selected as the scaffold, in which substituted 3-alkoxy groups were introduced that contained neutral terminal functional groups or charged terminal functional groups [17]. Additional substituents were introduced into the seven position. 4-Chloroisocoumarin scaffolds have been used in studies of serine protease inhibitors, [17] but with limited application to uPA [18]. The choice of the 4-chloroisocoumarin scaffold was based upon the potential of these compounds to function as mechanism-based inactivators [17]. In this study we demonstrate that introduction of bromine in place of a terminal charged functional group in the 3-alkoxy substituent provides uncharged uPA inhibitors with low micromolar dissociation constants. Further introduction of substituents at the seven position of these uncharged uPA inhibitors provides compounds with low nanomolar dissociation constants. These inhibitors may serve as lead compounds for the development of new uPA inhibitors. Molecular modeling with human uPA suggests that the bromine occupies the same site as the arginino mimetic functional groups.
Results and discussion
Chemistry
Compounds 4a-4e, which are 3-bromoalkoxy-4-chloroisocoumarins, were synthesized as shown in Figure 1. Two of the compounds, 4a and 4b, have a nitro group in position seven. These compounds have varying lengths of bromoalkoxy groups tethered in position three of the isocoumarin scaffold. 5-Nitrohomophthalic acid (1a) was prepared by regioselective nitration of homophthalic acid (1c) using fuming nitric acid [19]. 5-Nitrohomophthalic acid (1a) and homophthalic acid (1c) were monoesterified using bromoalcohols, compounds 2c-2e, in the presence of sulfuric acid to give moderate yields of bromoesters, 3a-3e. Monoesterification at the saturated acid has been attributed to the mesomeric effect of the carboxyl with the double bond in the aryl ring [20]. Cyclization of the esters, compounds 3a-3e, with phosphorus pentachloride in toluene gave 3-bromoalkoxy-4-chloroisocoumarins, 4a-4e, in moderate yields using a variation of a published method [17]. Compounds 4a-4e were synthesized to test the importance of an uncharged group in the three position of 4-chloroisocoumarins and the length of the tether of the alkoxy group.
Figure 1.
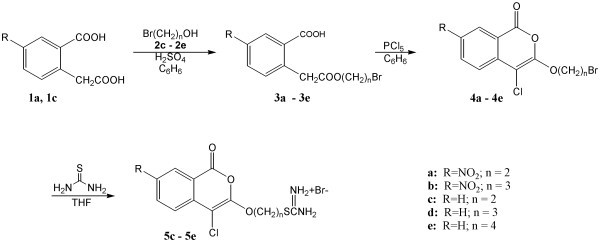
Scheme 1
Compounds 5c-5e are 3-isothioureidoalkoxy-4-chloroisocoumarin salts and were synthesized as shown in Figure 1. Nucleophilic substitution of the bromine in compounds 4c-4e was achieved by refluxing these compounds with thiourea in tetrahydrofuran to give the hydrobromide salts, compounds 5c-5e, in moderate yields. The 7-nitrosubstituted isocoumarins, compounds 4a and 4b, did not give any identifiable products when reacted with thiourea in tetrahydrofuran. Compounds 5c-5e were synthesized to test the importance of a charged alkoxy group in the three position of 4-chloroisocoumarins.
The synthesis of 3-bromo-4-chloro-7-aminoisocoumarins, compounds 6a and 6b, is shown in Figure 2. These 7-aminoisocoumarins were prepared by reduction of 3-bromoalkoxy-4-chloro-7-nitroisocoumarins, compounds 4a and 4b, using hydrogen in the presence of a catalytic amount of 10% palladium on charcoal under pressure in methanol. Compounds 6a and 6b were synthesized to test the importance of an amino group in the seven position and an uncharged alkoxy group in the three position of 4-chloroisocoumarins. Nucleophilic substitution of the bromine in compounds 6a and 6b by reaction with thiourea afforded hydrobromide salts, 7a and 7b. Compounds 7a and 7b were synthesized to test the importance of a charged alkoxy group in the three position of 4-chloro-7-aminoisocoumarins.
Figure 2.
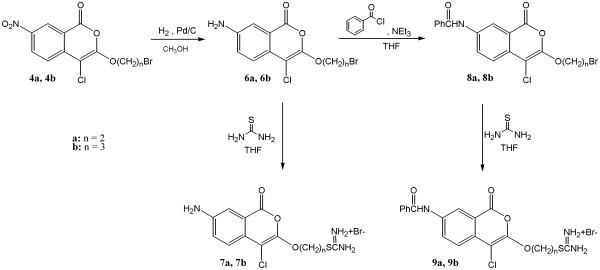
Scheme 2
3-Bromoalkoxy-4-chloro-7-benzamidoisocoumarins, compounds 8a and 8b, were synthesized by reaction of 6a and 6b with benzoyl chloride in the presence of triethylamine (Figure 2). Compounds 8a and 8b were synthesized to test the importance of an uncharged alkoxy group in the three position and a hydrophobic benzamide group in the seven position of 4-chloroisocoumarins. The two amides, 8a and 8b were reacted with thiourea to give the hydrobromide salts, 9a and 9b, in moderate yields. Compounds 9a and 9b were synthesized to test the importance of a charged alkoxy group in the three position and a hydrophobic benzamide group in the seven position of 4-chloroisocoumarins.
Figure 3 describes the synthesis of two 7-nitro-3-alkoxyisocoumarins, compounds 10a and 10b, which do not have a chlorine atom at position four. The synthesis of compound 10c, which is a 3-alkoxy-4-trifluoroacetylisocoumarin, is also described in Figure 3. Cyclization of compounds 3a and 3b, which have a nitro group in position seven, using trifluoroacetic anhydride gave compounds 10a and 10b, which contain a hydrogen at position four. On the other hand, cyclization of 3c which does not contain a nitro group in position seven gave compound 10c, which contains a trifluoroacetyl group in position four. This may be attributed to the fact that the intramolecular cyclization of the initially formed enol is faster when there is resonance stabilization by the electron withdrawing nitro group. When the nitro group is absent the initially formed enol reacts with trifluoroacetic anhydride giving compound 10c. Compounds 10a and 10b were synthesized to test the importance of a chlorine atom in position four and the presence of an electron withdrawing group in position seven of the isocoumarins. Compound 10c gives information on the importance of a trifluoroacetyl group in position four of the isocoumarins.
Figure 3.

Scheme 3
Compound 11, a 7-amino-3-alkoxyisocoumarin, was prepared by reduction of compound 10a using hydrogen in the presence of a catalytic amount of 10% palladium on charcoal under pressure in methanol (Figure 3). Compound 11 was synthesized to test the importance of a chlorine atom in position four and the presence of an electron donating group in position seven of the isocoumarins.
Structure activity relationships
On the basis of docking studies using the crystal structure of human uPA [15] we examined whether the 4-chloroisocoumarin scaffold containing 3-alkoxy substituents is predicted to be a good template for the design of uPA inhibitors. Our interest focused on compounds having an uncharged bromine group in place of the charged arginino mimetic group at the terminal position of the 3-alkoxy group. Specifically, we compared the experimentally determined dissociation constants with the docking orientations predicted for compounds with the isocoumarin scaffold containing a charged isothiourea group or an uncharged bromine atom in the terminal 3-alkoxy position.
The first series of compounds compared the 4-chloroisocoumarin scaffold with 3-alkoxy substituents where the terminal functional group was an isothiourea group or bromine atom. The distance between the terminal group and the alkoxy oxygen was varied by the insertion of methylene units (Table 1). The designed inhibitors were shown to dock in the area of the active site containing the catalytic triad (serine 195, histidine 57, aspartic acid 102) and aspartic acid 189, which is the residue in trypsin-like enzymes that forms a salt bridge with the arginino mimetic groups. All of the compounds were predicted to reside in the active site with the thiourea group and the bromine atom in the S1-subsite which contains aspartic acid 189. Compounds 4c-4e, which are 3-bromoalkoxy-4-chloroisocoumarins, are oriented in the active site with the uncharged bromine atom within 2.81–3.12 Å of aspartic acid 189. The best inhibitor in this series, compound 4d, exhibited Ki = 9 μM. Modeling of inhibitor 4d with uPA also showed that three methylene units between the bromine and oxygen of the 3-alkoxy substituent provided the closest interaction between the isocoumarin carbonyl and the active site residue serine 195. Nevertheless, compound 4d provided simple competitive inhibition with no evidence of rapid inactivation of uPA by 4d. For comparison, compounds 5c-5e were included in this series. These compounds contain the charged isothiourea group in place of the bromine atom and two, three or four methylene units between the isothiourea group and the oxygen atom. Modeling suggests that compounds 5c-5e are all oriented in the active site with the charged isothiourea group forming a salt bridge with aspartic acid 189. The predicted distance between the isothiourea group and aspartic acid 189 for compounds 5c-5e ranges from 2.49 – 2.58 Å. The best inhibitor, compound 5c, exhibited a Ki = 0.033 μM. Modeling suggested that the 3-alkoxy substituent with two methylenes between the isothiourea group and oxygen atom provided the closest interactions between the isocoumarin carbonyl and the active site serine. Clearly, the charged isothiourea group provides 3.5–4 kcal/mol more binding energy than the bromine atom in its interactions with aspartic 189 and surrounding residues. Thus, there is a large penalty in moving to the uncharged bromoalkoxy inhibitors. This may be the penalty that must be accepted if uncharged uPA inhibitors with improved bioavailability are to be developed in place of the charged uPA inhibitors that have been described [12-16].
Table 1.
Dissociation constants for inhibition of human uPA
| Structure | Number | Ki (μM) | Structure | Number | Ki (μM) |
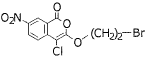 |
4a | 1.8 ± 0.2 | 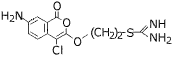 |
7a | 0.020 ± 0.003 |
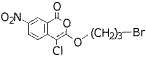 |
4b | 2.4 ± 0.2 | 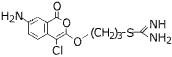 |
7b | 0.038 ± 0.005 |
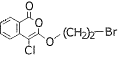 |
4c | 14 ± 1 | 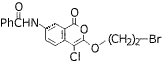 |
8a | 1.4 ± 0.2 |
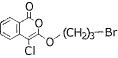 |
4d | 9 ± 1 | 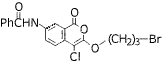 |
8b | 0.034 ± 0.002 |
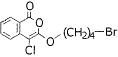 |
4e | 18 ± 2 | 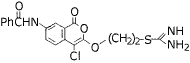 |
9a | 0.084 ± 0.007 |
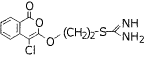 |
5c | 0.033 ± 0.004 | 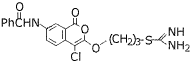 |
9b | 0.010 ± 0.008 |
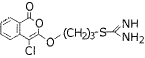 |
5d | 0.055 ± 0.003 | 10a | 4.2 ± 0.4 | |
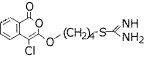 |
5e | 0.14 ± 0.02 | 10b | 4.3 ± 0.3 | |
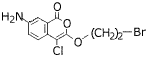 |
6a | 12 ± 1 | 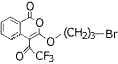 |
10c | 14 ± 2 |
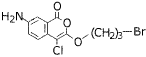 |
6b | 9.5 ± 1 | 11 | 65 ± 9 |
The second series of uPA inhibitors examined the effects of substituents in the seven position. Compounds 6a, 6b, 7a and 7b, which are 7-amino-4-chloroisocoumarins are attractive candidates because of the increased hydrogen bonding possibilities. Based upon modeling, compounds 6a and 6b, which have an uncharged bromine atom on the 3-alkoxy group, are within 2.80-3.15 Å of aspartic acid 189. Compounds 7a and 7b, which have a charged isothiourea group within 2.75–2.85 Å from aspartic acid 189. Compounds 8a, 8b, 9a and 9b are 7-substituted benzamides that were synthesized to investigate potential hydrophobic interactions from groups at the seven position. The uncharged bromine atoms of compounds 8a and 8b are within 2.93–3.32 Å of aspartic acid 189. The charged isothiourea group in compounds 9a and 9b are within 2.75 Å of aspartic acid 189. In the isothiourea series (7a, 7b, 9a and 9b), the 7-substituted compounds did not exhibit any significant improvement in binding to uPA (Table 1). By comparison, the compounds in the bromo series with hydrophobic groups at the seven position (8a and 8b) exhibited markedly improved binding; for 8b, Ki = 0.034 μM, which represents a 300-fold improvement over 4d. Inhibitor 8b was the best inhibitor in the bromo series and can be viewed as a promising lead compound for development of uncharged inhibitors of uPA. The improvement that the benzamide group in 8a and 8b of the bromo series provided compared to the lack of improvement that the benzamide group provided in the isothiourea series may relate to increased flexibility. The bromo group has weak interactions with aspartic acid 189; this may allow the inhibitors in the bromo series to slide within the active site and S1 sub-site to orientations that allow the 7-benzamide group to find additional energetically favorable interactions. By comparison, the strong salt bridge in the uPA complexes with inhibitors in the isothiourea series may lock the inhibitors into the S1 sub-site. The modeled uPA complexes with inhibitors 8b and 9b are shown in Figures 5 and 6 respectively. The additional binding energy for 8b appears to be the result of additional interactions of the aromatic ring of the benzamide group, especially with disulfide cysteine 58-cysteine 42.
Figure 5.
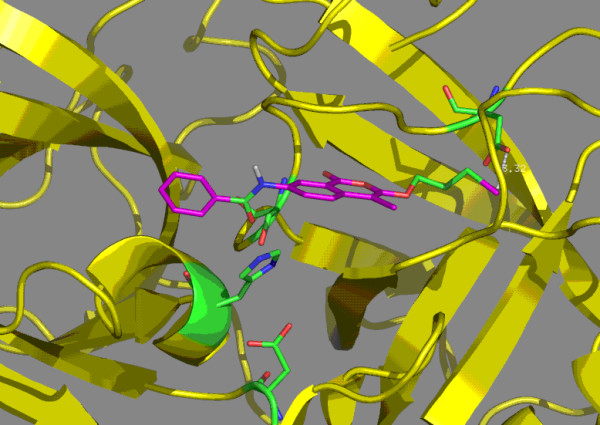
Molecular model of human uPA complexed with inhibitor 8b. The bromine is within 3.32 Å of aspartic acid 189.
Figure 6.
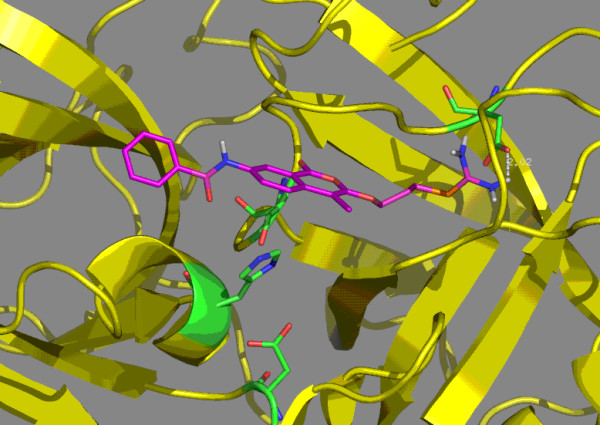
Molecular model of human uPA complexed with inhibitor 9b. The isothiourea is within 2.02 Å of aspartic acid 189.
The presence of a 7-nitro group also was beneficial. Compounds 4a and 4b, which are 3-bromoalkoxy-7-nitroisocoumarins, showed improved binding to uPA compared to the unsubstituted compounds. Compound 4b which has three methylene units between the bromine atom and the oxygen of the 3-alkoxy group has a Ki = 2.4 μM, which is an 4-fold improvement over the unsubstituted compound 4d.
Compounds 10a, 10b and 11 were synthesized to determine whether the chlorine atom in position four contributed to binding. 3-Bromoalkoxy-7-substituted isocoumarins without a chlorine atom in the four position show dissociation constants that are about 2 fold higher compared to their counterparts that have a chlorine atom in the four position suggesting a modest role for the chlorine atom (Table 1). Compound 10c which has trifluoroacetyl group in the four position did not show improved binding.
The isocoumarin-based inhibitors (Table 1) have the potential to function as suicide inhibitors or as substrates. However, simple reversible competitive inhibition was observed. The inhibitors were stable for several hours at neutral pH, as evidenced by no change in the spectral properties of the inhibitors. Addition of uPA for several hours at concentrations ten-fold higher than used in the kinetic studies did not produce any detectable changes in the inhibitors, suggesting that the inhibitors are not weak substrates of uPA. In addition, there was no loss of enzyme activity under these conditions, suggesting that the inhibitors are not functioning as suicide inhibitors. The observation of simple competitive inhibition is consistent with the modeling results in which the predicted orientations of the isocoumarin scaffold bound in the active site of uPA (figures 5 and 6) are not favorable for attack by serine 195.
Conclusion
Inhibition of uPA by uncharged inhibitor 8b represents a proof of concept that uPA inhibitors without a charged arginino mimetic group can be developed. Inhibitor 8b, which exhibits a dissociation constant in the low nanomolar range comparable to those of known arginino mimetic inhibitors, represents a lead compound for future development of uncharged inhibitors of uPA. The present study did not address the issue of specificity. Many previous studies of uPA inhibitors with arginino mimetic groups attached to various scaffold have resulted in the development of selective inhibitors of uPA. This information should be useful for developing selective uncharged inhibitors of uPA.
Methods
Modeling
The x-ray crystal structure of human urokinase (pdb code 1EJN) was obtained from the protein data bank. All compounds shown in Table 1 were docked to the enzyme using Autodock 3.0 [21,22] on a cluster of Silicon Graphics workstations consisting of Octanes and O2s. The compounds were prepared using Sybyl 7.0 (Tripos Inc., St. Louis, MO). The molecules were drawn in, assigned partial charges using the included Gasteiger-Hückel method and energy minimized using the BFGS method. Minimizations were run for 10,000 iterations and the rotatable bonds defined before docking. The protein was prepared before docking in Sybyl by removing non-native substrates and water molecules. Polar hydrogens and Kollman Uni charges were added to the protein as well. The molecules were docked in an area defined around the active site serine 195 by a cube of 60 × 60 × 60 Å.
Chemical synthesis
Reagent quality solvents were used without purification. Benzoyl chloride was distilled before use. Melting points were determined on a Thomas Hoover capillary melting point apparatus and are uncorrected. NMR spectra were recorded on a Bruker AC250 NMR spectrometer in CDCl3 unless noted. Chemical shifts are in ppm (δ) relative to TMS. High resolution mass spectra were recorded on a Waters/Micromass LCT- premier. Analytical data was obtained from Galbraith laboratories, Knoxville TN. 5-Nitrohomophthalic acid was prepared as reported [19]. Compounds 3a-3e, 4a-4e, 5c-5e, 6a, 6b, 8a, 8b 10a, 10b and 11 were prepared according to published procedures [23]. Compounds 7a, 7b, 9a, 9b were prepared as reported [24].
Kinetics
Human urokinase (Sigma/Aldrich, St. Louis, MO) and Spectrozyme UK (American Diagnostica, Stamford, CT) were used for the kinetic studies. Enzyme activity was routinely measured in 1 ml volumes of 0.1 M Tris, pH 8.8, Spectrozyme UK (10 μM to 150 μM) and 0.64 μg (3,770 units/mg protein) human urokinase. Reactions were monitored at 405 nm, 25°C, with a Perkin/Elmer Lambda S2 UV/vis spectrophotometer. Michaelis constants and Ki values were determined from initial rate data, measured at 8 to 10 substrate concentrations, by non-linear regression analysis with SigmaPlot's Enzyme Kinetics Module™ (Chicago, IL, USA).
Experimental
2- [2-(2-Bromoethoxy)-2-oxoethyl]-5-nitrobenzoic acid (3a) (66% yield) Tan crystals: mp 113–115°C (lit. [25] 90°C);1H NMR: δ 3.50 (t, 2H, J = 5.96 Hz) 4.21 (s, 2H) 4.43 (t, 2H, J = 6.06 Hz) 7.51 (d, 1H, J = 8.34 Hz) 8.39 (dd, 1H, J = 2.59 Hz, 8.39 Hz) 8.98 (d, J = 2.39 Hz);13C NMR: δ 28.40, 40.13, 64.17, 126.24, 126.45, 130.97, 133.32, 142.59, 146.84, 167.46, 169.63.
2- [2-(3-Bromopropoxy)-2-oxoethyl]-5-nitrobenzoic acid (3b) (51% yield) White crystals: mp 122–123°C; 1H NMR: δ 2.19 (m, 2H) 3.44 (t, 2H, J = 6.56 Hz) 4.18 (s, 2H) 4.28 (t, 2H, J = 5.96 Hz) 7.51 (d, 1H, J = 8.34 Hz) 8.39 (dd, 1H, J = 2.39 Hz, 8.35 Hz) 8.98 (d, 1H, J = 2.18 Hz) 9.78 (br s, 1H); 13C NMR: δ 29.18, 31.58, 40.51, 63.12, 126.84, 127.52, 129.77, 143.37, 147.47, 169.92, 170.05.
2- [2-(2-Bromoethoxy)-2-oxoethyl]benzoic acid (3c) (50% yield) Tan crystals: mp 82–83°C; 1H NMR: δ 3.50 (t, 2H, J = 6.25 Hz) 4.07 (s, 2H) 4.40 (t, 2H, J = 6.25 Hz) 7.27 (d, 1H, J = 7.75 Hz) 7.45 (t, 1H, J = 7.55 Hz) 7.54 (td, 1H, J = 1.39 Hz, 7.55 Hz) 8.14 (d, 1H, J = 7.74 Hz); 13C NMR: δ 28.50, 40.68, 64.08, 127.65, 128.32, 131.95, 132.43, 133.35, 136.40, 170.86, 172.43.
2- [2-(3-Bromopropoxy)-2-oxoethyl]benzoic acid (3d) (70% yield) White crystals: mp 79–80°C; 1H NMR: δ 2.16 (m, 2H, J = 6.31 Hz) 3.42 (t, 2H, J = 6.55 Hz) 4.05 (s, 2H) 4.24 (t, 2H, J = 5.96 Hz) 7.27 (d, 1H, J = 7.55 Hz) 7.40 (t, 1H, J = 7.65 Hz) 7.54 (td, 1H, J = 1.2, 7.35 Hz) 8.14 (dd, 1H, J = 0.99, 7.74 Hz); 13C NMR: δ 29.50, 31.80, 40.80, 62.52, 127.60, 128.35, 131.91, 132.41, 133.35, 136.86, 171.17, 172.47.
2- [2-(4-Bromobutoxy)-2-oxoethyl]benzoic acid (3e) A solution of 4-bromo-1-butanol (2e, 6.0 mL, 41.6 mmol), homophthalic acid (1c, 2.5 g, 13.8 mmol), and five drops of concentrated sulfuric acid was refluxed in benzene (50 mL) for four hours. The solution was cooled and washed with water (2 × 25 mL), brine (1 × 25 mL), and dried over magnesium sulfate. Filtration and evaporation of the solvent gave a dark oil that was triturated with hexane to afford a crude solid. Recrystallization from hexane/ethyl acetate gave 0.91 g (40%) of compound 3e as white crystals: mp 84–86°C; 1H NMR: δ 1.79 (m, 2H) 1.87 (m, 2H) 3.38 (t, 2H, J = 6.45 Hz) 4.04 (s, 2H) 4.13 (t, 2H, J = 6.15 Hz) 7.27 (d, 1H, J = 7.55 Hz) 7.39 (t, 2H, J = 7.65 Hz) 7.54 (td, 1H, J = 1.4, 7.55 Hz) 8.13 (dd, 1H, J = 1.19, 7.74 Hz); 13C NMR: δ 27.33, 29.32, 33.19, 63.85, 127.56, 128.41, 131.88, 132.42, 133.33, 136.77, 171.32, 172.58.
3-(2-Bromoethoxy)-4-chloro-7-nitro-1H-isochromen-1-one (4a) (43% yield) Yellow crystals: mp 126–128°C (lit. [25] 120°C); 1H NMR: δ 3.67 (t, 2H, J = 6.16 Hz) 4.74 (t, 2H, J = 6.06 Hz) 7.86 (d, 1H, J = 8.94 Hz) 8.53 (dd, 1H, J = 2.39 Hz, 8.94 Hz) 9.03 (d, 1H, J = 2.38 Hz); 13C NMR: δ 27.72, 69.63, 90.86, 117.17, 123.81, 126.32, 129.82, 142.71, 145.47, 154.77, 157.06.
3-(3-Bromopropoxy)-4-chloro-7-nitro-1H-isochromen-1-one (4b) (76% yield) Pale yellow crystals: mp 131–134°C; 1H NMR: δ 2.37 (m, 2H) 3.59 (t, 2H, J = 6.26 Hz) 4.61 (t, 2H, J = 5.86 Hz) 7.81 (d, 1H, J = 8.94 Hz) 8.50 (dd, 1H, J = 2.09 Hz, 8.84 Hz) 8.99 (d, 1H, J = 1.79 Hz); 13C NMR: δ 28.49, 31.92, 68.56, 90.46, 116.93, 123.52, 126.22, 129.68, 142.77, 145.77, 155.34, 157.18.
3-(2-Bromoethoxy)-4-chloro-1H-isochromen-1-one (4c) (30% yield) Yellow solid: mp 81–82°C; 1H NMR: δ 3.65 (t, 2H, J = 6.35 Hz) 4.64 (t, 2H, J = 6.35 Hz) 7.41 (t, 1H, J = 7.15 Hz) 7.74 (m, 2H) 8.20 (d, 1H, J = 7.75 Hz); 13C NMR: δ 28.07, 69.37, 92.09, 117.53, 122.47, 126.55, 130.06, 135.62, 137.35, 152.08, 159.01.
3-(3-Bromopropoxy)-4-chloro-1H-isochromen-1-one (4d) (53% yield) Yellow crystals: mp 95–97°C; 1H NMR: δ 2.33 (m, 2H) 3.60 (t, 2H, J = 6.35 Hz) 4.51 (t, 2H, J = 5.76 Hz) 7.40 (t, 1H, J = 6.75 Hz) 7.72 (m, 2H) 8.19 (d, 1H, J = 7.94 Hz); 13C NMR: δ 28.88, 32.26, 68.28, 91.91, 117.58, 122.39, 126.43, 130.12, 135.62, 137.57, 152.74, 159.33.
3-(4-Bromobutoxy)-4-chloro-1H-isochromen-1-one (4e) A solution of 3e (0.75 g, 2.3 mmol) and phosphorus pentachloride (1.23 g, 5.9 mmol) was refluxed in benzene (50 mL) for fourteen hours. The orange solution was cooled, washed with water (2 × 25 mL), saturated sodium bicarbonate (2 × 15 mL), brine (1 × 25 mL), and dried over magnesium sulfate. Filtration and evaporation of the solvent gave a yellow oil. Trituration with hexane gave 0.55 g (70%) of compound 4e as white crystals: mp 75–77°C; 1H NMR: δ 1.98 (m, 2H) 2.06 (m, 2H) 3.48 (t, 2H, J = 6.25 Hz) 4.40 (t, 2H, J = 5.96 Hz) 7.38 (td, 1H, J = 1.59 Hz, 7.50 Hz) 7.70 (m, 2H) 8.17 (d, 1H, J = 7.55 Hz); 13C NMR: δ 27.33, 29.32, 33.12, 40.80, 63.81, 127.55, 128.41. 131.86, 132.38, 133.30, 136.76, 171.26, 172.29. Exact mass calcd for C13H12BrClO3: 329.9658, observed (M+H) 330.9734.
2- [2-(4-Chloro-1-oxo-1H-isochromen-3-yloxy)ethyl]isothiourea hydrobromide (5c) (64% yield) Yellow solid: mp 168–170°C (lit. [24] 167–169°C); 1H NMR: (DMSO-d6) δ 3.65 (t, 2H, J = 5.66 Hz) 4.58 (t, 2H, J = 5.67 Hz) 7.53 (t, 1H, J = 7.65 Hz) 7.69 (d, 1H, J = 8.14 Hz) 7.92 (t, 1H, J = 7.05 Hz) 8.13 (d, 1H, J = 7.75 Hz) 9.15 (br s, 4H); 13C NMR: δ 29.73, 68.11, 90.48, 117.18, 121.72, 126.70, 129.48, 135.99, 136.56, 152.18, 158.35, 169.11.
2- [3-(4-Chloro-1-oxo-1H-isochromen-3-yloxy)propyl]isothiourea hydrobromide (5d) (40% yield) Yellow solid: mp 159–163°C (lit. [24] 165–167°C);1H NMR: (DMSO-d6) 2.21 (m, 2H) 3.40 (t, 2H, J = 7.18 Hz) 4.55 (t, 2H, J = 6.11 Hz) 7.62 (td, 1H, J = 0.95, 7.60 Hz) 7.79 (d, 1H, J = 7.70 Hz) 8.02 (td, 1H, J = 1.23, 7.70 Hz) 8.23 (dd, 1H, J = 1.25, 7.50 Hz) 10.09 (br s, 4H); 13C NMR: δ 26.66, 28.53, 68.68, 90.43, 117.13, 121.66, 126.59, 129.47, 135.96, 136.66, 152.63, 158.53, 169.36.
2- [4-(4-Chloro-1-oxo-1H-isochromen-3-yloxy)butyl]isothiourea hydrobromide (5e) A solution of 4e (0.25 g, 0.75 mmol) and thiourea (0.075 g, 0.98 mmol) in dry tetrahydrofuran (25 mL) was refluxed for forty-eight hours. The resulting pale yellow solid was filtered and washed with hot tetrahydrofuran (3 × 10 mL) to give 0.2 g (65%) of compound 5h as a pale yellow solid: mp 160–162°C; 1H NMR: (DMSO-d6) δ 1.82 (br s, 4H) 3.24 (t, 2H, J = 6.45 Hz) 4.39 (t, 2H, J = 5.75 Hz) 7.50 (t, 1H, J = 7.45 Hz) 7.65 (d, 1H, 7.45 Hz) 7.89 (t, 1H, J = 7.25 Hz) 8.09 (d, 1H, J = 7.75 Hz) 9.07 (br s, 4H); 13C NMR: δ 24.92, 27.35, 29.59, 69.96, 90.20, 116.95, 121.56, 126.48, 129.45, 135.94, 136.73, 152.80, 158.59, 169.56.
7-Amino-3-(2-bromoethoxy)-4-chloro-1H-isochromen-1-one (6a) Compound 4a (2.2 g, 6.3 mmol) was reduced on a Parr apparatus with hydrogen over 10% palladium on charcoal (50 mg) in ethanol (25 mL) until the reaction stopped absorbing hydrogen. The solution was filtered through celite and the filtrate was evaporated. The resulting crude solid was chromatographed (dichloromethane) to give 1.55 g (78%) of compound 6a as yellow crystals: mp 134–136°C, (lit. [26] 134–137°C); 1H NMR: δ 3.63 (t, 2H, J = 6.46 Hz) 3.95 (br s, 2H) 4.56 (t, 2H, J = 6.36 Hz) 7.10 (dd, 1H, J = 2.58 Hz, 8.54 Hz) 7.43 (d, 1H, J = 2.58 Hz) 7.54 (d, 1H, J = 8.74 Hz); 13C NMR: δ 28.18, 69.87, 93.59, 113.09, 119.21, 123.54, 124.04, 128.24, 145.63, 149.90, 159.47.
7-Amino-3-(3-bromopropoxy)-4-chloro-1H-isochromen-1-one (6b) (75% yield) Yellow crystals: mp 106–107°C (lit. [24,26] 98–100°C); 1H NMR (DMSO-d6) δ 2.29 (m, 2H) 3.60 (t, 2H, J = 6.36 Hz) 4.42 (t, 2H, J = 5.76 Hz) 7.09 (dd, 1H, J = 2.09 Hz, 8.44 Hz) 7.42 (d, 1H, J = 1.99 Hz) 7.51 (d, 1H, J = 8.54 Hz); 13C NMR: δ 29.11, 32.36, 68.71, 93.35, 113.11, 119.17, 123.58, 123.91, 128.42, 145.56, 150.49, 159.74.
2- [2-(7-amino-4-chloro-1-oxo-1H-isochromen-3-yloxy)ethyl]isothiourea hydrobromide (7a) (40% yield) Pale yellow solid: mp d 150°C; 1H NMR: (DMSO-d6) δ 3.59 (br s, 2H) 4.47 (br s, 2H) 5.81 (br s, 2H) 7.21 (d, 1H, J = 8.94 Hz) 7.26 (s, 1H) 7.44 (br d, 1H) 9.11 (br s, 4H);13C NMR: δ 29.73, 68.11, 90.48, 117.18, 121.72, 126.70, 129.48, 135.99, 136.56, 152.18, 158.35, 169.11.
2- [3-(7-Amino-4-chloro-1-oxo-1H-isochromen-3-yloxy)propyl]isothiourea hydrobromide (7b) A solution of 6b (0.25 g, 0.75 mmol), thiourea (0.071 g, 0.94 mmol) and tetrahydrofuran (25 mL) was refluxed for forty-eight hours to give a yellow precipitate. The precipitate was filtered and washed with hot tetrahydrofuran (3 × 25 mL), and recrystallized from methanol/ether to give 0.06 g (20%) of compound 7b as a pale yellow solid: mp 173°C; (lit. [24] 160–162°C); 1H NMR: (DMSO-d6) δ 2.07 (br s, 2H) 3.30 (br s, 2H) 4.32 (br s, 2H) 7.16 (d, 1H, J = 7.94 Hz) 7.26 (s, 1H) 7.41 (d, 1H, J = 7.95 Hz) 9.05 (br s, 4H);13C NMR: δ 26.73, 28.35, 69.26, 92.87, 110.88, 118.81, 122.84, 123.11, 124.66, 148.23, 149.41, 159.06, 169.37.
N- [3-(2-Bromoethoxy)-4-chloro-1-oxo-1H-isochromen-7-yl]benzamide (8a) To a solution of 6a (0.75 g, 2.4 mmol) in dry tetrahydrofuran (20 mL) was added benzoyl chloride (0.35 mL, 2.8 mmol) and triethylamine (0.33 mL, 2.3 mmol). The solution was stirred at room temperature for fourteen hours after which time the triethylamine hydrochloride was filtered off and washed with hot tetrahydrofuran (2 × 10 mL). The filtrate was evaporated to give a pale yellow solid that was recrystallized from tetrahydrofuran/hexane to afford 0.60 g (75%) of compound 8a as a pale yellow solid: mp 214–216°C; 1H NMR: (DMSO-d6) δ 3.83 (t, 2H, J = 5.46 Hz) 4.65 (t, 2H, J = 5.46 Hz) 7.56 (m, 3H) 7.71 (d, 1H, J = 8.93 Hz) 7.99 (d, 2H, J = 8.15) 8.29 (dd, 1H, J = 2.39 Hz, 8.74 Hz) 8.68 (d, 1H, J = 2.18 Hz) 10.63 (s, 1H); 13C NMR: δ 30.49, 69.98, 91.08, 117.64, 119.23, 122.38, 127.52, 127.86, 128.20, 131.62, 131.90, 134.12, 137.90, 151.40, 158.35, 165.43. Exact mass calcd for C18H13BrClNO4: 420.9716, observed (M+H) 421.9788.
N- [3-(3-Bromopropoxy)-4-chloro-1-oxo-1H-isochromen-7-yl]benzamide (8b) (82% yield) Pale yellow solid: mp 193–194°C; 1H NMR: (DMSO-d6) δ 2.28 (m, 2H) 3.66 (t, 2H, J = 6.56 Hz) 4.44 (t, 2H, J = 5.96 Hz) 7.55 (m, 3H) 7.68 (d, 1H, J = 8.74 Hz) 7.98 (d, 6.56 Hz) 8.25 (dd, 1H, J = 1.99 Hz, 8.74 Hz) 8.66 (d, 1H, J = 1.98 Hz) 10.62 (s, 1H); 13C NMR: δ 30.74, 32.09, 69.13, 91.36, 118.01, 119.74, 122.80, 127.88, 128.43, 128.67, 133.10, 132.46, 134.47, 138.15, 152.20, 158.95, 165.96. Exact mass calcd for C19H15BrClNO4: 434.9873, observed (M+H) 435.9959.
2- [2-(7-Benzamido-4-chloro-1-oxo-1H-isochromen-3-yloxy)ethyl]isothiourea hydrobromide (9a) A solution of 8a (0.3 g, 0.71 mmol) and thiourea (0.06 g, 0.78 mmol) in dry tetrahydrofuran (25 mL) was refluxed for twelve hours. The resulting pale yellow solids were filtered and washed with hot tetrahydrofuran (3 × 10 mL) to give 0.06 g (17%) of compound 9a as a pale yellow solid: mp 173–175°C. Evaporation of the filtrate afforded 8a (0.2 g). Yield based on recovered starting material is 51%; 1H NMR: (DMSO-d6) δ 3.68 (br s, 2H) 4.60 (br s, 2H) 7.59 (m, 3H) 7.74 (d, 1H, J = 8.54 Hz) 8.03 (d, 2H, J = 6.75 Hz) 8.32 (d, 1H, J = 8.14 Hz) 8.73 (s, 1H) 9.18 (br s, 4H) 10.69 (s, 1H); 13C NMR: δ 29.85, 68.32, 91.00, 117.72, 119.47, 122.53, 127.60, 128.22, 128.33, 131.79, 132.06, 134.12, 137.96, 151.44, 158.43, 165.63, 169.18.
2- [3-(7-Benzamido-4-chloro-1-oxo-1H-isochromen-3-yloxy]propyl)isothiourea hydrobromide (9b) (25% yield) Pale yellow solid: mp 203–204°C;1H NMR: (DMSO-d6) δ 2.12 (m, 2H) 3.31 (br s, 2H) 4.44 (t, 2H, J = 5.66 Hz) 7.56 (m, 3H) 7.71 (d, 1H, J = 8.74 Hz) 8.00 (d, 2H, J = 6.95 Hz) 8.28 (d, 1H, J = 8.54 Hz) 8.70 (s, 1H) 9.07 (br s, 4H) 10.65 (s, 1H); 13C NMR: δ 27.21, 28.79, 69.37, 91.38, 118.13, 119.85, 122.92, 128.04, 128.60, 128.77, 132.21, 132.57, 134.61, 138.33, 152.36, 159.05, 166.02, 169.83. Exact mass calcd for C20H18ClN3O4S: 431.0707, observed (M+H) 432.0780.
3-(2-Bromoethoxy)-7-nitro-1H-isochromen-1-one (10a) A solution of 3a (1.5 g, 4.5 mmol) and trifluoroacetic anhydride (0.64 mL, 5.0 mmol) in dichloromethane (50 mL) was stirred at room temperature for sixteen hours. The solution was evaporated, washed with water (1 × 25 mL), saturated sodium bicarbonate solution (1 × 25 mL), dried over magnesium sulfate, and evaporated to afford 1.23 g (87%) of a crude yellow solid. Recrystallization from isopropanol gave 0.66 g (47%) of compound 10a as yellow crystals: mp 95–97°C; 1H NMR: δ 3.65 (t, 2H, J = 5.96 Hz) 4.54 (t, 2H, J = 5.86 Hz) 5.77 (s, 1H) 7.42 (d, 1H, J = 8.74 Hz) 8.38 (d, 1H, J = 8.54 Hz) 8.96 (s, 1H); 13C NMR: δ 27.51, 68.87, 81.12, 117.12, 125.70, 126.17, 129.28, 144.97, 145.11, 158.81, 160.02.
3-(3-Bromopropoxy)-7-nitro-1H-isochromen-1-one (10b) (55% yield) Yellow crystals: mp 117–118°C; 1H NMR: δ 2.37 (m, 2H) 3.59 (t, 2H, J = 6.26 Hz) 4.38 (t, 2H, J = 5.86 Hz) 5.72 (s, 1H) 7.43 (d, 1H, J = 8.74 Hz) 8.41 (dd, 1H, J = 2.48 Hz, 8.84 Hz) 9.02 (d, 1H, J = 2.19 Hz);13C NMR: δ 28.80, 31.46, 67.29, 80.02, 117.02, 125.55, 126.07, 129.15, 144.79, 145.21, 158.94, 160.89.
3-(3-Bromopropoxy)-4-trifluoroacetyl-1H-isochromen-1-one (10c) A solution of 3d (0.60 g, 2.0 mmol) and trifluoroacetic anhydride (0.38 mL, 2.7 mmol) in dichloromethane (25 mL) was stirred at room temperature for fourteen hours. The solution was evaporated and the oil was chromatographed (chloroform) to afford 0.45 g (59%) of compound 10c as white crystals: mp 116–117°C; 1H NMR: δ 2.38 (m, 2H) 3.54 (t, 2H, J = 6.26 Hz) 4.70 (t, 2H, J = 5.96 Hz) 7.42 (m, 1H) 7.74 (m, 1H) 8.10 (d, 1H, J = 8.34 Hz) 8.22 (d, 1H, J = 7.95 Hz); 13C NMR: δ 28.24, 31.37, 68.94, 90.90, 115.81, 116.01, 123.40, 126.75, 130.26, 135.87, 136.27, 157.73, 162.08, 179.97. Anal. Calcd. for C14H10BrF3O4: C, 44.35; H, 2.66. Found: C, 44.25; H, 2.99.
7-Amino-3-(2-bromoethoxy)-1H-isochromen-1-one (11) A solution of 10a (1.5 g, 4.7 mmol) in methanol/ethyl acetate (1:1, 25 mL) was reduced on a Parr apparatus with hydrogen and 10% palladium on charcoal. After the reaction stopped absorbing hydrogen it was filtered through celite and the celite was washed with methylene chloride (3 × 50 mL). The filtrate was evaporated to near dryness keeping the temperature below 40°C. The semisolid was recrystallized from methylene chloride/methanol to afford 1.10 g (81%) of compound 11 as yellow crystals: mp > 280°C; 1H NMR: (DMSO-d6) 3.79 (t, 2H, J = 5.17 Hz) 4.36 (t, 2H, J = 4.97 Hz) 5.50 (s, 2H) 5.82 (s, 1H) 7.03 (d, 1H, J = 8.54 Hz) 7.19 (m, 2H); 13C NMR: δ 30.30, 68.71, 80.35, 110.20, 117.91, 123.17, 125.84, 128.03, 147.16, 154.71, 160.45.
Authors' contributions
JJH conducted the synthetic chemistry with the assistance of LMD. LAH and TAV conducted the kinetic studies. DLVJ and LDM conceived the study and wrote the manuscript with the assistance of JJH.
Acknowledgments
Acknowledgements
This work was supported by a grant from the National Institutes of Health, HL68598
Contributor Information
Justin J Heynekamp, Email: juster@unm.edu.
Lucy A Hunsaker, Email: lhunsaker@salud.unm.edu.
Thomas A Vander Jagt, Email: tavanderjagt@salud.unm.edu.
Lorraine M Deck, Email: ldeck@unm.edu.
David L Vander Jagt, Email: dlvanderjagt@salud.unm.edu.
References
- Kontogiorgis C, Papaioannou P, Hadjipavlou-Litina D. Matrix metalloproteinase inhibitors: a review on pharmacophore mapping and (Q)SARs results. Curr Med Chem. 2005;12:339–355. doi: 10.2174/0929867053363243. [DOI] [PubMed] [Google Scholar]
- Lee M, Fridman R, Mobashery S. Extracellular proteases as targets for treatment of cancer metastases. Chem Soc Rev. 2004;33:401–409. doi: 10.1039/b209224g. [DOI] [PubMed] [Google Scholar]
- McIntyre J, Matrisian L. Molecular imaging of proteolytic activity in cancer. J Cell Biochem. 2003;90:1087–1097. doi: 10.1002/jcb.10713. [DOI] [PubMed] [Google Scholar]
- Duffy M. The urokinase plasminogen activator system: role in malignancy. Curr Pharm Des. 2004;10:39–49. doi: 10.2174/1381612043453559. [DOI] [PubMed] [Google Scholar]
- Podgorski I, Sloane B. Cathepsin B and its role(s) in cancer progression. Biochem Soc Symp. 2003;70:263–276. doi: 10.1042/bss0700263. [DOI] [PubMed] [Google Scholar]
- Vihinen P, Kahari V. Matrix metalloproteinases in cancer: prognostic markers and therapeutic targets. Int J Cancer. 2002;99:157–166. doi: 10.1002/ijc.10329. [DOI] [PubMed] [Google Scholar]
- Wang Y. The role and regulation of urokinase-type plasminogen activator receptor gene expression in cancer invasion and matastasis. Med Res Rev. 2001;21:146–170. doi: 10.1002/1098-1128(200103)21:2<146::AID-MED1004>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Romer J, Neilsen B, Ploug M. The urokinase receptor as a potential target in cancer therapy. Curr Pharm Des. 2004;10:2359–2376. doi: 10.2174/1381612043383962. [DOI] [PubMed] [Google Scholar]
- Jo M, Thomas K, Marozkina N, Amin T, Silva C, Parsons S, Gonias S. Dynamic assembly of the urokinase-type plasminogen activator signaling receptor complex determines the mitogenic activity of urokinase-type plasminogen activator. J Biol Chem. 2005;280:17449–17457. doi: 10.1074/jbc.M413141200. [DOI] [PubMed] [Google Scholar]
- Steinmetzer T. Synthetic urokinase inhibitors as potential antitumor drugs. IDrugs. 2003;6:138–146. [PubMed] [Google Scholar]
- Rockway T, Giranda V. Inhibitors of the proteolytic activity of urokinase type plasminogen activator. Curr Pharm Des. 2003;9:1483–1498. doi: 10.2174/1381612033454649. [DOI] [PubMed] [Google Scholar]
- Renatus M, Bode W, Huber R, Sturzebecher J, Stubbs M. Structural and functional analyses of benzamidine-based inhibitors in complex with trypsin: implications for the inhibition of factor Xa, tPA, and urokinase. J Med Chem. 1998;41:5445–5456. doi: 10.1021/jm981068g. [DOI] [PubMed] [Google Scholar]
- Wendt M, Rockway T, Geyer A, McClellan W, Weitzberg M, Zhao X, Mantei R, Nienaber V, Stewart K, Klinghofer V, et al. Identification of novel binding interactions in the development of potent, selective 2-naphthamidine inhibitors of urokinase. Synthesis, structural analysis, and SAR of N-phenyl amide 6-substitution. J Med Chem. 2004;47:303–324. doi: 10.1021/jm0300072. [DOI] [PubMed] [Google Scholar]
- Mackman R, Katz B, Breitenbucher J, Hui H, Verner E, Luong C, Liu L, Sprengeler P. Exploiting subsite S1 of trypsin-like serine proteases for selectivity: potent and selective inhibitors of urokinase-type plasminogen activator. J Med Chem. 2001;44:3856–3871. doi: 10.1021/jm010244+. [DOI] [PubMed] [Google Scholar]
- Sperl S, Jacob U, Arroyo de Prada N, Sturzebecher J, Wilhelm O, Bode W, Magdolen V, Huber R, Moroder L. (4-aminomethyl)phenylguanidine derivatives as nonpeptidic highly selective inhibitors of human urokinase. Proc Natl Acad Sci USA. 2000;97:5113–5118. doi: 10.1073/pnas.97.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber C, Dickinson R, Fish P. Selective urokinase-type plasminogen activator (uPA) inhibitors. Part 3: 1-isoquinolinylguanidines. Bioorg Med Chem Lett. 2004;14:3227–3230. doi: 10.1016/j.bmcl.2004.03.094. [DOI] [PubMed] [Google Scholar]
- Kam C, Kerrigan J, Plaskon R, Duffy E, Lollar P, Suddath F, Powers J. Mechanism-based ioscoumarin inhibitors for blood coagulation serine proteases. Effect of the 7-substituent in 7-imino-4-chloro-3-(isothioureidoalkoxy)isocoumarins on inhibitory and anticoagulant potency. J Med Chem. 1994;37:1298–1306. doi: 10.1021/jm00035a009. [DOI] [PubMed] [Google Scholar]
- Bryson H, Bunning R, Feltell R, Kam C, Kerigan J, Powers J, Buttle D. A serine proteinase inactivator inhibits chondrocyte-mediated cartilege proteoglycan breakdown occurring in response to proinflammatory cytokines. Arch Biochem Biophys. 1998;355:15–25. doi: 10.1006/abbi.1998.0696. [DOI] [PubMed] [Google Scholar]
- Ungnade H, Nightingale D, French H. The synthesis of 7-methoxy-1-isoquinolone. J Org Chem. 1945;10:533. doi: 10.1021/jo01182a007. [DOI] [PubMed] [Google Scholar]
- Devi A, Rajaram S. An efficient and regiospecific esterification of dioic acids using PTSA. Indian J Chem. 2000;39:294–296. [Google Scholar]
- Morris G, Goodsell D, Halliday R, Huey R, Hart W, RK B, Olson A. Automated docking using a Lamarckian genetic alogithm and an empirical binding free energy function. J Comp Chem. 1998;19:1639–1662. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [DOI] [Google Scholar]
- Morris G, Goodsell D, Huey R, Olson A. Distributed automated docking of flexible ligands to proteins: parallel application of AutoDock 2.4. J Comput Aided Mol Des. 1996;10:293–304. doi: 10.1007/BF00124499. [DOI] [PubMed] [Google Scholar]
- Kerrigan J, Oleksyszyn J, Kam C, Selzler J, Powers J. Mechanism-based isocoumarin inhibitors for human leukocyte elastase. Effect of the 7-amino substituent and 3-alkoxy group in 3-alkoxy-7-amino-4-chloroisocoumarins on inhibitory potency. J Med Chem. 1995;38:544–552. doi: 10.1021/jm00003a017. [DOI] [PubMed] [Google Scholar]
- Kam C, Fujikawa K, Powers J. Mechanism-based isocoumarin inhibitors for trypsin and blood coagulation serine proteases" new anticoagulants. Biochemistry. 1988;27:2547–2557. doi: 10.1021/bi00407a042. [DOI] [PubMed] [Google Scholar]
- Bihel F, Quelever G, Lelouard H, Petit A, Alves de Costa C, Pourquie O, Checler F, Thellend A, Pierre P, Karaus J. Synthesis of new 3-alkoxy-7-amino-4-isocoumarin derivatives as new beta-amyloid peptide production inhibitors and their activities on various classes of protease. Bioorg Med Chem. 2003;11:3141–3152. doi: 10.1016/S0968-0896(03)00235-9. [DOI] [PubMed] [Google Scholar]
- Powers J, Oleksyszyn J, Narasimhan S, Kam C. Reaction of porcine pancreatic elastase with 7-substituted 3-alkoxy-4-chloroisocoumarins: design of potent inhibitors using the crystal structure of the complex formed with 4-chloro-3-ethoxy-7-guanidinoisocoumarin. Biochemistry. 1990;29:3108–3118. doi: 10.1021/bi00464a030. [DOI] [PubMed] [Google Scholar]