

Abstract
Modeling of the tetrahedral intermediate within the active site of Escherichia coli aspartate transcarbamoylase revealed a specific interaction with the side chain of Gln137, an interaction not previously observed in the structure of the X-ray enzyme in the presence of N-phosphonacetyl-L-aspartate (PALA). Previous site-specific mutagenesis experiments showed that when Gln137 was replaced by alanine, the resulting mutant enzyme (Q137A) exhibited approximately 50-fold less activity than the wild-type enzyme, exhibited no homotropic cooperativity, and the binding of both carbamoyl phosphate and aspartate were extremely compromised. To elucidate the structural alterations in the mutant enzyme that might lead to such pronounced changes in kinetic and binding properties, the Q137A enzyme was studied by time-resolved small-angle X-ray scattering and its structure was determined in the presence of PALA to 2.7Å resolution. Time-resolved small-angle X-ray scattering established that the natural substrates, carbamoyl phosphate and L-aspartate, do not induce in the Q137A enzyme the same conformational changes as observed for the wild-type enzyme, although the scattering pattern of the Q137A and wild-type enzymes in the presence of PALA were identical. The overall structure of the Q137A enzyme is similar to that of the R-state structure of wild-type enzyme with PALA bound. However, there are differences in the manner by which the Q137A enzyme coordinates PALA, especially in the side chain positions of Arg105 and His134. The replacement of Gln137 by Ala also has a dramatic effect on the electrostatics of the active site. These data taken together suggest that the side chain of Gln137 in the wild-type enzyme is required for the binding of carbamoyl phosphate in the proper orientation so as to induce conformational changes required for the creation of the high-affinity aspartate binding site. The inability of carbamoyl phosphate to create the high-affinity binding site in the Q137A enzyme results in an enzyme locked in the low activity low affinity T state. These results emphasize the absolute requirement of the binding of carbamoyl phosphate for the creation of the high-affinity aspartate binding site and for inducing the homotropic cooperativity in aspartate transcarbamoylase.
Keywords: allosteric enzyme, polar contacts, electrostatics, Poisson-Boltzmann equation, small-angle X-ray scattering, domain closure, allosteric transition, intersubunit interactions
Introduction
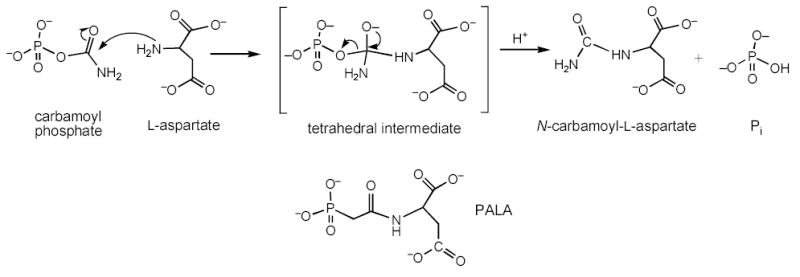
Escherichia coli aspartate transcarbamoylase (EC 2.1.3.2) catalyzes the committed step of the pyrimidine biosynthesis pathway, the formation of N-carbamoyl-l-aspartate from CP and L-aspartate via a tetrahedral intermediate (see Scheme I).
Scheme I.
The E. coli enzyme is composed of two trimeric catalytic subunits (Mr 34,000/chain) and three dimeric regulatory subunits (Mr 17,000/chain).1 The enzyme is inhibited allosterically by CTP,2 and UTP in the presence of CTP,3 and it is activated allosterically by ATP.2 Consequently, when the concentration of pyrimidines is high, the activity of the enzyme is reduced and when the concentration of purines is high the activity of the enzyme is enhanced. Regulation is achieved by a switch in the structure of the enzyme from an “off” or T state to an “on” or R state. The three-dimensional structures of the aspartate transcarbamoylase in both the T and R states are available and provide detailed structural information concerning the two extreme states of this enzyme.4–6 The T to R transition involves an expansion of the enzyme by 11Å along the 3-fold axis, with rotations of the catalytic subunits about the 3-fold axis, and rotations of the regulatory subunits about their respective 2-fold axes. The change in quaternary structure is accompanied by alterations in the tertiary structure, the most significant of which involves movements of domains and reorientations of loops,7–9 resulting in the formation of the R state and the catalytically competent active sites.
The binding of aspartate to the enzyme-CP complex induces the transition from the T to R state. This transition can also be induced by succinate, an analog of aspartate, binding to the enzyme-CP complex or by the binding of the bisubstrate analog N-phosphonacetyl-L-aspartate (PALA) to the unligated enzyme.10
A high resolution structure of the enzyme in the presence of PALA6 has defined the active site and many of the interactions that specifically stabilize the binding of PALA. Using the structure of the enzyme-PALA complex, it was possible to model the structure of the tetrahedral intermediate into the active site.6 This modeling required essentially no alterations in the side change positions observed in the PALA structure. Close examination of the model of the enzyme-tetrahedral intermediate revealed three additional interactions toward the tetrahedral intermediate, not observed in the PALA structure. One of these entailed the side chain of Gln137 which interacted with the amino group, derived from CP, connected to the tetrahedral carbon of the intermediate (see Figure 1).
Figure 1.
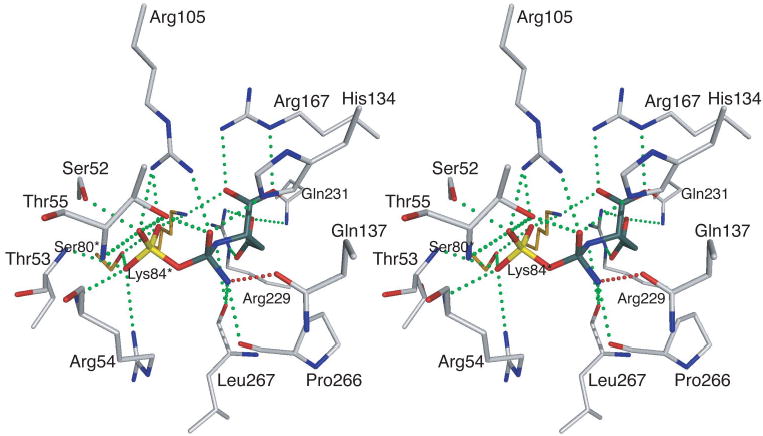
Stereoview of the active site of aspartate transcarbamoylase based upon the structure of the enzyme in the presence of PALA.6 In place of PALA the tetrahedral intermediate has been modeled into the active site.6 All the interactions observed between the enzyme and PALA are also observed between the enzyme and the tetrahedral intermediate. Three new interactions are observed between the enzyme and the tetrahedral intermediate including an interaction, shown in red, between the Gln137 and the amino group on the tetrahedral carbon derived from CP. The asterisk after the residue number indicates that the side chain is donated into the active site from the adjacent chain.
The interaction between Gln137 and CP was observed in the 2.6Å structure of the ternary complex of the enzyme with CP and succinate, an analog of aspartate.11. In addition a similar interaction was observed in the 2.8Å structure of the ternary complex of the enzyme with phosphonacetamide, an analog of CP, and malonate, an analog of aspartate.4
To determine the structural and functional effects of specific residues at the CP binding site of aspartate transcarbamoylase, a set of mutations was constructed, each eliminating an interaction between the enzyme and CP. Gln137 was selected for replacement to Ala/Asn by site-directed mutagenesis. The Q137N enzyme exhibited only small changes in activity, whereas the Q137A enzyme was approximately 50-fold less active than the wild-type enzyme.12 In addition, the binding of CP was so compromised in the Q137A enzyme, that extremely high concentrations of substrates were required to attain maximal activity such that only estimates of [Asp]0.5 and [CP]0.5 could be obtained.12 In the case of the Q137A enzyme, the lack of cooperativity and the low affinity for substrates and low catalytic activity suggested that the binding of the substrates could not convert the enzyme into the R state. To understand the structural alterations in the enzyme that could lead to changes in kinetic and binding properties relative to wild-type enzyme, the Q137A enzyme was studied by time-resolved small-angle X-ray scattering and its crystal structure was determined in the presence of PALA.
Results
Three-dimensional structure of the PALA bound Q137A enzyme
The unit cell dimensions of the Q137A crystals in the P321 space group (a = b = 122.12Å, c = 155.93Å) are similar to the unit cell dimensions of the R state of the wild-type enzyme (a = b = 122.24Å, c = 156.36Å) in the same space group.6 For crystals of aspartate transcarbamoylase in this space group, the c-axis unit cell dimension is indicative of the quaternary state. The T-state of the unligated wild-type enzyme has a c-axis dimension of 142Å, while the a and b axes dimensions are approximately the same in the T and R states.5 The length of the c-axis of the Q137A crystals suggests that the mutant enzyme is in the R quaternary structure in the presence of PALA. Therefore, the initial model used for the refinement of the Q137A structure was the structure of the wild-type enzyme in the presence of PALA (PDB code: 1D09).6 The data were refined to a Rfactor and Rfree of 17.0% and 22.4 % respectively with 250 water molecules in the asymmetric unit.
In order to evaluate the conformational states of specific structures of aspartate transcarbamoylase, the vertical separation between the upper and lower catalytic subunits (vertical separation along the three-fold axis) can be compared.13 The vertical separation for the wild-type enzyme in the T and R states is 45.6Å and 56.6Å respectively. The difference between the R- and T-state vertical separations corresponds to the vertical expansion of the enzyme observed during in the T to R transition (56.6 – 45.6 = 11Å). For the Q137A structure, the vertical separation was 56.3 Å, which is 0.3 Å different from the vertical separation of the R state of the wild-type enzyme.
Analysis of active site of the Q137A structure
The overall structure of the Q137A enzyme is similar to that of the wild-type enzyme with PALA bound (PDB code: 1D09). The RMS deviation of the backbone between the two structures was only 0.23Å. Shown in Figure 2 is the active site of the Q137A structure showing PALA bound. Overlaid onto this are the active site residues and PALA as determined for the wild-type enzyme.6 The occupancy of PALA is 100% with an average B-factor of 23 Å2.
Figure 2.
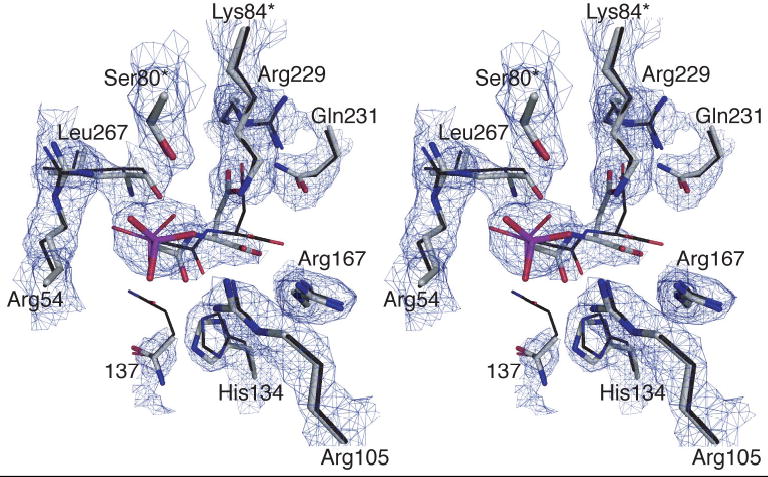
Stereoview of the active site of the Q137A enzyme. The 2Fo-Fc electron density map was contoured at 2.0σ. Both the mutant residue 137 and PALA have been omitted from the map calculation. Overlaid onto the electron density map is the refined positions of the residues in the Q137A structure shown as thick lines. Also overlaid is the structure of the wild-type enzyme in the presence of PALA (PDB code: 1D09) shown with thin lines. The active site is oriented so that the carbamoyl phosphate domain is on the left and aspartate domain is on the right. The asterisk after the residue number indicates that the side chain is donated into the active site from the adjacent chain.
As shown in Table 1, PALA has almost all the same interactions with the Q137A enzyme that it has with the wild-type enzyme. However, there are differences in the manner by which the Q137A enzyme coordinates PALA compared to the wild-type enzyme. The NE2 of His134 interacts with the β-carboxylate of PALA in the wild-type structure, whereas there is no interaction between His134 and PALA in the Q137A structure. Ser80 loses one of its interactions with a phosphonate oxygen in the Q137A structure. However, the major change that occurs in the interaction of the Q137A enzyme with PALA involves the side chain of Arg105. Five new interactions are observed between the side chain of Arg105 and PALA in the Q137A structure that are not observed in the wild-type structure with PALA, and two interactions involving Arg105 with PALA in the wild-type structure are not observed in the Q137A structure (see Table 1).
Table 1.
Interactions to PALA in the active site of the wild-type and the Q137A enzymes
| Residue | Atom | PALA atoma | C1 distanceb (Å) Wild-typec | C6 distanceb (Å) Wild-typec | C1 distanceb (Å) Q137A | C6 distanceb (Å) Q137A |
|---|---|---|---|---|---|---|
| Ser52 | OG | O3P | 2.47 | 2.46 | 2.71 | 2.80 |
| Thr53 | N | O2P | 2.88 | 3.01 | 3.22 | 3.08 |
| Arg54 | N | O2P | 3.14 | 2.75 | 2.96 | 2.79 |
| Arg54 | NE | O2P | --- | 3.06 | --- | 2.84 |
| Arg54 | NH1 | O2P | 3.12 | --- | 3.10 | --- |
| Arg54 | NH2 | O2P | --- | 2.89 | --- | 3.09 |
| Thr55 | N | O3P | 2.82 | 2.76 | 3.09 | 2.55 |
| Thr55 | OG1 | O3P | 2.82 | 2.63 | 2.78 | 2.65 |
| OG1 | O1 | --- | --- | 3.34 | 3.23 | |
| Arg105 | NH1 | O1 | 2.64 | 2.98 | --- | --- |
| NH1 | O1P | --- | --- | 2.73 | 2.78 | |
| NH1 | O3P | --- | --- | 2.87 | --- | |
| NH1 | O3 | --- | --- | 2.91 | --- | |
| Arg105 | NH2 | O1P | 2.63 | 3.35 | --- | |
| NH2 | O3P | --- | --- | 2.95 | --- | |
| NH2 | O1 | --- | --- | 2.54 | 2.85 | |
| His134 | NE2 | O1 | 2.57 | 2.90 | --- | --- |
| Arg167 | NE | O2 | 2.73 | 2.75 | 2.60 | 2.70 |
| Arg167 | NH2 | O3 | 2.97 | 3.16 | 3.23 | 3.09 |
| Arg229 | NE | O5 | 2.71 | 2.86 | 2.87 | 2.82 |
| Arg229 | NH2 | O4 | 2.87 | 2.94 | 2.88 | 3.10 |
| Gln231 | NE2 | O5 | 3.12 | --- | 3.12 | 3.27 |
| Leu267 | O | N2 | 3.06 | 2.93 | 2.97 | 2.61 |
| Ser80d | OG | O1P | 3.30 | 2.97 | 2.92 | 3.12 |
| Ser80d | OG | O2P | 2.77 | 3.16 | --- | --- |
| Lys84d | NZ | O3 | 3.02 | 2.91 | 2.75 | 2.78 |
| Lys84d | NZ | 04 | 2.67 | 2.99 | 2.82 | 2.62 |
| Lys84d | NZ | O1P | 2.90 | 2.77 | 2.52 | 3.26 |
Scheme II.
C1 and C6 refer to the two independent catalytic chains in the asymmetric unit.
The data for the wild-type structure determined in the presence of PALA to 2.1Å resolution (PDB code: 1D09).
Ser80 and Lys84 are contributed into the active site from the adjacent catalytic chain.
The replacement of Gln137 by Ala also has a dramatic effect on the electrostatics of the active site. Shown in Figure 3 is a comparison of the electrostatic potential maps of the active site area for the wild-type6 and the Q137A enzymes. These calculations were performed first by removing PALA from the structure. There is a striking difference in the electrostatic environment of the active site between the Q137A and wild-type enzymes. The presence of an anionic “hole” over the carbamoyl phosphate domain in the Q137A structure helps to explain the compromised binding of CP to the Q137A enzyme.
Figure 3.
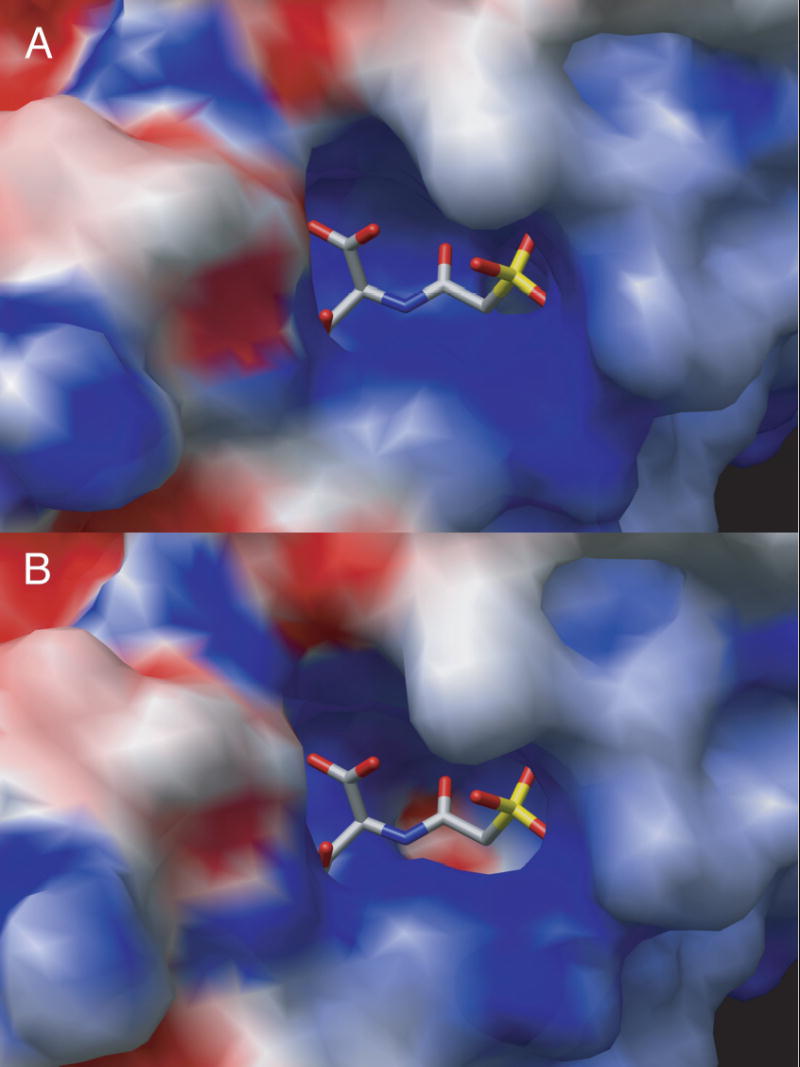
GRASP34 surface electrostatic map of overlay of the 1D09 and Q137A structures with catalytic chain C1. In (A) the surface and map are calculated from 1D09 and PALA in the active site is docked after the electrostatics are calculated. In (B) the surface and map are calculated from Q137A.
Comparison of the subunit interfaces of the Q137A and wild type PALA-ligated structures
Since the Q137A mutation occurs near the interface of the CP and aspartate domains, planar angles between these adjacent domains were calculated. This calculation was performed in order to compare the relative positions of the domains in the mutant structure compared to the values previously reported for wild-type enzyme in the R state (PDB code: 1D09).6 The angle between these two domains of the catalytic chain was 126.8° in C1# and 127.2° in C6, approximately 1° less than what was observed in wild-type R structure at 127.7° and 128.4° for both C1 and C6 chains (PDB code: 1D09).6 This decrease in the planar angle means that the domains are slightly more closed in the Q137A structure. There is an increase in the angle between the carbamoyl phosphate and zinc domains in both C1 and C6. In the wild-type R structure (PDB code; 1D09) the angles between the carbamoyl phosphate domain of the catalytic chain and adjacent zinc domain of the regulatory chain are 111.2° and 110.8° degrees for C1–R1 and C6–R6. In the Q137A structure, these values increase to 112.9° and 111.9°, indicating that these adjacent domains are more open. The angle between the R1-R6 allosteric domains for in the Q137A structure is 154.9°, 1° less than the wild-type R structure (155.8°) (PDB code; 1D09). These observations indicate that the Q137A mutation in the active site of the catalytic chains causes a subtle but important adjustment in the overall structure of the enzyme.
Small-Angle X-ray Scattering
The X-ray structure of the Q137A enzyme in the presence of PALA shows the enzyme to be in the R state. However, the original kinetic data on the Q137A enzyme12 suggested that the enzyme could not attain the R state. The enzyme exhibited no cooperativity, with extremely high [Asp]0.5 and [CP]0.5 values. Here we employed stopped flow X-ray scattering to determine the quaternary conformational changes of the Q137A enzyme induced either by the addition of PALA, or by the natural substrates CP and aspartate.
Shown in Figure 4 are the results of the stopped-flow X-ray scattering experiments with wild-type and the Q137A enzymes. For these experiments, the wild-type or the Q137A enzyme (~80 mg/ml) was mixed with an equal volume of 10 mM PALA at 5°C. Upon mixing the final concentration of enzyme and PALA was ~40 mg/ml and 5 mM respectively. The differential integrated intensity over the subsidiary scattering peak (h = 0.085 Å−1 to h = 0.152 Å−1) shows the kinetics of the quaternary structural change most clearly because of the large changes that occur in this region. In order for the curves in Figure 4A to start at zero, the integrated intensity of the enzyme in the absence of PALA was subtracted. The rate of change of the quaternary structure for both the wild-type and Q137A enzymes was virtually identical, as was the extent of the structural transition.
Figure 4.
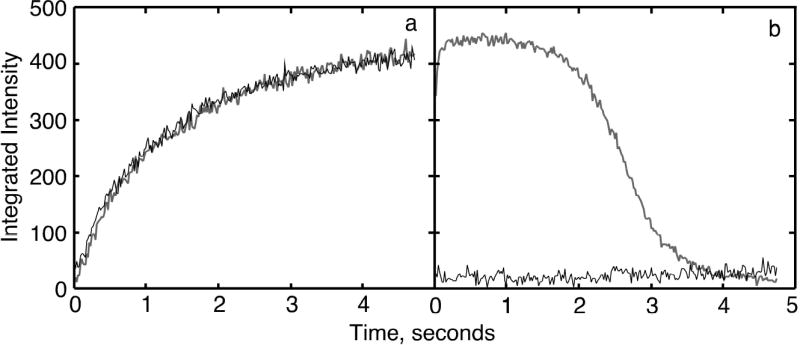
Time-resolved X-ray scattering of the wild-type and the Q137A enzymes. (A) Reaction of PALA (5 mM final) with the Q137A and wild type enzymes, and (B) reaction the Q137A and wild-type enzymes with CP (50 mM final) mixed with l-aspartate (50 mM final). The data were fit to a first order reaction.
Using time-resolved small-angle X-ray scattering it was also possible to determine whether the natural substrates, CP and L-aspartate could induce the same conformational changes in the Q137A enzyme as are observed for the wild-type. Shown in Figure 4B are the results observed upon addition of CP and L-aspartate to the wild-type and Q137A enzymes. In contrast to the behavior seen with the wild-type enzyme, the natural substrates do not cause a structural transition for the Q137A as they do for the wild-type enzyme. Repeating the experiment with higher concentrations of substrates did not induce a change in the Q137A structure to the R state.
The SAXS patterns of the wild-type and Q137A enzyme were also compared in the absence and presence of either substrates or PALA. For the wild-type enzyme, shown in Figure 5A, there is a significant change in the SAXS pattern upon addition of the PALA, corresponding to both a change in the peak position and an increase in relative intensity,14 as compared to a control with CP and D-aspartate. For the wild-type enzyme, the curve observed in the presence of PALA was essentially identical to one obtained in the presence of CP and L-aspartate averaged between 0.6 and 1.3 sec after mixing (see Fig. 3B).
Figure 5.
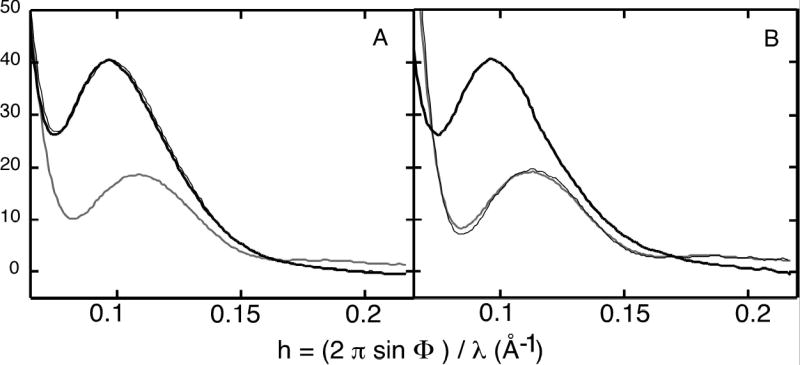
X-ray scattering patterns of the wild-type and Q137A enzymes. (A) The wild-type enzyme in the absence and presence of 5 mM PALA, and (B) the Q137A enzyme in the absence and presence of 5 mM PALA, and in the presence of 50 mM CP and L-aspartate. The curves were obtained by averaging 10 frames of the time-solved data starting at 1.68 sec.
The SAXS pattern for the Q137A enzyme (see Figure 5B) in the absence of ligands is almost identical to that observed for the unligated wild-type enzyme. However, in the presence of CP and L-aspartate, averaged between 0.6 and 1.3 sec after mixing, the quaternary structure of the enzyme has not changed when compared to a control using the same concentrations of CP and D-aspartate. The similarity of the structures of the wild-type and Q137A enzymes in the presence of PALA as determined by small-angle X-ray scattering agrees with the crystallographic data, which also show that in the presence of PALA the two enzymes have the same quaternary structure.
Discussion
The Q137A enzyme is in the R quaternary structure in the presence of PALA
The loss of cooperativity and low activity along with the extremely low affinity of the Q137A enzyme for both CP and aspartate,12 suggested that this mutation resulted in a enzyme that was stabilized in the T state. In order to understand the role of Gln-137 for substrate binding and catalysis, the X-ray structure of the Q137A enzyme was determined in the presence of PALA, the bisubstrate analog known to convert the wild-type enzyme to the R state.6,10 The X-ray structure of the Q137A enzyme in the presence of PALA, reported here, conclusively shows that the Q137A enzyme is in the R quaternary structure. The vertical separation between the upper and lower catalytic subunits is almost identical for the Q137A (56.3Å) and the wild-type R-state (56.6Å) structures. In the wild-type enzyme, Gln137 hydrogen bonds to the amino group of CP.11 No corresponding functional group is available in PALA, and no interaction between Glu137 and PALA is observed in the wild-type structure.6 For this reason, the replacement of Gln137 by Ala does not have a significant influence on the free energy of PALA binding, and thus PALA is able to induce the T to R transition in the Q137A enzyme.
The PALA bound active site of the Q137A and wild-type enzymes are similar
As shown in Figure 2, the active site of the Q137A mutant is quite similar to the wild-type PALA bound R-state (PDB code: 1D09).6 A comparison of the active site structures of the Q137A and wild-type enzymes does not provide an explanation for the extremely low activity and low affinity of the mutant enzyme. One possible explanation is that PALA is able to induce the T to R transition in the Q137A enzyme, while the combination of the natural substates, CP and aspartate, cannot. This exact situation has been observed previously with the T-state stabilized E50A aspartate transcarbamoylase. In this case, stopped-flow X-ray scattering revealed that PALA could convert this enzyme to the R structure, whereas the natural substrates can only convert the enzyme partially to the R structure.15 In the E50A enzyme, the R state is destabilized by the loss of interdomain bridging interactions caused by the E50A mutation. However, PALA was able to bridge the carbamoyl phosphate and aspartate domains and cause domain closure resulting in the R quaternary structure, whereas CP and aspartate could not.15
CP and aspartate cannot convert the Q137A enzyme to the R state
Time-resolved X-ray scattering experiments performed on the Q137A enzyme showed conclusively that the natural substrates were unable to induce the structural transition from the T to the R state (see Figure 4). No structural change was observed even in the presence of 100 mM concentrations of both CP and aspartate. The SAXS experiments indicate that in the absence of ligands the Q137A enzyme has a T-like quaternary structure, and in the presence of PALA the Q137A enzyme has a R-like quaternary structure. In the absence of ligands and in the presence of PALA, the SAXS curves for the Q137A enzyme are very similar to those observed for the wild-type enzyme (see Figure 5). In the presence of PALA, the crystal structure and the SAXS data for the Q137A are in agreement, both indicating a R structural state. A comparison of the time-evolution of the structural transition for the wild-type and the Q137A enzymes induced by PALA binding further reveals that both enzymes not only reach the same final structure, but also that the rate of the T to R transition for both enzymes is identical. These results indicate that the Q137A mutation has no influence on the ability of PALA to convert the enzyme from the T to the R state, proving that under the proper conditions the Q137A enzyme can attain the R structure.
Even though the combination of CP and aspartate (or the aspartate analog succinate) convert the wild-type enzyme into the R structure, no such conformational change is observed for the Q137A enzyme. For aspartate transcarbamoylase the binding of the two substrates is ordered with the binding of CP required for the binding of aspartate.16 CP alone promotes only a subtle change in the quaternary structure of the enzyme,17 however the binding of CP can be detected in the aromatic region by circular dichroism.18 These results taken together suggest that CP does not induce a quaternary conformational change from the T state, but does cause local structural changes in the active site that create the aspartate binding site.
The structure of the aspartate transcarbamoylase in the presence of phosphonoacetamide indicates a number of interactions between the enzyme and this non-reactive analog of CP, including an interaction between Gln137 and the amide.4 The severe effect that the Q137A mutation causes on catalytic activity and substrate affinity indicates that the Gln137 side chain must play a critical role in correctly orienting CP in the binding site. The properties of the Q137A enzyme are mimicked when the wild-type enzyme utilizes acetyl phosphate as substrate. The interaction between the side chain of Gln137 and the nitrogen of the carbamoyl group is lost when acetyl phosphate replaces CP as substrate. For the wild-type enzyme, the dissociation constant of acetyl phosphate is 35-fold higher than for CP.19 However, the affinity of the wild-type and the Q137A enzymes for acetyl phosphate are almost identical.12 Evidence from ultraviolet difference spectroscopy also shows that acetyl phosphate and succinate do not produce the same change as does CP and succinate suggesting that acetyl phosphate and aspartate do not cause the structural transition to the R state.20 We have verified by SAXS experiments that the combination of acetyl phosphate and succinate does not shift the structure of the wild-type enzyme from the T to the R state (data not shown). Thus, when the interaction between the enzyme and the carbamoyl nitrogen is removed either by the mutation of Gln137 or by the substitution of acetyl phosphate for CP, the enzyme is unable to undergo the allosteric transition to the R structure.
An intriguing question remains, how does this relatively benign substitution have such a large impact on the function of the enzyme? The low affinity of aspartate observed for the Q137A enzyme must be related to the inability of the bound CP to induce the necessary conformational changes required to form the high affinity aspartate binding site. The poor binding of CP alone is an insufficient explanation, since at high concentrations of CP, where the CP site should be saturated, aspartate still binds weakly. This suggests that the orientation of CP in its binding site is altered by the replacement of Gln137 by Ala in such a way that the aspartate binding site does not form correctly. Therefore, when CP binds to the Q137A enzyme, it does not induce the conformational changes required for its own tight binding, and therefore cannot preorganize the enzyme correctly for aspartate binding. The incorrect binding of CP prevents the enzyme from undergoing the T to R transition upon aspartate binding. The kinetic characteristics of the Q137A enzyme are what would be expected for an enzyme unable to transit to the R state.
The side chain of Arg105 coordinates PALA differently in the wild-type and the Q137A R-state structures. In the wild-type R-state structure the NH1 and NH2 of Arg105 coordinate O1 and O1P of PALA (Table 1). These interactions are very similar to the way Arg105 coordinates phosphonacetamide, a CP analogue.4 In contrast, new interactions are formed between Arg105 and PALA in the Q137A structure reported here. In the C1 catalytic chain, NH1 coordinates O1P, O3P, and O3 of PALA, while NH2 coordinates O3P and O1 (Table 1). This additional “bridging” of Arg105 in the Q137A structure is not analogous to what is seen in the T state phosphonoacetamide ligated structure.4 This difference in coordination suggests that the orientation of CP bound to the Q137A enzyme is different from that in the wild-type enzyme. This altered orientation has reduced affinity and prevents the proper formation of the high affinity aspartate binding site.
An explanation for the dramatic alteration in the binding of CP to the Q137A enzyme was suggested by studying the changes in the electrostatics of the active site induced by the mutation, which results in the formation of an anionic “hole” (see Figure 3). This anionic “hole” in the CP binding site is most likely due to shift in the positions of several residues near the site of the mutation, resulting in the elimination of some polar contacts in the CP binding site. Using DELHPI21 the protonation states of charged active site residues were calculated. In the Q137A enzyme, arginine side chains of both the CP and ASP domains as well as His134 were more deprotonated then in the wild-type active site. This can be explained in part by positional changes of the supporting networks of hydrophobic and ionic interactions disrupted by the Q137A mutation. Disruption of local interactions in the active site has a “ripple” or “domino” effect throughout the entire catalytic chain and outward to the interfaces between domains. The alteration in the planar angles between domains in the structure of the Q137A enzyme supports this hypothesis.
More specifically, an example of how alteration in the salt bridge network can change the charge gradient can be seen in the Q137A structure near the crucial interdomain bridging interactions that involve Glu50. A network of interactions between Glu50c1‡, Lys83c2, Asp129c1 and Arg167c1 is not present in wild-type R-state structure (PDB code: 1D09)6 mainly because Lys83c2 points toward the active site and Asp129c1 is “free” to draw charge from Arg167c1. In addition, Arg54 which exhibits the largest differences in the electrostatic calculations is more deprotonated in the Q137A enzyme because Glu86c2 is not “neutralized “ by Ser80c2 as observed in R-state structure (PDB code: 1D09).6
The loss of Gln137 disrupts a salt bridge network between Asp141-Gln137 and His134. This network is present in both the wild-type R-state structure (PDB code: 1D09) and the T-state structure with phosphonacetamide (PDB code: 3AT1). In the absence of Q137, His134 turns out of position such that it no longer interacts with the substrate or substrate analogs. The disruption in the Asp141-Gln137-His134 salt-bridge network may in part be responsible for the anionic hole in the active site observed in the Q137A enzyme. This alteration in the electrostatics of the active site prevents proper neutralization of substrates. This underscores the importance of Gln137 in the wild-type enzyme for the proper neutralization and positioning the substrates for the induction of the allosteric transition.
In summary, the Q137A enzyme can undergo the transition between the T and R structural states. However, this transition can be induced by the bisubstrate analog PALA, but not the natural substrates. Therefore, the low activity and low substrate affinity of the Q137A enzyme is not due to the inability of the enzyme to be transformed to the R state, but rather the inability of the natural substrates, CP and L-aspartate to induce the transition. The reorientation of several amino acid side chains in the active site observed in the structure of the Q137A enzyme in the presence of PALA, along with the reduced changes in ellipticity when CP binds to the Q137A enzyme,12 as measured by circular dichroism, suggests that CP does not bind in the same orientation to the Q137A enzyme as it does to the wild-type enzyme. Thus, the interactions between the side chain of Gln137 and CP in the active site of aspartate transcarbamoylase are not only critical for the formation of the high affinity binding of CP, but also for CP to bind in the proper orientation to create the aspartate binding site. When CP binds incorrectly, aspartate affinity and enzymatic activity are reduced dramatically, and homotropic cooperativity is lost because the mutant enzyme is unable to transit from the T to the R state. The Q137A enzyme demonstrates the novelty of an active site residue whose role appears to be directed towards the proper orientation of the substrate upon binding and in the stabilization of the tetrahedral intermediate of the reaction.
Materials and Methods
Materials
Agar, L-aspartate, ampicillin, carbamoyl phosphate, N-carbamoyl-L-aspartate, 2-mercaptoethanol, polyethyleneglycol 1450, potassium dihydrogen phosphate, Bis Tris, sodium ethylenediaminetetracetic acid, sodium acetate, sodium azide, uracil, and cholesterol myristate were obtained from Sigma. Q-Sepharose Fast Flow resin was obtained from Amersham Pharmacia Biosciences. Casamino acids, yeast extract and tryptone were obtained from Difco (Detroit, MI). Sodium dodecyl sulfate and Protein Assay Dye was purchased from Bio-Rad Laboratories. Enzyme grade ammonium sulfate, Tris, electrophoresis grade acrylamide and agarose were purchased from ICN Biomedicals (Costa Mesa, CA). Carbamoyl phosphate dilithium salt was purified before use by precipitation from 50% (v/v) ethanol and was stored dessicated at −20°C. Crystal cryo-mounting loops and microdialysis buttons were obtained from Hampton Research (Laguna Niguel, CA).
Methods
Overexpression and Purification of the Mutant Holoenzymes
The wild-type and Q137A enzymes were overexpressed utilizing E. coli strain EK110422 containing plasmids pEK15223 and pEK89,12 respectively and purified as previously described.22 After concentration, the purity of the enzymes was checked by SDS-PAGE 24 and nondenaturing PAGE.25,26
Determination of Enzyme Concentration
The concentration of pure wild-type enzyme was determined by absorbance measurements at 280 nm with an extinction coefficient of 0.59 cm2 • mg−1.27 The protein concentration of the Q137A enzyme was determined by the Bio-Rad version of Bradford’s dye-binding assay using the wild-type enzyme as standard.28
Solution X-ray Scattering
Solution x-ray scattering experiments were conducted at the Beam Line 4-2, the Stanford Synchrotron Radiation Laboratory, Menlo Park, CA. Synchrotron Radiation from a 20-pole 2 Tesla wiggler was focused by a bent cylinder mirror and monochromatized (x-ray wavelength 1.38Å) by a pair of synthetic Mo/B4C multilayer diffraction elements. A stopped-flow mixer injected 0.1 ml of enzyme solution and an equal volume of another solution, typically containing substrates or substrate analogs, into an observation cell via a mixing chamber within ~10 ms. The observation cell was kept at 5±0.5°C. A series of successive measurements of two-dimensional scattering data and corresponding beam intensities was synchronized with the completion of sample mixing. The scattering data were recorded by an image-intensified interline CCD x-ray detector system (Hamamatsu Photonics C4880-80-14A & V5445P), 29 located at ~85 cm from the observation cell. The beam intensities incident on sample were integrated during a series of CCD exposures by the EMBL data collection system.29 The detector pixels were converted to Q=2π·sinθ/λ, where 2θ is a scattering angle and λ is the x-ray wavelength, by recording the position of (100) reflection of a cholesterol myristate power sample placed at the sample position. Image distortion correction and radial integration of 2D data were performed using the program Fit2D.30 Intensity scaling, background subtraction and correction for non-uniformity of detector response were done by a program developed in-house.
Crystallization
Crystals of the Q137A enzyme were grown in 50 μL microdialysis buttons. Prior to crystallization 15 mg/ml of protein stock was dialyzed into 40 mM KH2PO4, 0.2 mM EDTA, 2 mM 2-mercaptoethanol pH 7.0. The microdialysis buttons were equilibrated against crystallization buffer containing 50 mM maleic acid, 3 mM sodium azide and 1 mM PALA at 20° C.7 Typically, crystals (~0.6 x 0.3 x 1.5 mm) grew in approximately one week.
Data Collection and Structure Refinement
Crystals of the Q137A enzyme were mounted in glass capillaries in preparation for data collection. The crystallographic data for the Q137A enzyme were collected at room temperature at the Crystallographic Facility in the Chemistry Department of Boston College on two multiwire area detectors from Area Detector Systems mounted on a Rigaku RU-200 rotating anode generator operated at 50 kV and 150 mA. A DEC-Alpha 3300 computer controlled the data collection. Diffraction data were processed using software provided by Area Detector Systems.
The initial model for the Q137A structure was derived from the coordinates of the wild-type enzyme crystallized in the presence of PALA (R-state structure) (PDB code: 1D09) to 2.1 Å 6 with all of the waters, PALA and Zn removed. The refinement was carried out using CNS.31 After initial rigid body, simulated annealing, minimization and B-factor refinement a round of manual rebuilding was performed in XtalView.32 Waters added in CNS, were checked and retained when they could be justified by hydrogen bonds and Fo–Fc electron density at or above the 2.5σ level. The details of data processing and refinement statistics for the Q137A structure are given in Table I. The model was checked for correctness using the program PROCHECK.33
Table 2.
Data collection and Refinement Summary of the Q137A Structure
| Data collection | |
| Space Group | P321 |
| dmin (Å) | 2.70 |
| Total Reflections | 100,265 |
| Unique reflections | 35,809 |
| Redundancy | 2.80 |
| Completeness (%) (All/outer shell) | 89.0/85.0 |
| Unit cell (Å) | |
| a = b | 122.116 |
| c | 155.928 |
| Angles | α = β = 90°, γ= 120° |
| Rmergea % (All/outer shell) | 8.75/32.9 |
| Refinement | |
| Resolution range (Å) | 7.00 - 2.70 |
| Sigma cutoff () | 0 |
| Reflections | 35,200 |
| Average (I/sigma) | 10.5 |
| Working R-factor | |
| Beginning | 0.280 |
| End | 0.170 |
| Free R-factor | |
| Beginning | 0.320 |
| End | 0.224 |
| Statistics (RMS deviations) | |
| Bonds (Å) | 0.007 |
| Angles (degrees) | 1.310 |
| Impropers (degrees) | 1.150 |
| Dihedrals (degrees) | 24.38 |
Acknowledgments
This work was supported by Grant GM26237 from the National Institutes of Health. Stanford Synchrotron Radiation Laboratory (SSRL) is operated by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Biology Resource is supported by the National Institutes of Health, National Center for Research Resources (P41RR01209), and by the Department of Energy, Office of Biological and Environmental Research. We would like to thank Dr. Neelima Alam and Ms. Kelly Dusinberre for preparing the Q137A enzyme used in the X-ray scattering experiments.
Footnotes
Abbreviations used: c, catalytic chain; r, regulatory chain; PALA, N-(phosphonoacetyl)-L-aspartate; [Asp]0.5, the aspartate concentration at one-half the maximal observed specific activity; [CP]0.5, the carbamoyl phosphate concentration at one-half the maximal observed specific activity; CP, carbamoyl phosphate; CP domain, carbamoyl phosphate binding domain of the catalytic chain; ASP domain, aspartate binding domain of the catalytic chain; AL domain, allosteric domain of the regulatory chain; ZN domain, zinc domain of the regulatory chain; SAXS, small-angle X-ray scattering; Q137A, the mutant aspartate transcarbamoylase with Gln137 replaced by Ala; 80’s loop, a loop in the catalytic chain of aspartate transcarbamoylase comprised of residues 73-88; 240’s loop, a loop in the catalytic chain of aspartate transcarbamoylase comprised of residues 230-245.
Within the E. coli holoenzyme, the catalytic chains of the upper catalytic trimer are numbered C1, C2 and C3, whereas the catalytic chains of the lower catalytic trimer are numbered C4, C5 and C6, with C4 under C1. The regulatory dimers contain chains R1-R6, R2-R4, and R3-R5. A regulatory chain is in direct contact with the catalytic chain, carrying the same number. In the crystal the asymmetric unit is composed of one catalytic chain from the upper trimer, the regulatory dimer connected to it and one catalytic chain from the lower catalytic trimer (e.g. C1-R1-R6-C6). These four polypeptide chains are labeled A, B, D, C respectively in the coordinate data file.
To distinguish amino acids in the catalytic and regulatory chains of aspartate transcarbamoylase a c (catalytic) or r (regulatory) is appended to the residue number (e.g. Asp236c and Lys143r). To clarify a particular catalytic or regulatory chain, the chain number is also appended to the residue number (e.g. Asp236c4 and Lys143r1).
References
- 1.Lipscomb, W. N. (1992). Activity and Allosteric Regulation in Aspartate Transcarbamoylase. In Proc. Robert A. Welch Found. Conf. Chem. Res., 36th (Regulation of Proteins by Ligands), pp. 103–143. The Robert A. Welch Foundation, Houston.
- 2.Gerhart JC, Pardee AB. Enzymology of control by feedback inhibition. J Biol Chem. 1962;237:891–896. [PubMed] [Google Scholar]
- 3.Wild JR, Loughrey-Chen SJ, Corder TS. In the presence of CTP, UTP becomes an allosteric inhibitor of aspartate transcarbamylase. Proc Natl Acad Sci USA. 1989;86:46–50. doi: 10.1073/pnas.86.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gouaux JE, Lipscomb WN. Crystal structures of phosphonoacetamide ligated T and phosphonoacetamide and malonate ligated R states of aspartate carbamoyltransferase at 2.8 Å resolution and neutral pH. Biochemistry. 1990;29:389–402. doi: 10.1021/bi00454a013. [DOI] [PubMed] [Google Scholar]
- 5.Stevens RC, Gouaux JE, Lipscomb WN. Structural consequences of effector binding to the T state of aspartate carbamoyltransferase: Crystal structures of the unligated and ATP- and CTP-complexed enzymes at 2.6 Å Resolution. Biochemistry. 1990;29:7691–7701. doi: 10.1021/bi00485a019. [DOI] [PubMed] [Google Scholar]
- 6.Jin L, Stec B, Lipscomb WN, Kantrowitz ER. Insights into the mechanism of catalysis and heterotropic regulation of E. coli aspartate transcarbamoylase based upon a structure of enzyme complexed with the bisubstrate analog N-phosphonacetyl-L-aspartate at 2.1 Å. Proteins: Struct Funct Genet. 1999;37:729–742. [PubMed] [Google Scholar]
- 7.Ke HM, Lipscomb WN, Cho Y, Honzatko RB. Complex of N-phosphonacetyl-L-aspartate with aspartate carbamoyltransferase: X-ray refinement, analysis of conformational changes and catalytic and allosteric mechanisms. J Mol Biol. 1988;204:725–747. doi: 10.1016/0022-2836(88)90365-8. [DOI] [PubMed] [Google Scholar]
- 8.Kantrowitz ER, Lipscomb WN. Escherichia coli aspartate transcarbamylase: The relations between structure and function. Science. 1988;241:669–674. doi: 10.1126/science.3041592. [DOI] [PubMed] [Google Scholar]
- 9.Kantrowitz ER, Lipscomb WN. Escherichia coli aspartate transcarbamoylase: The molecular basis for a concerted allosteric transition. Trends Biochem Sci. 1990;15:53–59. doi: 10.1016/0968-0004(90)90176-c. [DOI] [PubMed] [Google Scholar]
- 10.Collins KD, Stark GR. Aspartate transcarbamylase: Interaction with the transition state analogue N-(phosphonacetyl)-L-aspartate. J Biol Chem. 1971;246:6599–6605. [PubMed] [Google Scholar]
- 11.Gouaux JE, Lipscomb WN. Three-dimensional structure of carbamyl phosphate and succinate bound to aspartate carbamyltransferase. Proc Natl Acad Sci USA. 1988;85:4205–4208. doi: 10.1073/pnas.85.12.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stebbins JW, Xu W, Kantrowitz ER. Three residues involved in binding and catalysis in the carbamyl phosphate binding site of Escherichia coli aspartate transcarbamylase. Biochemistry. 1989;28:2592–2600. doi: 10.1021/bi00432a037. [DOI] [PubMed] [Google Scholar]
- 13.Stieglitz K, Stec B, Baker DP, Kantrowitz ER. Monitoring the transition from the T to the R state in E. coli aspartate transcarbamoylase by X-ray crystallography: Crystal structures of the E50A mutant in four distinct allosteric states. J Mol Biol. 2004;341:853–868. doi: 10.1016/j.jmb.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Moody MF, Vachette P, Foote AM. Changes in the X-ray solution scattering of aspartate transcarbamylase following the allosteric transition. J Mol Biol. 1979;133:517–532. doi: 10.1016/0022-2836(79)90405-4. [DOI] [PubMed] [Google Scholar]
- 15.Tsuruta H, Vachette P, Kantrowitz ER. Direct Observation of an Altered Quaternary Structural Transition in a Mutant Aspartate Transcarbamoylase. Proteins: Struct Funct Genet. 1998;31:383–390. [PubMed] [Google Scholar]
- 16.Wedler FC, Gasser FJ. Ordered Substrate Binding and Evidence for a Thermally Induced Change in Mechanism for E. coli Aspartate Transcarbamylase. Arch Biochem Biophys. 1974;163:57–68. doi: 10.1016/0003-9861(74)90454-8. [DOI] [PubMed] [Google Scholar]
- 17.Fetler L, Tauc P, Vachette P. Carbamyl phosphate modifies the T quaternary structure of aspartate transcarbamylase, thereby facilitating the structural transition associated with cooperativity. J Appl Cryst. 1997;30:781–786. [Google Scholar]
- 18.Griffin JH, Rosenbusch JP, Weber KK, Blout ER. Conformational changes in aspartate transcarbamylase. I Studies of ligand binding and of subunit interactions by circular dichroism spectroscopy. J Biol Chem. 1972;247:6482–6490. [PubMed] [Google Scholar]
- 19.Porter RW, Modebe MO, Stark GR. Aspartate transcarbamylase: Kinetic studies of the catalytic subunit. J Biol Chem. 1969;244:1846–1859. [PubMed] [Google Scholar]
- 20.Collins KD, Stark GR. Aspartate transcarbamylase: Studies of the catalytic subunit by ultraviolet difference spectroscopy. J Biol Chem. 1969;244:1869–1877. [PubMed] [Google Scholar]
- 21.Yang AS, Gunner MR, Sampogna R, Sharp K, Honig B. On the calculation of pKas in proteins. Proteins. 1993;15:252–265. doi: 10.1002/prot.340150304. [DOI] [PubMed] [Google Scholar]
- 22.Nowlan SF, Kantrowitz ER. Superproduction and rapid purification of E. coli aspartate transcarbamoylase and its catalytic subunit under extreme derepression of the pyrimidine pathway. J Biol Chem. 1985;260:14712–14716. [PubMed] [Google Scholar]
- 23.Baker DP, Kantrowitz ER. The conserved residues glutamate-37, asparate-100 and arginine-269 are important for the structural stabilization of Escherichia coli aspartate transcarbamoylase. Biochemistry. 1993;32:10150–10158. doi: 10.1021/bi00089a034. [DOI] [PubMed] [Google Scholar]
- 24.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 25.Davis BJ. Disc electrophoresis-II Method and application to human serum proteins. Ann N Y Acad Sci. 1964;121:680–685. doi: 10.1111/j.1749-6632.1964.tb14213.x. [DOI] [PubMed] [Google Scholar]
- 26.Ornstein L. Ann N Y Acad Sci. 1964;121:321–349. doi: 10.1111/j.1749-6632.1964.tb14207.x. [DOI] [PubMed] [Google Scholar]
- 27.Gerhart JC, Holoubek H. The purification of aspartate transcarbamylase of Escherichia coli and separation of its protein subunits. J Biol Chem. 1967;242:2886–2892. [PubMed] [Google Scholar]
- 28.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 29.Boulin CJ, Dainton D, Dorrington E, Elsner G, Gabriel A, Bordas J, Koch MHJ. Systems for time resolved x-ray measurements using one-dimensional and two-dimensional detectors - Requirements and pratical experience. Nuc Instru Meth Phys Res. 1982;201:209–220. [Google Scholar]
- 30.Hammersley AP, Svensson SO, Thompson A, Graafsma H, Kvick A, Moy JP. Calibration and correction of distortions in 2-dimensional detector systems. Rev Sci Instru. 1995;66:2729–2733. [Google Scholar]
- 31.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 32.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 34.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]