

Abstract
There is a great interest in determining the impact of p53 on ageing and, for this, it is important to discriminate among the known causes of ageing. Telomere loss is a well-established source of age-associated damage, which by itself can recapitulate ageing in mouse models. Here, we have used a genetic approach to interrogate whether p53 contributes to the elimination of telomere-damaged cells and its impact on telomere-driven ageing. We have generated compound mice carrying three functional copies of the p53 gene (super-p53) in a telomerase-deficient background and we have measured the presence of chromosomal abnormalities and DNA damage in several tissues. We have found that the in vivo load of telomere-derived chromosomal damage is significantly decreased in super-p53/telomerase-null mice compared with normal-p53/telomerase-null mice. Interestingly, the presence of extra p53 activity neither accelerates nor delays telomere-driven ageing. From these observations, we conclude that p53 has an active role in eliminating telomere-damaged cells, and we exclude the possibility of an age-promoting effect of p53 on telomere-driven ageing.
Keywords: p53, telomeres, ageing, DNA damage, tumour suppression
Introduction
Ageing is produced by the accumulation of cellular damage that eventually results in tissue dysfunction (Hasty et al, 2003). Telomeres have emerged in recent years as an important source of damage, the harmful potential of which increases progressively with time (Blasco, 2003). Mammalian telomeres protect chromosomal ends from being recognized and processed as DNA double-strand breaks (de Lange, 2002). Telomeres are shortened with each cell division owing to incomplete replication of the DNA ends, and this progressive loss of telomere length is compensated in germline cells by the activity of telomerase (Blackburn, 2001). However, in adult somatic tissues, telomerase activity is insufficient and, accordingly, most cells and tissues experience telomere shortening in association with cell division and ageing (Blackburn, 2001). In this manner, telomeres are cell division counting devices that, on reaching a critically short length, impose an end to the replicative lifespan of individual cells. In addition to this, the rate of telomere loss per cell division is influenced by the presence of cellular stressors, such as oxidative damage, an aspect that further reinforces the involvement of telomeres in the onset of ageing (Von Zglinicki, 2003).
Critically short telomeres are dysfunctional and trigger DNA damage signalling pathways that may provoke senescence or apoptosis (Smogorzewska & de Lange, 2002; d'Adda di Fagagna et al, 2003; Takai et al, 2003). DNA repair mechanisms may incur in the infliction of further damage by ligating dysfunctional telomeres and, thus, generating interchromosomal end-to-end fusions and other chromosomal aberrations (Goytisolo & Blasco, 2002). The relevance of telomere dysfunction for ageing is supported by studies in telomerase-deficient mice, which show accelerated ageing (Herrera et al, 1999; Rudolph et al, 1999; Blasco, 2003; Leri et al, 2003). In addition, germline mutations in telomerase components underlie the human progeroid syndrome dyskeratosis congenita (Vulliamy et al, 2001), and it has been reported for humans and worms that there is a correlation between the telomere length of a given individual and its lifespan (Cawthon et al, 2003; Joeng et al, 2004). Altogether, present evidence strongly suggests that telomere damage is a relevant determinant of ageing.
The tumour suppressor p53 is an important sensor of dysfunctional telomeres (Chin et al, 1999; Gonzalez-Suarez et al, 2000; Smogorzewska & de Lange, 2002; Leri et al, 2003; Sharpless & DePinho, 2004). Absence of p53 enables proliferation in the face of telomere dysfunction and this translates into delayed telomere-driven phenotypes in p53-null/telomerase-null mice, such as testicular atrophy, and in higher incidence of spontaneous carcinomas compared with p53-null/telomerase-proficient mice (Chin et al, 1999; Artandi et al, 2000). Notably, however, the impact of p53 on telomere-driven ageing has remained unexplored. The analysis of ageing in p53-null or p53-heterozygous mice is not possible because these mice die of cancer at an early age. To circumvent this limitation, we have taken advantage of our recently generated super-p53 mice (Garcia-Cao et al, 2002). These mice carry a large genomic transgene containing an intact p53 gene that recapitulates in a quantitative manner the activity of a single endogenous allele. The availability of wild-type p53 and super-p53 mice, carrying respectively two and three gene doses of p53, allows the analysis of the impact of p53 on phenotypes that require long observation periods, such as ageing.
Results
The p53 transgene is functional in aged mice
The rapid and robust apoptotic response of lymphocytes to ionizing radiation is among the best-characterized biological readouts for p53 function in vivo (Lowe et al, 1993; Midgley et al, 1995). In this regard, we have previously reported that young super-p53 mice present enhanced thymocyte and splenocyte apoptosis following irradiation (Garcia-Cao et al, 2002). In the light of reports on ageing-associated silencing of certain transgenes (Cohn et al, 1991; Robertson et al, 1996; Sutherland et al, 1997; Guglielmi et al, 2003), we considered of relevance to assess the functionality of our p53 transgene in aged animals. To address this, we have examined the apoptotic response to γ-radiation in the thymus and spleen of wild-type and super-p53 mice of advanced age. As shown in Fig 1, radiation-induced apoptosis was higher in super-p53 mice (both p53+/+;tg/• and p53+/+;tg/tg) compared with wild-type (p53+/+) mice regardless of age. These data show that the p53 transgene is not subject to ageing-associated silencing.
Figure 1.
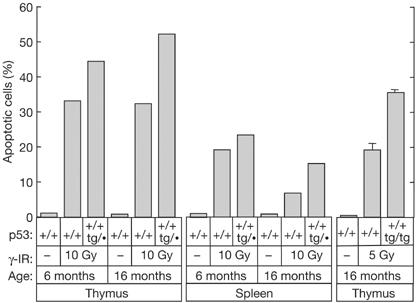
Functionality of the p53 transgene in aged mice. Young (6 months) and old (16 months) mice of the indicated genotypes were left untreated or exposed to 5 or 10 Gy whole-body γ-irradiation (γ-IR). At 3 h post-irradiation, the thymus was extracted and thymocytes were analysed by flow cytometry to measure their DNA content and evaluate the percentage of apoptosis. Data correspond to individual mice, except the rightmost columns, which correspond to the average±s.d. of three mice.
p53 reduces telomere-derived damage in MEFs
To explore the effect of increased p53 gene dosage in the particular context of telomere-derived damage, we crossed super-p53 mice (Garcia-Cao et al, 2002) into a genetic background lacking the RNA component of telomerase (Terc−/−; Blasco et al, 1997). In human and mouse cells, telomere uncapping triggers p53 activation (Chin et al, 1999; Gonzalez-Suarez et al, 2000; Smogorzewska & de Lange, 2002; Leri et al, 2003). Accordingly, we found that super-p53 mouse embryo fibroblasts (MEFs) derived from late-generation (G3) telomerase-null mice (Terc-G3−/−;p53+/+;tg/•) have higher basal levels of p53 and p21 than their p53 wild-type (Terc-G3−/−;p53+/+) counterparts (Fig 2A,B). Chromosomal fusions through the telomeres constitute the most prevalent and characteristic type of damage in telomerase-deficient cells (Blasco et al, 1997). To determine the consequences of p53 activity on telomere-derived damage, we compared the incidence of telomere fusions in primary Terc-G3−/− MEFs with wild-type or super-p53 gene dosage. As shown in Table 1, combined telomere fluorescence in situ hybridization (FISH) and cytogenetic analyses showed that the p53 transgene significantly reduced both the number of chromosomes with critically short telomeres (‘signal-free ends') and the incidence of chromosomal fusions through the telomeres (‘end-to-end fusions'). From these data, we conclude that genetically increased p53 function translates into a significant reduction in the amount of viable cells with telomere-derived DNA damage.
Figure 2.
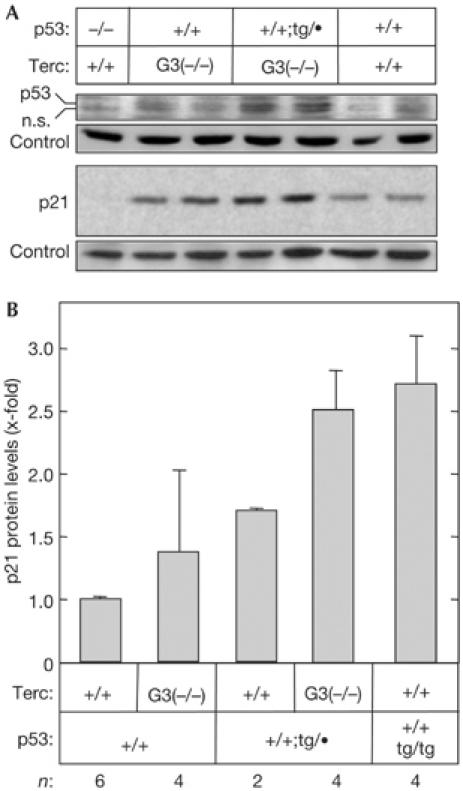
Super-p53 mouse embryo fibroblasts respond to telomere dysfunction by activating the p53/p21 pathway. (A) Representative example of the levels of p53 and p21 in cell extracts from early-passage primary mouse embryo fibroblasts (MEFs) of the indicated genotypes. The band labelled as n.s. is nonspecific for p53. Bands from Ponceau S-stained western blots were used as loading control. The blots shown are representative of three experiments. (B) Quantification of p21 protein levels. Data are relative to p21 levels in wild-type MEFs. For each genotype, several independent MEF cultures (derived from different embryos) were assayed, indicated at the bottom of the figure (n). Data correspond to the average±s.d.
Table 1.
Increased p53 gene dosage reduces chromosomal aberrations in late-generation telomerase-null mice
| p53(+/+) Terc-G3(−/−) | p53(+/+;tg/•) Terc-G3(−/−) | P-value* | |
|---|---|---|---|
| Primary MEFs† | |||
| Signal-free ends/telomeres‡ | 369/3,160 (11.7%) | 233/3,196 (7.3%) | <0.001 |
| p–p fusions/metaphases (Robertsonian-like) | 17/200 (8.5%) | 5/200 (2.5%) | <0.01 |
| q–q fusions/metaphases (dicentrics) | 7/200 (3.5%) | 1/200 (0.5%) | <0.05 |
| p–q fusions/metaphases | 2/200 (1.0%) | 1/200 (0.5%) | NS |
| Total number of end-to-end fusions/metaphases | 26/200 (13.0%) | 7/200 (3.5%) | <0.001 |
| 7-month-old mice—splenocytes† | |||
| Signal-free ends/telomeres‡ | 246/3,012 (8.2%) | 238/3,192 (7.5%) | NS |
| p–p fusions/metaphases (Robertsonian-like) | 12/100 (12.0%) | 1/100 (1.0%) | <0.001 |
| q–q fusions/metaphases (dicentrics) | 1/100 (1.0%) | 0/100 (0%) | NS |
| p–q fusions/metaphases | 0/100 (0%) | 0/100 (0%) | NS |
| Total number of end-to-end fusions/metaphases | 13/100 (13.0%) | 1/100 (1.0%) | <0.001 |
| 12-month old mice—splenocytes† | |||
| Signal-free ends/telomeres‡ | 431/3,136 (13.7%) | 364/3,084 (11.8%) | 0.01 |
| p–p fusions/metaphases (Robertsonian-like) | 55/94 (58.5%) | 23/95 (24.2%) | <0.001 |
| q–q fusions/metaphases (dicentrics) | 0/94 (0%) | 1/95 (1.1%) | NS |
| p–q fusions/metaphases | 0/100 (0%) | 0/100 (0%) | NS |
| Total number of end-to-end fusions/metaphases | 55/94 (58.5%) | 24/95 (26.3%) | <0.001 |
*Fisher's exact test; NS, not significant (P>0.05).
†For each genotype, data for chromosome fusions were obtained from two independent cultures of primary MEFs, or from two littermate mice of the indicated age, as corresponds.
‡Data for signal-free ends are given as the frequency of chromosome ends without detectable telomere signal relative to the total number of chromosome ends analysed.
MEFs, mouse embryo fibroblasts.
p53 reduces telomere-derived damage in the organism
To extend the above observations, we obtained splenocytes from late-generation (G3) telomerase-null mice and we examined the integrity of telomeres and chromosomes in ex vivo metaphases. For these analyses, we used Terc-G3−/− mice of two different ages: 7 months of age, when ∼75% of the Terc-G3−/− mouse colony is alive; and 12 months of age, when less than 50% of the Terc-G3−/− mouse colony is alive (Fig 4). Telomere length was significantly shorter in 12-month-old than in 7-month-old Terc-G3−/− mice, regardless of the presence or absence of the extra gene copy of p53 (data not shown). Terc-G3−/− mice with wild-type p53 (Terc-G3−/−;p53+/+) presented a considerable amount of telomere-derived damage in the spleen (‘signal-free ends' and ‘end-to-end fusions') at 7 months of age and this was remarkably exacerbated at 12 months of age (Table 1). Importantly, Terc-G3−/−;p53+/+;tg/• splenocytes showed a clear reduction in the amount of telomere-derived damage, and this was particularly significant at 12 months of age, as reflected by a diminished incidence of critically short telomeres and chromosomal fusions (Table 1). These data indicate that a modest increase in the activity of p53 (from the normal complement of two copies to three copies) has a measurable and significant effect reducing the load of telomere-damaged cells in the spleen.
Figure 4.
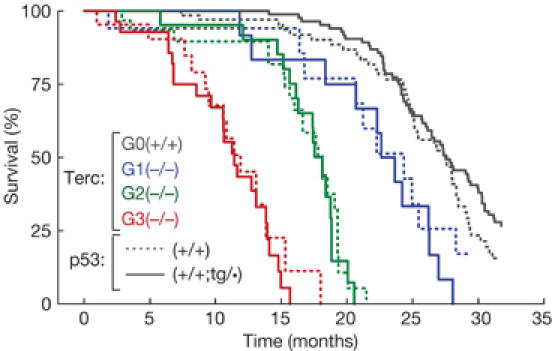
Increased p53 function does not affect telomere-driven ageing. Cohorts of successive generations of telomerase-deficient mice (G0, black lines; G1, blue lines; G2, green lines; G3, red lines) either wild-type for p53 (p53(+/+), dashed lines) or super-p53 (p53(+/+;tg/•), solid lines) were followed up for a period of 32 months. The figure shows a Kaplan–Meier representation of the survival of the following groups of mice: G0-Terc(+/+)p53(+/+), n=68; G0-Terc(+/+)p53(+/+;tg/•), n=105; G1-Terc(−/−)p53(+/+), n=17; G1-Terc(−/−)p53(+/+;tg/•), n=15; G2-Terc(−/−)p53(+/+), n=31; G2-Terc(−/−)p53(+/+;tg/•), n=21; G3-Terc(−/−) p53(+/+), n=22; G3-Terc(−/−)p53(+/+;tg/•), n=28.
We wished to extend the above concept to other adult cell types. For this, we measured γ-H2AX foci as a surrogate marker of telomere-derived damage. Previous investigators have shown that dysfunctional telomeres trigger the formation of γ-H2AX foci (d'Adda di Fagagna et al, 2003; Takai et al, 2003; Hao et al, 2004) and, moreover, that there is an age-dependent increase in the number of cells containing γ-H2AX foci (Sedelnikova et al, 2004). We analysed intestinal sections from 9-month-old mice of the relevant genotypes and we measured the percentage of γ-H2AX-positive epithelial cells (Fig 3A,B). Telomerase-null (Terc-G3−/−;p53+/+) mice showed a significant increase in the percentage of γ-H2AX-positive cells compared with telomerase-proficient (Terc+/+;p53+/+) mice, thus validating the use of this marker as a measurement of telomere-derived damage. Importantly, telomerase-null mice carrying an extra copy of p53 (Terc-G3−/−;p53+/+;tg/•) had levels of γ-H2AX-positive cells similar to those in telomerase-proficient (Terc+/+;p53+/+ and Terc+/+;p53+/+;tg/•) mice (Fig 3B). These observations were corroborated in epidermal cells (Fig 3E,F), thus confirming and extending the role of p53 in eliminating telomere-damaged cells. The cells of the intestinal epithelium are prone to undergo apoptosis on infliction of damage. A prediction from the above observations is that the number of apoptotic cells should be increased in super-p53/telomerase-null (Terc-G3−/−;p53+/+;tg/•) mice compared with normal-p53/telomerase-null (Terc-G3−/−;p53+/+) mice. Indeed, the levels of activated caspase 3 in the intestine of Terc-G3−/−;p53+/+;tg/• mice were significantly higher than those in Terc-G3−/−;p53+/+ mice (Fig 3C,D), thus providing a mechanistic explanation for the decreased number of γ-H2AX-positive cells in super-p53/telomerase-null mice. Together, our data on splenocytes, intestinal epithelium and epidermis indicate that an increment of p53 function mitigates the accumulation of telomere-derived DNA damage in the organism.
Figure 3.
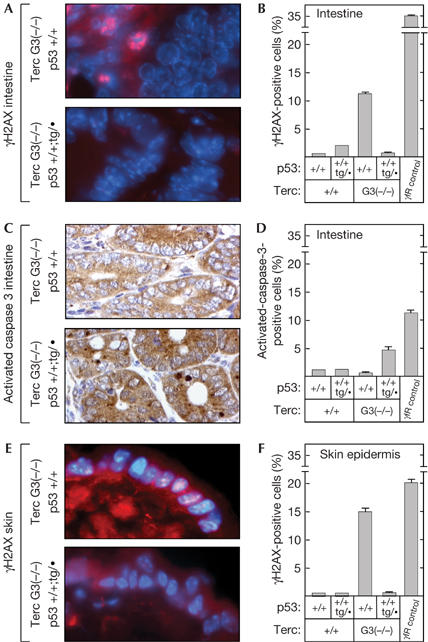
Super-p53 mice with short telomeres have reduced levels of DNA-damaged cells and present higher levels of apoptosis. (A) Illustrative example of cryostat sections of the small intestine from 9-month-old mice of the indicated genotypes assayed for the presence of γ-H2AX foci. (B) Quantification of the percentage of γ-H2AX-positive cells. (C) Illustrative example of paraffin-embedded sections of the small intestine from 9-month-old mice of the indicated genotypes assayed for the presence of active caspase 3. (D) Quantification of the percentage of active caspase 3-positive cells. (E) Illustrative example of cryostat sections of the skin from 9-month-old mice of the indicated genotypes assayed for the presence of γ-H2AX foci. (F) Quantification of the percentage of γ-H2AX-positive cells. Quantifications shown in (B,D,F) correspond to the average±s.d. of two mice. As a positive control, two wild-type mice were γ-irradiated with 10 Gy and killed 3 h after irradiation.
p53 has no impact on telomere-driven ageing
The biological end point of telomere dysfunction in somatic cells is a generalized proliferative defect culminating in diverse symptoms of tissue atrophy that become particularly evident in highly proliferative tissues such as the intestine, spleen and testis (Lee et al, 1998; Herrera et al, 1999; Rudolph et al, 1999). Histopathological analysis of moribund mice of successive generations (G1–G3) of telomerase-deficient wild-type and super-p53 genotypes showed essentially the same incidence and spectrum of degenerative pathologies (Table 2; a complete histopathological analysis is available as supplementary information online). The only exception to this was the incidence of testicular atrophies, which were moderately, but significantly, increased in the case of old super-p53 mice in a telomerase-proficient background. This effect of the p53 transgene was not noticeable in telomerase-deficient mice, probably owing to the dominant effect of telomerase deficiency, which results in high incidence of testicular atrophies (Table 2). Together, these data indicate that increased p53 function does not have an appreciable impact on the tissue renewal defects associated to telomere dysfunction.
Table 2.
Increased p53 function does not affect atrophic lesions associated with telomere-driven ageing*
|
Terc genotype |
||||||||
|---|---|---|---|---|---|---|---|---|
|
Terc(+/+) |
G1 Terc(−/−) |
G2 Terc(−/−) |
G3 Terc(−/−) |
|||||
|
p53 genotype |
||||||||
| Atrophic tissue | (+/+) | (+/+; tg/•) | (+/+) | (+/+; tg/•) | (+/+) | (+/+; tg/•) | (+/+) | (+/+; tg/•) |
| Spleen | 5/32 (16%) | 11/65 (17%) | 3/9 (33%) | 1/9 (11%) | 4/15 (27%) | 4/15 (27%) | 5/12 (42%) | 3/14 (21%) |
| Intestine | 0/32 (0%) | 0/65 (0%) | 7/9 (78%) | 5/9 (56%) | 13/15 (87%) | 13/15 (87%) | 10/12 (83%) | 12/14 (86%) |
| Testis | 0/18 (0%) | 5/29† (17%) | 2/4 (50%) | 4/5 (80%) | 6/8 (75%) | 8/11 (73%) | 5/6 (83%) | 6/9 (67%) |
| Ovary | 0/14 (0%) | 7/37 (19%) | 1/5 (20%) | 0/4 (0%) | 1/7 (14%) | 0/4 (0%) | 1/6 (17%) | 2/5 (40%) |
*Mice of the indicated genotype were killed when they showed signs of poor health, such as reduced activity or cachexia, and subjected to exhaustive histopathological analysis. The table shows data only for those tissues that presented signs of atrophy with high frequency (for more details, see the supplementary information online).
†P<0.05 versus wild-type controls (Fisher's exact test).
Finally, we measured the effect of increased p53 function on the lifespan in the context of telomere-driven ageing. Observation of telomerase-null cohorts for more than two and half years confirmed, as described in previous work (Herrera et al, 1999; Espejel et al, 2002a, 2002b), that progressive telomere shortening along successive generations (G1–G3) was paralleled by a corresponding progressive decrease in lifespan (Fig 4). Also, in the case of telomerase-proficient mice, we confirmed that increased p53 gene dosage has no effect on lifespan (Fig 4; Garcia-Cao et al, 2002). Remarkably, the gene dose of p53 did not affect the lifespan of the different mouse colonies at increasing generations in the absence of telomerase (Fig 4). Regarding the impact of p53 on spontaneous tumour development, super-p53 mice have a significantly lower incidence of spontaneous malignant tumours (supplementary Table 2 online; Garcia-Cao et al, 2002). In this regard, it should be noted that the lower incidence of malignant tumours is nonetheless insufficient to extend the lifespan, suggesting that spontaneous malignancies are probably not the primary cause of death in these aged mice. In the case of telomerase-deficient mice, the incidence of spontaneous carcinomas in our colonies was too low to detect significant effects (supplementary Table 2 online). In summary, we conclude that extra p53 activity does not accelerate telomere-driven atrophies or ageing.
Discussion
Here, we provide evidence that super-p53 mice have an enhanced response to telomere dysfunction. We have observed that late-generation super-p53/telomerase-null MEFs express higher protein levels of p53 and p21 than their normal-p53/telomerase-null counterparts. This observation reinforces the concept that p53 is a sensor of telomere damage (Introduction). An important issue of relevance for ageing is whether the activation of p53 in response to telomere damage results in the elimination of damaged cells or, alternatively, in the accumulation of damaged cells. We have observed that increased p53 function correlates with reduced presence of damaged cells in the spleen, intestine and skin, while at the same time we could demonstrate an increased apoptosis in the intestine. Together, these data demonstrate that, in the context of telomere dysfunction, the primary consequence of enhancing p53 function is a more efficient elimination of telomere-damaged cells.
We have previously shown that ageing in telomerase-proficient mice is not affected by increasing either the number of p53 alleles (Garcia-Cao et al, 2002) or the number of alleles of ARF, a p53 activator (Matheu et al, 2004). In the present work, we have further challenged the putative impact of p53 on ageing by focusing on telomere-driven ageing, thus minimizing the contribution of other types of damage that could operate in the case of spontaneous ageing. Importantly, in the absence of telomerase, super-p53 mice aged with the same kinetics and pathological spectrum as p53 wild-type mice. These results, together with our previous data on super-p53 mice and recent data on mice with extra ARF or hypomorphic Mdm2, negate a role for p53 in ageing, both spontaneous and telomere driven (Garcia-Cao et al, 2002; Matheu et al, 2004; Mendrysa et al, 2006). Although increased gene dosage of p53 has no impact on telomere-driven ageing, it is important to note that it has a detectable impact, decreasing the load of telomere-derived damage. This suggests that the reduction in damaged cells, although significant at a cellular level, is not of sufficient magnitude to delay the multi-organ failure characteristic of ageing. Finally, it must be emphasized that our data are not in conflict with the fact that aberrant, non-regulated, p53 activity could accelerate ageing. Indeed, mouse models vastly overexpressing p53 or carrying truncated forms of p53 present severe organ atrophies (Nakamura et al, 1995; Godley et al, 1996) or premature ageing (Tyner et al, 2002; Maier et al, 2004).
In summary, we demonstrate here that p53 has an active role in eliminating telomere-damaged cells in vivo, and that moderately increased levels of regulated p53 do not affect telomere-driven ageing.
Methods
Mice and primary mouse cells. The generation of super-p53 mice carrying one (p53+/+;tg/•) or two (p53+/+;tg/tg) extra copies of p53 has been described previously (Garcia-Cao et al, 2002). Successive generations of telomerase-proficient and telomerase-deficient mice were generated by crosses of super-p53 (p53+/+; tg/•) mice with a mouse strain deficient for the RNA component of telomerase (Terc−/−; Blasco et al, 1997). From these crosses, the two basic genotypes compared in this study were obtained: Terc−/−;p53+/+;tg/• and Terc−/−;p53+/+. Their genetic background was largely (>75%) C57BL6. Mice were housed in a pathogen-free barrier area. On signs of morbidity, mice were killed in accordance with the Guidelines for Humane Endpoints for Animals Used in Biomedical Research. Primary MEFs were prepared from day 13.5 embryos, as previously described (Palmero & Serrano, 2001). Cells were cultured under atmospheric oxygen pressure in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Sigma) and 1% penicillin G/streptomycin sulphate (Sigma).
Immunoblots. Immunoblots were carried out according to standard procedures. The antibodies used were as follows: for detection of p53, monoclonal antibody PAb240 (Calbiochem); for p21, polyclonal antibody C-19 (Santa Cruz). Secondary antibodies were either horseradish peroxidase-linked anti-rabbit (Amersham) or anti-mouse (DAKO). Detection was carried out by chemiluminescence using the ECL detection system (Amersham).
Chromosome analysis. Metaphase spreads from MEFs prepared from littermate embryos were scored for end-to-end fusions by superimposing the telomere image (quantitative-FISH (Q-FISH)) on the chromosome image (4,6-diamidino-2-phenylindole (DAPI)) using the TFL-telo software as described (Samper et al, 2000). Telomere fluorescence was quantified using the TFL-telo software kindly provided by Peter Lansdorp (British Columbia Cancer Center, Vancouver, Canada). For Q-FISH analysis of spleen sections, whole spleens were fixed in 10% formaldehyde for 2 h and paraffin embedded following standard methods. Spleen sections were hybridized with a PNA-tel probe and the telomere signals in meiotic nuclei were captured at × 100 magnification and analysed using the TFL-telo program.
Immunohistochemical analysis. Histological sections were carried out according to standard procedures. Apoptotic cells were identified using monoclonal antibodies against the active form of caspase 3 (AF835, R&D Systems) and visualized using peroxidase-coupled secondary antibodies and 3,3′-diaminobenzidine tetrahydrochloride as chromogen. Nuclei were counterstained with haematoxylin. Photomicrographs were obtained with a Leica DM LB microscope.
For the detection of γ-H2AX-positive cells in intact tissue, OCT-embedded cryostat sections of small intestine (6 μm thick) were placed on slides, air-dried at ambient temperature and fixed in acetone for 2 min. Slides were then air-dried, permeabilized with 0.1% Triton X-100 and blocked for 45 min at 37°C with 5% BSA. Subsequently, samples were incubated for 1 h with an anti-phospho-histone H2AX monoclonal antibody (1:500 in 2% BSA; Upstate Biotechnology), rinsed with PBS and incubated with a Cy3-labelled secondary antibody (Molecular Probes) according to standard procedures. Nuclei were counterstained with DAPI. Slides were mounted with coverslips using Vectashield/DAPI (Vector Laboratories) and examined under a fluorescence microscope (Leitz DMRB; Leica).
Quantifications were carried out, in a blind manner, by one of the authors (A.T.-L.). A minimum of 200 epithelial cells, intestinal or epidermal, were counted per mouse, distributed in at least two randomly chosen microscope fields.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Acknowledgments
We are indebted to M. Muñoz, R. Serrano and S. Rodriguez for excellent mouse colony management and animal care, and to E. Santos for mouse genotyping. M.A.B.'s laboratory has been funded by the Spanish Ministry of Science and Technology (SAF2001-1869, GEN2001-4856-C13-08), Autonomous Community of Madrid (CAM08.1/0054/01), European Union (TELOSENS, INTACT, ZINCAGE, RISC-RAD) and the Josef Steiner Award 2003. M.S.'s laboratory has been funded by the Spanish Ministry of Science and Technology (SAF2002-03402) and by the European Union (INTACT, PROTEOMAGE).
References
- Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA (2000) Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406: 641–645 [DOI] [PubMed] [Google Scholar]
- Blackburn EH (2001) Switching and signaling at the telomere. Cell 106: 661–673 [DOI] [PubMed] [Google Scholar]
- Blasco MA (2003) Telomeres in cancer and aging: lessons from the mouse. Cancer Lett 194: 183–188 [DOI] [PubMed] [Google Scholar]
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW (1997) Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91: 25–34 [DOI] [PubMed] [Google Scholar]
- Cawthon RM, Smith KR, O'Brien E, Sivatchenko A, Kerber RA (2003) Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 361: 393–395 [DOI] [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA (1999) p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97: 527–538 [DOI] [PubMed] [Google Scholar]
- Cohn SM, Roth KA, Birkenmeier EH, Gordon JI (1991) Temporal and spatial patterns of transgene expression in aging adult mice provide insights about the origins, organization, and differentiation of the intestinal epithelium. Proc Natl Acad Sci USA 88: 1034–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194–198 [DOI] [PubMed] [Google Scholar]
- de Lange T (2002) Protection of mammalian telomeres. Oncogene 21: 532–540 [DOI] [PubMed] [Google Scholar]
- Espejel S, Franco S, Rodriguez-Perales S, Bouffler SD, Cigudosa JC, Blasco MA (2002a) Mammalian Ku86 mediates chromosomal fusions and apoptosis caused by critically short telomeres. EMBO J 21: 2207–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espejel S, Franco S, Sgura A, Gae D, Bailey SM, Taccioli GE, Blasco MA (2002b) Functional interaction between DNA–PKcs and telomerase in telomere length maintenance. EMBO J 21: 6275–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM, Weill JC, Blasco MA, Serrano M (2002) ‘Super p53' mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J 21: 6225–6235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godley LA, Kopp JB, Eckhaus M, Paglino JJ, Owens J, Varmus HE (1996) Wild-type p53 transgenic mice exhibit altered differentiation of the ureteric bud and possess small kidneys. Genes Dev 10: 836–850 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Suarez E, Samper E, Flores JM, Blasco MA (2000) Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat Genet 26: 114–117 [DOI] [PubMed] [Google Scholar]
- Goytisolo FA, Blasco MA (2002) Many ways to telomere dysfunction: in vivo studies using mouse models. Oncogene 21: 584–591 [DOI] [PubMed] [Google Scholar]
- Guglielmi L, Le Bert M, Truffinet V, Cogne M, Denizot Y (2003) Insulators to improve expression of a 3(′)IgH LCR-driven reporter gene in transgenic mouse models. Biochem Biophys Res Commun 307: 466–471 [DOI] [PubMed] [Google Scholar]
- Hao LY, Strong MA, Greider CW (2004) Phosphorylation of H2AX at short telomeres in T cells and fibroblasts. J Biol Chem 279: 45148–45154 [DOI] [PubMed] [Google Scholar]
- Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J (2003) Aging and genome maintenance: lessons from the mouse? Science 299: 1355–1359 [DOI] [PubMed] [Google Scholar]
- Herrera E, Samper E, Martin-Caballero J, Flores JM, Lee HW, Blasco MA (1999) Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J 18: 2950–2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joeng KS, Song EJ, Lee KJ, Lee J (2004) Long lifespan in worms with long telomeric DNA. Nat Genet 36: 607–611 [DOI] [PubMed] [Google Scholar]
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW II, Greider CW, DePinho RA (1998) Essential role of mouse telomerase in highly proliferative organs. Nature 392: 569–574 [DOI] [PubMed] [Google Scholar]
- Leri A, Franco S, Zacheo A, Barlucchi L, Chimenti S, Limana F, Nadal-Ginard B, Kajstura J, Anversa P, Blasco MA (2003) Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J 22: 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T (1993) p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 362: 847–849 [DOI] [PubMed] [Google Scholar]
- Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M, Scrable H (2004) Modulation of mammalian life span by the short isoform of p53. Genes Dev 18: 306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheu A, Pantoja C, Efeyan A, Criado LM, Martin-Caballero J, Flores JM, Klatt P, Serrano M (2004) Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev 18: 2736–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendrysa SM, O'Leary KA, McElwee MK, Michalowski J, Eisenman RN, Powell DA, Perry ME (2006) Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev 20: 16–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley CA, Owens B, Briscoe CV, Thomas DB, Lane DP, Hall PA (1995) Coupling between gamma irradiation, p53 induction and the apoptotic response depends upon cell type in vivo. J Cell Sci 108: 1843–1848 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Pichel JG, Williams-Simons L, Westphal H (1995) An apoptotic defect in lens differentiation caused by human p53 is rescued by a mutant allele. Proc Natl Acad Sci USA 92: 6142–6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmero I, Serrano M (2001) Induction of senescence by oncogenic Ras. Methods Enzymol 333: 247–256 [DOI] [PubMed] [Google Scholar]
- Robertson G, Garrick D, Wilson M, Martin DI, Whitelaw E (1996) Age-dependent silencing of globin transgenes in the mouse. Nucleic Acids Res 24: 1465–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA (1999) Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 96: 701–712 [DOI] [PubMed] [Google Scholar]
- Samper E, Goytisolo FA, Slijepcevic P, van Buul PP, Blasco MA (2000) Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Rep 1: 244–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC (2004) Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol 6: 168–170 [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA (2004) Telomeres, stem cells, senescence, and cancer. J Clin Invest 113: 160–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, de Lange T (2002) Different telomere damage signaling pathways in human and mouse cells. EMBO J 21: 4338–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland HG, Martin DI, Whitelaw E (1997) A globin enhancer acts by increasing the proportion of erythrocytes expressing a linked transgene. Mol Cell Biol 17: 1607–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T (2003) DNA damage foci at dysfunctional telomeres. Curr Biol 13: 1549–1556 [DOI] [PubMed] [Google Scholar]
- Tyner SD et al. (2002) p53 mutant mice that display early ageing-associated phenotypes. Nature 415: 45–53 [DOI] [PubMed] [Google Scholar]
- Von Zglinicki T (2003) Replicative senescence and the art of counting. Exp Gerontol 38: 1259–1264 [DOI] [PubMed] [Google Scholar]
- Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I (2001) The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413: 432–435 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information