

Abstract
The Wiskott–Aldrich syndrome (WAS) is an X-chromosome-linked immunodeficiency disorder. The most common symptom seen in WAS patients is bleeding. One of the main causes of bleeding is defective platelet aggregation. The causative gene of WAS encodes WAS protein (WASP). Here, we show that WASP binds to the calcium- and integrin-binding protein (CIB) in platelets. CIB was originally identified as a protein binding to the αIIb cytoplasmic tail of platelet integrin αIIbβ3, which has a primary role in platelet aggregation. We also show that the WASP–CIB complex is important in αIIbβ3-mediated cell adhesion, and that in patients mutant forms of WASP are expressed at reduced levels or show lower affinities for CIB than wild-type WASP. Our results indicate that impaired complex formation between mutant WASPs and CIB reduces αIIbβ3-mediated cell adhesion and causes defective platelet aggregation, resulting in bleeding.
Keywords: Wiskott–Aldrich syndrome, Wiskott–Aldrich syndrome protein, WASP, platelets
Introduction
The Wiskott–Aldrich syndrome (WAS) is a rare X-chromosome-linked immunodeficiency disorder characterized by thrombocytopenia, small-sized platelets, eczema, recurrent infections and increased risk for autoimmunity and malignancy (Wiskott, 1937; Aldrich et al, 1954; Nonoyama & Ochs, 1998; Thrasher, 2002). The causative gene encodes the Wiskott–Aldrich syndrome protein (WASP; Derry et al, 1994), which contains several functional domains (Thrasher, 2002). The basic region and the GTPase-binding domain have a crucial role in activation of the WASP carboxy-terminal region (VCA domain; Fig 1A), stimulating actin polymerization (Kim et al, 2000; Prehoda et al, 2000).
Figure 1.
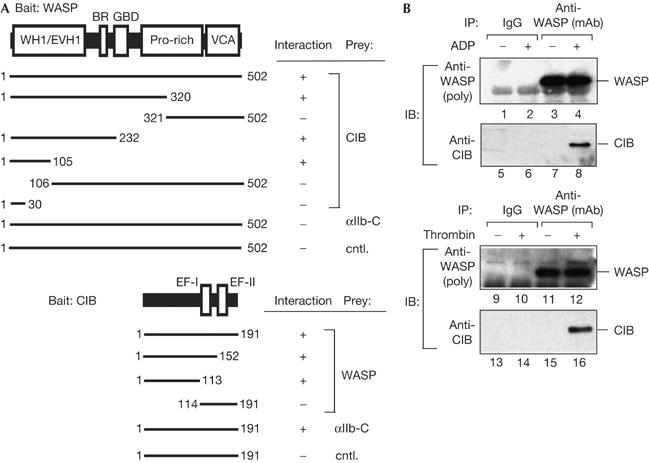
Wiskott–Aldrich syndrome protein binds to calcium- and integrin-binding protein in platelets. (A) Domain organizations of Wiskott–Aldrich syndrome protein (WASP) and calcium- and integrin-binding protein (CIB). WH1, WASP-homology 1 domain; EVH1, ENA/vasodilator-stimulated phosphoprotein (VASP)-homology 1 domain (the WH1 domain is homologous to the EVH1 domain); BR, basic region; GBD, GTPase-binding domain; Pro-rich, proline-rich domain; VCA, verprolin/cofilin/acidic domain. CIB has two putative Ca2+-binding domains, EF hand-I and EF hand-II (EF-I and EF-II). Each binding site was determined by yeast two-hybrid assay. Calcyclin was used as a control protein (cntl.). (B) Binding of WASP to CIB in platelets. Platelets were stimulated with 100 μM of ADP (top panel) or 1.0 U/ml of thrombin (bottom panel). WASP was immunoprecipitated from platelet lysates with anti-WASP monoclonal antibody followed by immunoblotting for WASP (lanes 1–4,9–12) and CIB (lanes 5–8,13–16).
Several distinct phenotypes caused by mutations in the WAS gene have been reported, including classic WAS, X-linked thrombocytopenia (XLT), X-linked neutropenia (XLN) or X-linked neutropenia myelodysplasia (XLM; Devriendt et al, 2001; Thrasher, 2002). In classic WAS patients, WASP expression is not detected. Most XLT patients have missense mutations in the WASP amino terminus (within residues 1–137), and express the mutant proteins at a lower concentration than normal subjects (Zhu et al, 1997; Imai et al, 2004).
Bleeding is the most consistent symptom seen in WAS patients, except for two families with XLN and XLM. Several clinical observations indicate that in addition to a low platelet count, a functional defect in platelet aggregation is also an important cause of bleeding in WAS patients. For example, platelet counts increase to nearly normal levels in patients after splenectomy, although the bleeding tendency is not completely resolved (Mullen et al, 1993). We suggested that such defects in platelet aggregation are probably caused by a functional deficiency of the WASP N-terminal region (residues 1–137), as bleeding is observed in both WASP-deficient patients (classic WAS) and XLT patients with missense mutations occurring in the WASP N terminus. However, no biological function of the WASP N-terminal region associated with platelet aggregation has been identified.
Results and Discussion
WASP binds to CIB in platelets
To determine the role of the WASP N terminus in platelet aggregation, we undertook a yeast two-hybrid screen using the WASP N-terminal domain as bait and isolated a complementary DNA encoding the calcium- and integrin-binding protein (CIB). CIB was originally cloned as a protein binding to the αIIb cytoplasmic tail of platelet integrin αIIbβ3 (Naik et al, 1997) and shown to increase αIIbβ3 affinity for its ligand (Tsuboi, 2002). We found that the binding site for CIB on WASP resided in the WASP N terminus (residues 1–105) and that WASP binding required the CIB N terminus (residues 1–113; Fig 1A).
Analysis of WASP–CIB binding using the surface plasmon resonance-based biosensor, BIACORE 3000 (BIACORE, Piscataway, NJ, USA), indicated that the WASP N terminus bound directly to the CIB N terminus (supplementary Fig S1 online). As a control, we used calcyclin because it is a small calcium-binding protein, similar to CIB (Murphy et al, 1988). No significant binding was observed between WASP and calcyclin.
We examined whether WASP binds to CIB in platelets, as platelets are the only cells that express the three factors WASP, CIB and αIIbβ3. Platelets were stimulated with thrombin or ADP without stirring in the presence of the αIIbβ3 integrin antagonist, RGD-containing peptide (GRGDSP peptide (Gibco/BRL)), to prevent αIIbβ3 clustering and platelet aggregation. WASP was immunoprecipitated from platelet lysates with anti-WASP antibody (Fig 1B, lanes 3,4,11,12). CIB was detected only in immunoprecipitates obtained from lysates of stimulated platelets in a time-dependent manner, but not resting platelets (Fig 1B, lanes 8,16; supplementary Fig S2 online). WASP was also detected only in anti-CIB immunoprecipitates obtained from lysates of stimulated platelets (supplementary Fig S10 online). These results indicate that WASP is not associated with CIB in resting platelets and that WASP binds to CIB following agonist stimulation.
WASP–CIB binding increases cell adhesion
We suggested that WASP and CIB are involved in αIIbβ3-mediated cell adhesion in stimulated platelets, as WASP binds to CIB (Fig 1) and CIB increases αIIbβ3 affinity for its ligand (Tsuboi, 2002). To determine the role of WASP and CIB in cell adhesion, we used a human megakaryoblastoid cell line, MEG-01 cells, that expresses αIIbβ3 and WASP but not CIB. When cells were transfected with CIB, CIB co-immunoprecipitated with WASP (Fig 2A, lane 14), indicating that WASP binds to CIB in transfected cells. Adhesion of MEG-01 cells expressing CIB to immobilized fibrinogen was increased, compared with mock-transfected MEG-01 cells (Fig 2B). The cell adhesion to fibrinogen was inhibited by RGD peptide, but not by RGE (GRGESP), confirming that the cell adhesion was mediated by αIIbβ3.
Figure 2.
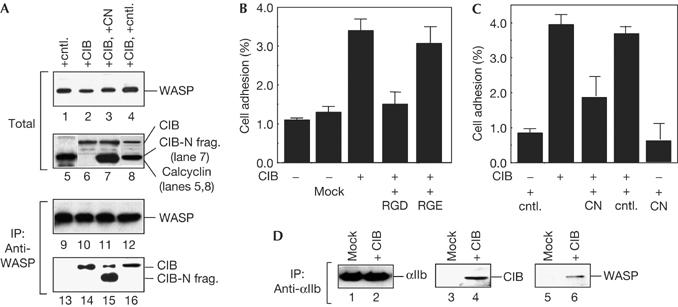
Wiskott–Aldrich syndrome protein binding to calcium- and integrin-binding protein increases αIIbβ3-medaited cell adhesion. (A) MEG-01 cells were transfected with pcDNA3-CIB (calcium- and integrin-binding protein), FLAG-tagged CIB amino-terminal fragment (CN) and/or FLAG-tagged calcyclin. Cell lysates were immunoblotted with anti-Wiskott–Aldrich syndrome protein (WASP; lanes 1–4), CIB (lanes 5–8) and FLAG (anti-FLAG conjugated with horseradish peroxidase; lanes 5–8). WASP was immunoprecipitated from cell lysates with anti-WASP antibody followed by immunoblotting for WASP (lanes 9–12), CIB and the CN (lanes 13–16). (B) MEG-01 cells transfected with CIB were tested for adhesion to fibrinogen in the presence of 100 μM of GRGDSP or GRGESP peptide. (C) MEG-01 cells transfected with calcyclin, CIB and CN were tested for adhesion to fibrinogen. All cell adhesion data are the mean±s.e. of three experiments. (D) MEG-01 cells were transfected with CIB and αIIb was immunoprecipitated from cell lysates with anti-αIIb followed by immunoblotting for αIIb (lanes 1,2), CIB (lanes 3,4) and WASP (lanes 5,6).
In cells coexpressing the CIB N-terminal fragment with CIB, the amount of co-immunoprecipitated CIB with WASP was decreased (Fig 2A, lane 15), indicating that WASP binding to CIB was inhibited by the CIB N-terminal fragment in transfected cells. Adhesion of such cells was significantly decreased, compared with cells expressing CIB and control (Fig 2C). These results indicate that the WASP–CIB complex is involved in αIIbβ3-mediated cell adhesion.
Binding of the CIB N-terminal fragment (residues 1–113) to WASP was detected (Fig 1), but not to the αIIb tail (Tsuboi, 2002). No significant adhesion of cells expressing only CIB N-terminal fragment was observed (Fig 2C). This result, taken together with Fig 2B, suggests that both WASP binding and αIIb binding to CIB are necessary for αIIbβ3-mediated cell adhesion. In fact, blocking αIIb binding to CIB reduced αIIbβ3 affinity for its ligand in platelets (Tsuboi, 2002).
In cells expressing CIB, both CIB and WASP co-immunoprecipitated with αIIb (Fig 2D, lanes 4,6). CIB binds to WASP in cells (Fig 2A, lane 11), and WASP binding to αIIb was not detected (Fig 1A). These results indicate that the WASP–CIB complex binds to the αIIb tail by means of CIB in transfected cells.
Blocking WASP–CIB binding reduces αIIbβ3 affinity
To examine whether WASP binding to CIB is important for αIIbβ3-mediated cell adhesion in platelets, we asked whether blocking WASP binding to CIB affects the affinity of αIIbβ3 for fibrinogen, as an increase in αIIbβ3 affinity is a prerequisite for αIIbβ3-mediated cell adhesion. To block binding, we permeabilized platelets with streptolysin O (SLO) and then introduced the CIB N-terminal fragment (residues 1–113) into SLO-permeabilized platelets. This method was used because platelets are not amenable to transfection. The CIB N-terminal fragment is predicted to block WASP binding to CIB, because the CIB N-terminal fragment binds directly to the WASP N terminus (Fig 1). In the absence of exogenous fragments, endogenous CIB co-immunoprecipitated with WASP (Fig 3A, lane 4). When permeabilized platelets were incubated with the CIB N-terminal fragment, CIB failed to co-immunoprecipitate with WASP (Fig 3A, lane 5), indicating that WASP binding to CIB is blocked by the CIB N-terminal fragments in SLO-permeabilized platelets.
Figure 3.
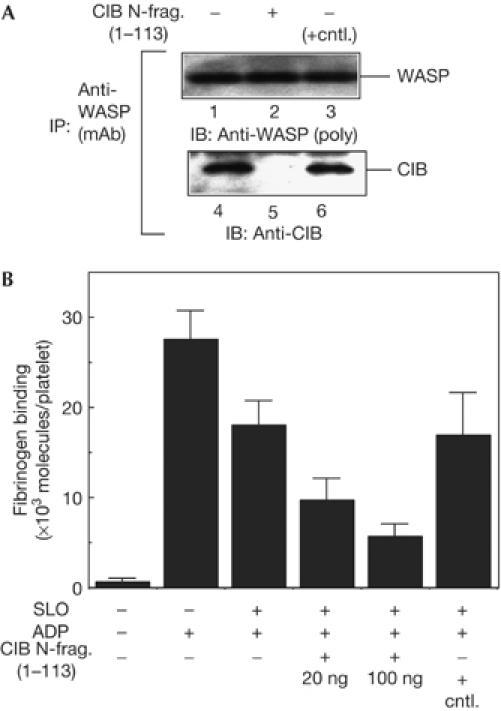
Blocking Wiskott–Aldrich syndrome protein binding to calcium- and integrin-binding protein reduces αIIbβ3 affinity for its ligand. (A) Streptolysin O (SLO)-permeabilized platelets were incubated with the calcium- and integrin-binding protein (CIB) amino-terminal fragment or a control protein and then stimulated with ADP without stirring in the presence of 100 μM of GRGDSP peptide. Wiskott–Aldrich syndrome protein (WASP) was immunoprecipitated from platelet lysates with anti-WASP monoclonal antibody followed by immunoblotting for WASP (lanes 1–3) and CIB (lanes 4–6). (B) SLO-permeabilized platelets (8 × 107 cells) were incubated with the CIB N-terminal fragment or 100 ng of FLAG–calcyclin as a control protein (cntl.) and then stimulated with ADP without stirring. Fibrinogen binding was measured as described in Methods. Data are the means±s.e. of three experiments.
We then examined affinity of αIIbβ3 for fibrinogen. To measure binding of soluble fibrinogen to αIIbβ3, platelets were stimulated with ADP without stirring. We prevented integrin clustering and platelet aggregation without interfering with fibrinogen binding to αIIbβ3 by measuring binding in the absence of stirring (Shattil et al, 1994). To determine the specific binding, we measured fibrinogen binding in the presence or absence of an αIIbβ3 antagonist, GRGDSP peptide. Specific fibrinogen binding was defined as binding inhibited by GRGDSP peptide.
Fibrinogen bound to stimulated intact platelets and stimulated SLO-permeabilized platelets at about 27,000 and 19,000 molecules per platelet, respectively, indicating that SLO-permeabilized platelets showed roughly 70% of signalling capacity to activate αIIbβ3 (Fig 3B). When WASP binding to CIB was blocked by the CIB N-terminal fragment, fibrinogen binding to platelets in response to ADP decreased in a dose-dependent manner (Fig 3B). The control had no effect on fibrinogen binding to platelets.
Conformation of αIIbβ3 is converted from a low-affinity state for its ligand to a high-affinity state following agonist stimulation, and a monoclonal antibody, PAC-1, binds specifically to the high-affinity state of αIIbβ3 (Shattil et al, 1985). We confirmed that such a conformational change of αIIbβ3 was inhibited by blocking WASP binding to CIB (supplementary Fig S3 online). These results indicate that blocking WASP binding to CIB inhibits the increase in the affinity of αIIbβ3 for fibrinogen. Blocking WASP binding to CIB had no effect on the affinity of another platelet integrin, α2β1, for collagen (supplementary Fig S4 online), calcium mobilization and α-granule secretion in platelets (supplementary Fig S5 online). When WASP binding to CIB was blocked by the WASP N-terminal fragment (residues 1–105), results similar to Fig 3 were obtained (supplementary Fig S6 online). This result confirms that WASP binding to CIB is important for αIIbβ3-mediated cell adhesion in platelets.
Role of WASP in cell adhesion
When WASP expression in CIB-expressing MEG-01 cells was silenced by short interfering RNA, cell adhesion to fibrinogen was significantly decreased (see the supplementary information online and supplementary Fig S11 online), indicating that WASP is important in αIIbβ3-mediated cell adhesion.
To further investigate the role of WASP in αIIbβ3-mediated cell adhesion, we next evaluated the importance of the WASP C-terminal VCA domain, which stimulates actin polymerization. The WASP lacking the VCA domain (WASP-dVCA or WdV) does not stimulate actin polymerization, but binds to CIB (Fig 1A). In cells expressing WASP-dVCA, CIB bound to both endogenous WASP and WASP-dVCA (Fig 4A, lanes 15,20). Adhesion of cells expressing WASP-dVCA was significantly decreased (Fig 4B). The decrease in adhesion is probably caused by the decrease in the WASP–CIB complex due to the WASP-dVCA–CIB complex. This result suggests that in addition to WASP binding to CIB, the WASP C terminus stimulating actin polymerization is necessary for cell adhesion. Actin polymerization and association of actin cytoskeleton to αIIbβ3 are essential for αIIbβ3-mediated cell adhesion (Schoenwaelder & Burridge, 1999). As blocking WASP binding to CIB had no effect on actin polymerization itself in platelets (supplementary Fig S7A online), our results indicate that WASP binding to CIB may be important in the concentration and association of actin cytoskeleton to αIIbβ3 sites. For further discussion, see the supplementary information online.
Figure 4.
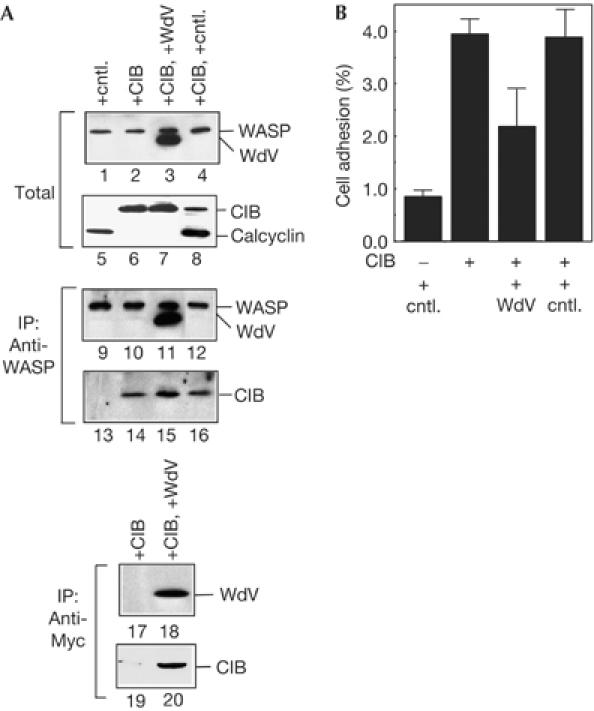
Role of Wiskott–Aldrich syndrome protein in cell adhesion. (A) MEG-01 cells were transfected with calcium- and integrin-binding protein (CIB), FLAG-tagged calcyclin and Myc-tagged Wiskott–Aldrich syndrome protein (WASP)-dVCA (residues 1–435). Transfected cells were lysed and immunoblotted with anti-WASP antibody (lanes 1–4), anti-FLAG (anti-FLAG conjugated with horseradish peroxidase) and CIB antibodies (lanes 5–8). WASP was immunoprecipitated from cell lysates with anti-WASP antibody followed by immunoblotting for WASP (lanes 9–12) and CIB and FLAG–calcyclin (lanes 13–16). Myc-tagged WASP-dVCA was also immunoprecipitated with anti-Myc monoclonal antibody followed by immunoblotting with anti-Myc antibody (lanes 17,18) and anti-CIB antibody (lanes 19,20). (B) Cells transfected with calcyclin, CIB and WASP-dVCA were tested for adhesion to fibrinogen in the presence of GRGDSP or GRGESP peptide. Data are the means±s.e. of three experiments.
We discuss αIIb binding to CIB during stimulation in supplementary information online.
Potential mechanism underlying bleeding in patients
Bleeding is the most common symptom seen in WAS/XLT patients, and defective platelet aggregation is an important cause of bleeding. However, the molecular basis underlying defective platelet aggregation in WAS/XLT patients is not understood. We showed that WASP binding to CIB is important for αIIbβ3-mediated cell adhesion (Figs 2 and 3). We suggested that αIIbβ3-mediated cell adhesion is altered by mutations in patients, as adhesion of platelets to fibrinogen is a key process in platelet aggregation. To determine whether the affinity of αIIbβ3 for its ligand is affected in stimulated platelets from WAS/XLT patients, we measured fibrinogen binding to platelets. We examined four XLT patients expressing mutant WASP with missense mutations in the WASP N-terminal region and one classic WAS patient (211delT) expressing no detectable WASP. In all cases tested, fibrinogen binding to stimulated platelets was reduced, compared with a normal control (Fig 5A). PAC-1 binding to stimulated patients' platelets was also reduced, and there was no significant difference in the surface expression level of αIIbβ3 between the normal control and patients (supplementary Fig S8 online). These results indicate that the affinity of αIIbβ3 for its ligand is reduced in stimulated platelets from XLT and classic WAS patients. This evidence indicates that there is a functional defect in the process of platelet aggregation in WAS/XLT patients' platelets. Some other patients' information is provided in supplementary Fig S12 online.
Figure 5.
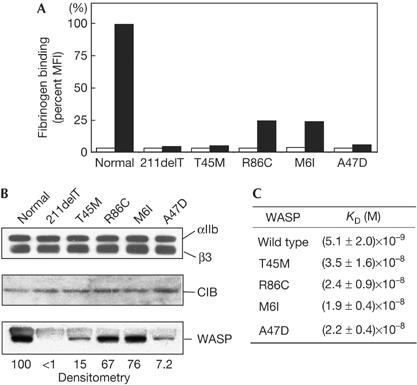
αIIbβ3, calcium- and integrin-binding protein and Wiskott–Aldrich syndrome protein in Wiskott–Aldrich syndrome/X-linked thrombocytopenia patients. (A) Fluorescein isothiocyanate-labelled fibrinogen binding to platelets from one classic Wiskott–Aldrich syndrome patient (211delT) and four X-linked thrombocytopenia patients (T45M, R85C, M6I and A47D) was analysed with FACSort. As a semiquantitative estimate of fluorescence, binding was expressed as per cent mean fluorescence intensity (MFI). Open and closed bars indicate fibrinogen binding to resting and ADP-stimulated platelets, respectively. (B) Expression levels of αIIbβ3, calcium- and integrin-binding protein (CIB) and Wiskott–Aldrich syndrome protein (WASP) in WAS patients. Lysates from platelets derived from one classic WAS patient and four XLT patients were subjected to immunoblotting. Relative amounts of WASP mutant were determined by densitometric analysis using Alpha Imager 2200. (C) The binding parameters of wild-type and WASP mutants for CIB were determined by BIACORE 3000. Myc–WASP and its mutants were passed over a chip with immobilized FLAG-tagged CIB. Each analyte was injected at concentrations of 250, 125, 62.5, 37.5, 18.8 and 9.38 nM. The dissociation affinity constants (KD) of WASP for CIB were estimated from these data using BIAevaluation 2.0.
We assumed that the reduced affinity of αIIbβ3 in selected WAS/XLT patients (Fig 5A) was the direct result of inefficient formation of the WASP–CIB complex due to mutations within the CIB-binding site of WASP. We, therefore, asked how mutations affect WASP–CIB complex formation.
First, we determined whether WASP mutations affect the expression level of WASP protein in patients' platelets. By immunoblotting, we observed no significant difference in the expression levels of αIIbβ3 or CIB in platelets between WAS/XLT patients and normal control (Fig 5B, top two panels). We detected no WASP in platelets from this classic WAS patient because of the frameshift (Fig 5B, bottom panel). By contrast, platelets from each of the four XLT patients with missense mutations within the CIB-binding site of the WASP N terminus (residues 1–105) expressed reduced levels of normal-sized mutant WASPs (Fig 5B, bottom panel). The relative amounts of WASP as determined by densitometric analysis (normal control=100) were 15 (T45M), 67 (R86C), 76 (M6I) and 7.2 (A47D).
Second, we asked whether the affinity of mutant WASP protein for CIB was altered as a result of mutations. We determined the binding parameters of mutant WASPs to CIB by surface plasmon resonance. Myc-tagged WASP and mutant forms were injected at various concentrations onto the chip with immobilized FLAG-tagged CIB. Curves were fitted to a 1:1 binding model, indicating that the stoichiometry of WASP binding to CIB is 1:1. The dissociation constants (KD) of wild-type WASP and the WASP mutants for CIB were determined from these data (Fig 5C). The KD values indicate that the binding affinity of the four WASP mutants with XLT mutations to CIB was markedly lower than that of wild-type WASP.
These results indicate that in patients the levels of WASP–CIB complex are not sufficient to associate actin cytoskeleton to αIIbβ3 sites and optimize the αIIbβ3 affinity for its ligand because of reduced WASP expression and/or lower affinity of WASP mutants for CIB (Fig 5B,C). These defects in affinity modulation of αIIbβ3 most probably result in reduced αIIbβ3-mediated cell adhesion in patients. Reduced αIIbβ3-mediated cell adhesion would then account for defective platelet aggregation, contributing to increased bleeding seen in classic WAS and XLT patients.
Gross et al (1999) reported that platelets from two WAS patients underwent full or slightly enhanced aggregation compared with a normal control. In this study, in vitro aggregation of platelets from two brothers of a family with a missense mutation (V75M) was measured using aggregometer. This is the first measurement of platelet aggregation in patients after the molecular cloning of WASP. Thus, their study clearly demonstrated that a cause of bleeding in these two brothers is low platelet count. Our study demonstrated that in addition to low platelet count, impaired activation of a specific platelet integrin, αIIbβ3, is also a cause of bleeding by examining five patients with five different mutations (Fig 5). Gross et al measured in vitro platelet aggregation, which results from gross adhesion mediated by several integrins (for example, αIIbβ3, α2β1 and αVβ3) to their ligands (Andrews & Berndt, 2004). We focused on αIIbβ3 among platelet integrins, as our results showed that WASP is involved specifically in αIIbβ3-mediated cell adhesion through binding to CIB (Figs 1, 2, 3 and 4; supplementary Fig S4 online). We showed that the increase in the affinity of αIIbβ3 following agonist stimulation in patients is impaired because of mutations (Fig 5).
In platelets from WASP-deficient mice, defective platelet aggregation is not observed. However, WASP-deficient WAS patients show defective platelet aggregation. For further discussion of this difference, see the supplementary information online.
The WASP interacting protein (WIP) was also identified as a protein binding to the WASP N-terminal region. We discuss WIP in the supplementary information online.
Speculation
We showed that reduced expression of mutant WASPs and/or their lower affinity for CIB affected affinity of αIIbβ3 for its ligand (Fig 5) and that these defects most probably result in defective platelet aggregation observed in patients. Our findings not only show the linkage of WASP to platelet aggregation but also indicate a potential mechanism underlying bleeding in WAS and XLT patients.
Methods
Platelets. After informed consent was obtained, blood was collected from healthy volunteers and WAS/XLT patients. Platelet isolation, permeabilization, stimulation and fluorescence-activated cell sorting analysis were performed as described in the supplementary information online. The Internal Review Boards of The Burnham Institute and University of Washington approved these experiments.
Binding of fibrinogen. 125I-labelled fibrinogen (1 μM; Amersham, Uppsala, Sweden) was added to platelets and incubated at 22°C for 15 min without stirring. The platelet suspension was layered on top of 0.4 ml of 20% (w/v) sucrose in Tyrode's buffer in a microcentrifuge tube and centrifuged at 12,000g at 22°C for 10 min to remove unbound 125I-labelled fibrinogen. The pellet was separated from the supernatant and counted in a gamma counter (20/20 Series). The number of molecules bound per platelet was calculated from the radioactivity in the pellet fraction. In some experiments, fluorescein isothiocyanate (FITC)-labelled fibrinogen (300 nM) was used. FITC–fibrinogen binding was analysed by a flow cytometer.
Adhesion assay. MEG-01 cells were labelled with 35S-labelled methionine (NEN) and resuspended in RPMI 1640 medium containing 5% fetal calf serum (adhesion buffer). Microtitre plates were coated with human fibrinogen (Calbiochem, La Jolla, CA, USA) at 10 μg/ml. MEG-01 cells were incubated in the microtitre plates (5 × 104 cells/well) at 37°C for 15 min and unbound cells were removed by washing with adhesion buffer, and then bound cells were counted.
Other experiments. Yeast two-hybrid screening, cell culture, transfection, recombinant protein preparation, surface plasmon resonance and immunoprecipitation were performed as described in the supplementary information online.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Acknowledgments
We thank Dr E. Ruoslahti and Dr M. Fukuda for critical comments. S.T. was supported by the National Institute of Health (R01HD42752). H.D.O. was supported by the National Institute of Health (R01HD17427-33), the Jeffery Modell Foundation and the Immunodeficiency Foundation.
References
- Aldrich RA, Steinberg AG, Campbell DC (1954) Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. Pediatrics 13: 133–139 [PubMed] [Google Scholar]
- Andrews RK, Berndt MC (2004) Platelet physiology and thrombosis. Thromb Res 114: 447–453 [DOI] [PubMed] [Google Scholar]
- Derry JM, Ochs HD, Francke U (1994) Isolation of a novel gene mutated in Wiskott–Aldrich syndrome. Cell 78: 635–644. Erratum in Cell 79: 922 [DOI] [PubMed] [Google Scholar]
- Devriendt K et al. (2001) Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet 27: 313–317 [DOI] [PubMed] [Google Scholar]
- Gross BS, Wilde JI, Quek L, Chapel H, Nelson DL, Watson SP (1999) Regulation and function of WASp in platelets by the collagen receptor, glycoprotein VI. Blood 94: 4166–4176 [PubMed] [Google Scholar]
- Imai K, Morio T, Zhu Y, Jin Y, Itoh S, Kajiwara M, Yata J, Mizutani S, Ochs HD, Nonoyama S (2004) Clinical course of patients with WASP gene mutations. Blood 103: 456–464 [DOI] [PubMed] [Google Scholar]
- Kim AS, Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK (2000) Autoinhibition and activation mechanisms of the Wiskott–Aldrich syndrome protein. Nature 404: 151–158 [DOI] [PubMed] [Google Scholar]
- Mullen CA, Anderson KD, Blaese RM (1993) Splenectomy and/or bone marrow transplantation in the management of the Wiskott–Aldrich syndrome: long-term follow-up of 62 cases. Blood 82: 2961–2966 [PubMed] [Google Scholar]
- Murphy LC, Murphy LJ, Tsuyuki D, Duckworth ML, Shiu RP (1988) Cloning and characterization of a cDNA encoding a highly conserved, putative calcium binding protein, identified by an anti-prolactin receptor antiserum. J Biol Chem 263: 2397–2401 [PubMed] [Google Scholar]
- Naik UP, Patel PM, Parise LV (1997) Identification of a novel calcium-binding protein that interacts with the integrin αIIb cytoplasmic domain. J Biol Chem 272: 4651–4654 [DOI] [PubMed] [Google Scholar]
- Nonoyama S, Ochs HD (1998) Characterization of the Wiskott–Aldrich syndrome protein and its role in the disease. Curr Opin Immunol 10: 407–412 [DOI] [PubMed] [Google Scholar]
- Prehoda KE, Scott JA, Mullins RD, Lim WA (2000) Integration of multiple signals through cooperative regulation of the N-WASP–Arp2/3 complex. Science 290: 801–806 [DOI] [PubMed] [Google Scholar]
- Schoenwaelder SM, Burridge K (1999) Bidirectional signaling between the cytoskeleton and integrins. Curr Opin Cell Biol 11: 274–286 [DOI] [PubMed] [Google Scholar]
- Shattil SJ, Hoxie JA, Cunningham M, Brass LF (1985) Changes in the platelet membrane glycoprotein Iib.IIIa complex during platelet activation. J Biol Chem 260: 11107–11114 [PubMed] [Google Scholar]
- Shattil SJ, Ginsberg MH, Brugge JS (1994) Adhesive signaling in platelets. Curr Opin Cell Biol 6: 695–704 [DOI] [PubMed] [Google Scholar]
- Thrasher AJ (2002) WASp in immune-system organization and function. Nat Rev Immunol 2: 635–646 [DOI] [PubMed] [Google Scholar]
- Tsuboi S (2002) Calcium integrin-binding protein activates platelet integrin αIIbβ3. J Biol Chem 277: 1919–1923 [DOI] [PubMed] [Google Scholar]
- Wiskott A (1937) Familiarer, angeborener Morbus Welhofii? Monatsschr Kinderheilkd 68: 212–216 [Google Scholar]
- Zhu Q, Watanabe C, Liu T, Hollenbaugh D, Blaese RM, Kanner SB, Aruffo A, Ochs HD (1997) Wiskott–Aldrich syndrome/X-linked thrombocytopenia: WASP gene mutations, protein expression, and phenotype. Blood 90: 2680–2689 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information