

Abstract
Binding of high risk human papillomavirus (HPV) E6 protein to E6-associated protein (E6AP), a cellular ubiquitin-protein ligase, enables E6AP to ubiquitinate p53, leading to p53 degradation in cervical cancer cells such as HeLa cells. Here we report that Pitx2a, a bicoid-type homeodomain transcription factor, can bind to HPV E6 protein and inhibit E6/E6AP-mediated p53 degradation. Deletion of the Pitx2a homeodomain abrogates its ability to bind to HPV E6 protein and to induce p53 accumulation in HeLa cells, suggesting that the homeodomain of Pitx2a is essential for inhibition of E6/E6AP-mediated p53 degradation. Recombinant Pitx2a can also block E6/E6AP-mediated p53 degradation in vitro, indicating that this function of Pitx2a is independent of its transcription activity. Pitx2a does not regulate Hdm2-mediated p53 degradation, because Pitx2a does not affect p53 protein levels in HPV-negative cells, such as HCT116, U2OS, and C33A cells. In addition, Pitx2a-induced p53 is transcriptionally active and maintains its specific DNA binding activity in HeLa cells. Taken together, these findings suggest that, by binding to E6, Pitx2a interferes with E6/E6AP-mediated p53 degradation, leading to the accumulation of functional p53 protein in HeLa cells.
Pitx2 is a bicoid-type homeodomain transcription factor, which was originally identified as a candidate gene for Rieger syndrome, an autosomal dominant genetic disease characterized by craniofacial dysmorphologies as well as defects in the heart, limb, and pituitary (1). Ablation of Pitx2 in mice resulted in abnormal left-right development, such as lung isomerism, as well as defects in the heart, teeth, pituitary, and eyes (2–5). Pitx2 has also been suggested as an important regulator of GABAergic neuron differentiation (6, 7), as well as a downstream target for the acute leukemia ALL1 gene (8). Recently, Pitx2 was identified as a component of the Wnt signaling pathway, controlling cell proliferation in a tissue-specific manner via regulation of its downstream target genes, such as cyclin D1, D2, and Myc (9, 10). Therefore, Pitx2 is likely to execute its multibiological functions by regulating cell proliferation and differentiation.
Infection by high risk human papilloma viruses (HPVs),2 such as HPV types 16 and 18, is associated with >90% of cervical carcinomas, the second leading cause of cancer-related deaths among women worldwide. HeLa cells, derived from cervical carcinoma, express wild-type p53 but only at a very low level, because p53 is targeted for ubiquitination and degradation by HPV E6 protein. Each HPV E6 protein consists of ~150 amino acids and has a molecular mass of ~18 kDa. E6 is the initiator of p53 degradation in cervical cancer cells. Integration of HPVs into the genome and continuous expression of HPV E6 protein are required for the maintenance of the transformed phenotype of cell lines derived from cervical carcinoma. Binding of HPV E6 protein to E6-associated protein (E6AP), a cellular ubiquitin-protein ligase, enables E6AP to ubiquitinate p53, leading to p53 degradation. E6/E6AP-mediated p53 degradation was believed to be the major mechanism leading to cervical carcinoma associated with high risk HPVs (11–14). However, it has been shown recently that the PDZ domain of E6 is responsible for the development of cancer in transgenic mice (15), indicating that not only does E6-mediated p53 degradation, but also E6-PDZ partner interaction, play critical roles in the development of HPV E6-mediated cancer.
We have previously reported that expression of Pitx2a in HeLa cells resulted in the accumulation of p53 and p21, leading to cell cycle arrest at G1/G0 (16). Here we demonstrate that Pitx2a can bind to HPV type 18 E6 protein and inhibit p53 degradation in vitro. The homeodomain of Pitx2a is important for Pitx2a-induced p53 accumulation as well as the E6-Pitx2a interaction in HeLa cells. Pitx2a-induced p53 is transcriptionally active and maintains its specific DNA binding activity. These findings indicate that binding of Pitx2a to HPV type 18 E6 protein interferes with E6/E6AP-mediated p53 degradation, leading to the accumulation of functional p53 protein and the induction of cell cycle arrest in HeLa cells.
MATERIALS AND METHODS
Cell Culture and Transfection
The inducible HeLa cell line expressing GFP-tagged Pitx2a has been described previously (16). We will refer to this stable cell line as Pitx2a cells in this article. The Pitx2a cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum in the presence of hygromycin B and geneticin (each 100 μg/ml) (Invitrogen). The Pitx2a cells express GFP-Pitx2a in the presence (but not in the absence) of doxycycline. The HeLa Tet-On cells (BD Biosciences) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum in the presence of geneticin (100 μg/ml) (both from Invitrogen). The expression plasmids were transfected into HeLa Tet-On cells using a Lipofectamine 2000 kit (Invitrogen) according to the manufacturer’s instructions.
Plasmids
GFP-Pitx2a plasmid has been described previously (16). Pitx2aΔN-(39–271) and Pitx2aΔHD-(99–271) fragments were generated by PCR using GFP-Pitx2a as a template. Both fragments were then inserted into pEGFP-C3 and pCMV-Tag3B (Myc-tagged vector) to generate pEGFP-Pitx2aΔN, pEGFP-Pitx2aΔHD, pCMV-Tag3B-Pitx2aΔN, and pCMV-Tag3B-Pitx2aΔHD, respectively. Hemagglutinin-tagged E6AP II plasmids were kindly provided by Dr. P. M. Howley (17). E6AP II full-length cDNA was also cloned into pEGFP-C3 (BD Biosciences) to generate GFP-tagged E6AP II. The DNA fragment encoding HPV type 18 E6 protein was cloned into pCS3+MT (provided Dr. Y. S. Kim, NHLBI, National Institutes of Health) and pEGFP-C3 (BD Biosciences) to generate Myc-tagged and GFP-tagged HPV 18 E6, respectively. pcDNA3-F:11E6 was kindly provided by Dr. Cheng-Ming Chiang (18).
p53 RNA interference (RNAi) expression plasmid was purchased from Imgenex Corporation (San Diego, CA). Mouse Pitx2a RNAi expression plasmid was constructed using the “PCR-SHAGging” strategy as described in katahdin.cshl.org:9331/RNAi/html/rnai.html (19, 20). The DNA sequence for the mouse Pitx2a RNAi is 5′-GAGAGGACAGGGGATTGACGTTCATGGAGGAAGCTTGCTCCGTGGACGTCGATCCCTTGTCCTCTC-3′ (the underlined sequence will become the loop when the short hairpin is formed).
The human E6AP small interfering RNA oligo was purchased from Dharmacon (Chicago, IL). The sequence for its sense oligo is 5′-CUUUCUCAAUGCACUUGUAUU-3′. Transfection of E6AP small interfering RNA into U2OS cells was done using a Lipofectamine 2000 kit (Invitrogen) according to the manufacturer’s instructions.
GST Fusion Proteins and in Vitro Translated Proteins
cDNA fragments encoding mouse Pitx2a was cloned into pGEX-5x-1 expression vector, and the GST-Pitx2a protein was expressed in BL21 cells. Baculoviruses expressing GST-E6AP and GST-p53 were purchased from Orbigen, Inc. (San Diego, CA). The GST fusion proteins were purified using glutathione beads (Sigma). Myc-E6 was translated in vitro using a TnT kit (Promega, Madison, WI), and the in vitro translated proteins were confirmed by immunoblot analysis using anti-Myc (9E10) antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
In Vitro Binding Assay
GST-Pitx2a immobilized on glutathione agarose beads was incubated with 5 μl of in vitro translated HPV type 18 E6 rabbit reticulocyte lysate in binding buffer (0.15 m NaCl, 50 mm Tris, pH 7.4, 1% Nonidet P-40, 1.0 mm dithiothreitol, and 0.5 mm phenylmethylsulfonyl fluoride) at 4 °C overnight. The glutathione-agarose beads were then washed five times with binding buffer. The washed glutathione-agarose beads were boiled in SDS loading buffer for 10 min, and proteins were resolved on 10% SDS-polyacrylamide gels, followed by immunoblot to detect Myc-tagged HPV E6 protein using Myc (9E10) antibody (Santa Cruz).
Co-immunoprecipitation and Immunoblot Analysis
The transfected cells were lysed in radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, leupeptin, and pepstatin) on ice for 10 min. The cell lysates were clarified at 11,750 × g at 4 °C for 12 min and were incubated with protein G-agarose at 4 °C for 30 min, followed by collection of the supernatant. Antibodies were added to the supernatant and incubated at 4 °C for 2 h followed by the addition of protein G-agarose. After another 12 h of incubation at 4 °C, the beads were washed in radioimmune precipitation assay buffer four times, boiled in SDS loading buffer, and proteins were resolved on 10% SDS-PAGE. Proteins were then transferred to an Immobilon-P transfer membrane (Millipore, Bedford, MA), blocked with 5% nonfat milk for 1 h at 23 °C, and incubated with primary antibodies overnight at 4 °C, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (1:5000; Santa Cruz Biotechnology) for 1 h at 23 °C. The blots were visualized by SuperSignal West Pico Luminol/Enhancer solution (Pierce).
The following primary antibodies were used: polyclonal rabbit antibody to GFP (1:1000, Santa Cruz Biotechnology); polyclonal rabbit antibody to E6AP (1:1000, Upstate Biotechnology); monoclonal antibodies to p53 (DO-1, 1:1000, Santa Cruz Biotechnology), and Myc (9E10, 1:1000, Santa Cruz Biotechnology); polyclonal goat antibody to p53 (1:1000, Santa Cruz Biotechnology); and polyclonal rabbit antibody to Pitx2 (21).
p53 in Vitro Degradation Assay
p53 degradation assay was performed as described previously (22). Briefly, in vitro translated Myc-E6 and GST-p53 were incubated at 23 °C for 0 and 90 min with or without GST-Pitx2a. 10 μg of ubiquitin was also included in the degradation assay mix. The incubation was done in 25 mm Tris (pH 7.5), 100 mm NaCl, and 3 mm dithiothreitol. The mixed proteins were resolved on a 10% SDS-polyacrylamide gel, followed by immunoblot analysis to assess the extent of p53 degradation.
Luciferase Assay
The Pitx2a cells were transfected with reporter plasmids p53-TA-Luc or ppTA-Luc (BD Biosciences), together with the β-galactosidase expression plasmid. 24 h after the transfection, the transfected cells were split and cultured for another 24 h in the absence or presence of doxocycline. The cells were lysed in reporter lysis buffer, followed by centrifugation to remove the cell debris. The luciferase activity was assessed using a TR717 microplate luminometer (Tropix). The light units were normalized by β-galactosidase activity. The p53 response element in p53-TA-Luc is as follows: 5′-ACGTTTGCCTTGCCTGGACTTGCCTGGCCTTGCCTTGGACATGCCCGGGCTGTC-3′.
Assay of p53 Binding to Its Consensus Site
The Pitx2a cells were cultured in the presence or absence of doxycycline for 2 days. The cells were lysed in binding buffer (10 mm Tris, pH 7.5, 50 mm NaCl, 1 mm dithiothreitol, 1 mm EDTA, 5% glycerol, 1 μg/ml poly(dI-dC)) and clarified by centrifugation to remove cell debris. The clarified supernatant was incubated with agarose-conjugated double-stranded DNA oligos with wild-type or mutant p53 consensus binding sites at 4 °C overnight. The agarose beads were washed with binding buffer five times and boiled in SDS loading buffer, and the eluted proteins were resolved on 10% SDS-polyacrylamide gels, followed by immunoblot analysis using an antibody specific for p53 (Santa Cruz Biotechnology).
RESULTS
We previously generated a stable HeLa cell line inducibly expressing GFP-tagged Pitx2a. The cell line expresses Pitx2a in the presence (but not in the absence) of doxycycline (16). We will refer to this stable cell line as Pitx2a cells in this article. As shown in Fig. 1A, expression of Pitx2a in HeLa cells resulted in the accumulation of p53. In addition, transient expression of GFP-Pitx2a (Fig. 1B, a–c) (but not GFP-vector (Fig. 1B, d–f)) in HeLa cells induced p53 accumulation in the nucleus.
FIGURE 1. Effect of Pitx2a on the expression levels of p53, E6AP, E6, and p21 in HeLa cells.

A, inducible expression of GFP-Pitx2a in HeLa cells induces p53 accumulation. Immunoblot analysis for the protein levels of p53, GFP-Pitx2a, and β-tubulin using specific antibodies. Note that the addition of doxycyclin (+Dox) leads to the expression of GFP-Pitx2a and the accumulation of p53 protein. B, transient expression of GFP-Pitx2a (a– c), but not GFP-vector (d–f), in HeLa cells induces p53 accumulation in the nucleus. Scale bar, 20 μm. C, reverse transcription-PCR analysis of p53, p21, E6AP, and E6 mRNA expression in the presence (+Dox) or absence (−Dox) of doxycycline. D, Northern-blot analysis of E6AP mRNA expression in the presence or absence of doxycycline.
Because Pitx2a is a transcription factor, we also tested whether Pitx2a up-regulated p53 expression at the transcriptional level. Total RNA was isolated from the Pitx2a cells cultured in the presence or absence of doxycycline, and reverse transcription-PCR was performed to examine the p53 mRNA level. The result showed that the p53 mRNA level was not significantly affected by Pitx2a, suggesting that Pitx2a does not regulate p53 expression at the transcriptional level (Fig. 1C). This result is consistent with the notion that the p53 level is mainly regulated by ubiquitination and the proteasome-dependent proteolysis pathway (23). In addition, the mRNA level for one of the p53 downstream target genes, p21, was up-regulated in the presence of doxycycline (Fig. 1C).
High risk HPV E6 protein and ubiquitin-protein ligase E6AP play an important role in the regulation of p53 degradation in HPV-positive cells such as HeLa cells. In the presence of HPV E6 protein, E6AP acts as a p53 ubiquitin-protein ligase to target p53 protein for degradation (24). It is therefore conceivable that down-regulation of E6AP or E6 would result in p53 accumulation in HeLa cells. Pitx2a is a transcription factor. We therefore asked whether Pitx2a could down-regulate the expression of HPV E6 and E6AP in HeLa cells. Reverse transcription-PCR indicated that E6 and E6AP mRNA levels in HeLa cells were not significantly affected by Pitx2a (Fig. 1C). Northern blot also revealed that the E6AP mRNA level did not significantly change (Fig. 1D). Therefore, expression of Pitx2a in HeLa cells did not affect the mRNA levels of E6AP and HPV E6, suggesting that Pitx2a-induced p53 accumulation was not because of the repression of E6 and E6AP transcription.
HPV E6 can bind to E6AP, forming an E6·E6AP complex, which in turn binds to and ubiquitinates p53 (24). Recently, it was also shown that E6 protein can bind to p53 in an E6AP-independent fashion to regulate p53-dependent gene activation. Interestingly, this E6-p53 interaction was not involved in p53 degradation (18). Expression of Pitx2a in HeLa cells did not affect the mRNA levels of HPV E6 and E6AP (Fig. 1, C and D). We therefore hypothesized that Pitx2a could bind to HPV 18 E6 in HeLa cells, interfering with E6/E6AP-mediated p53 ubiquitination and degradation. As indicated in Fig. 2A, immobilized GST-Pitx2a protein (but not GST alone) could pull down the in vitro translated Myc-tagged HPV E6 protein (compare lane 2 with lane 3). This in vitro E6-Pitx2a interaction was also confirmed by a co-immunoprecipitation experiment. As shown in Fig. 2B, Myc-E6 could pull down GFP-Pitx2a in transfected HeLa cells (compare lane 2 with lane 3). Additionally, the HPV E6-Pitx2a interaction was further supported by the similar intra-cellular localization of HPV E6 and Pitx2a proteins. GFP-tagged Pitx2a was only expressed in the nucleus, and GFP-tagged HPV 18E6 also localized predominantly to the nucleus of this HeLa cell line (data not shown), consistent with previous reports (21, 25, 26). Another possibility is that Pitx2a can bind to E6AP and directly inhibit the activity of E6AP ubiquitin-protein ligase toward p53. To test this possibility, we transfected GFP-E6AP into Saos-2 cells, and GST pull-down assay was performed to test whether GST-Pitx2a interacted with GFP-E6AP from transfected Saos-2 cells. The Saos-2 cell line was used because it does not express E6 and p53 proteins. We were not able to detect the interactions between GST-Pitx2a and GFP-tagged full-length E6AP in Saos-2 cells (Fig. 2C), suggesting that Pitx2a did not interact with full-length E6AP protein.
FIGURE 2. Pitx2a can binds to HPV and E6 but not E6AP.
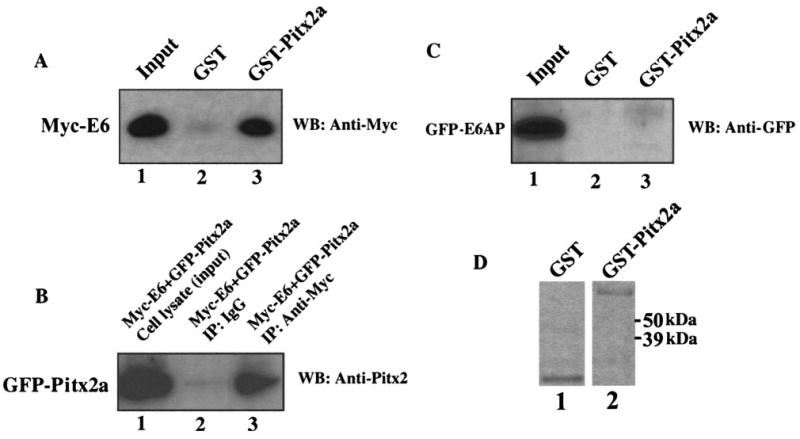
A, immobilized GST-Pitx2a can pull down in vitro translated Myc-tagged E6 protein. B, plasmid encoding GFP-Pitx2a was co-transfected with plasmid encoding Myc-E6. Co-immunoprecipitation (IP) was done using anti-Myc antibody, followed by immunoblot (WB) analysis using antibody specific for Pitx2. C, immobilized GST-Pitx2a cannot pull down GFP-E6AP protein from transfected Saos-2 cell lysate. D, Coomassie Blue staining gel of GST and GST-Pitx2a proteins used in A and C.
The Pitx2a homeodomain contains both DNA binding and transcriptional activation domains, which can bind to bicoid DNA binding sites and regulate the transcription of its target genes. We therefore asked whether the homeodomain is required for Pitx2a-induced p53 accumulation. We deleted amino acids 1–38 and 1–98 from the amino terminus of Pitx2a protein, generating truncated Pitx2a polypeptides, Pitx2aΔN (containing amino acids 39–271, with the homeodomain) and Pitx2aΔHD (containing amino acids 99–271, without the homeodomain) (Fig. 3A). The expression plasmids encoding Pitx2a, Pitx2aΔN, and Pitx2aΔHD polypeptides were transfected into HeLa cells, and the p53 protein level was examined 24 h following the transfection. As shown in Fig. 3B, deletion of 38 amino acids from the amino terminus of Pitx2a, i.e. Pitx2aΔN, did not affect the ability of Pitx2a to induce p53 accumulation in HeLa cells. However, deletion of 98 amino acids (including the homeodomain) from the amino terminus of Pitx2a, i.e. Pitx2aΔHD, almost completely abrogated the ability of Pitx2a to induce p53 accumulation, suggesting that the homeodomain of Pitx2a is essential for Pitx2a-induced p53 accumulation.
FIGURE 3. The homeodomain of Pitx2a is essential for p53 accumulation.

A, schematic diagrams for GFP-tagged full-length Pitx2a (GFP-Pitx2a) and truncated Pitx2a polypeptides (GFP-Pitx2aΔN and Pitx2aΔHD). The number represents the amino acids in Pitx2a. B, HeLa cells were transfected with vector alone (lane 1) and plasmids encoding GFP-Pitx2a (lane 2), GFP-Pitx2aΔN (lane 3), and GFP-Pitx2aΔHD (lane 4). Immunoblot analysis of the transfected cell lysates was conducted to assess the p53 protein levels. β-tubulin was used as a loading control. C, plasmids encoding Myc-Pitx2a (lane 1), Myc-Pitx2aΔN (lane 2), and Myc-Pitx2aΔHD (lane 3) were co-transfected with plasmid encoding GFP-E6, respectively. Co-immunoprecipitation was done using anti-Myc antibody, followed by immunoblot analysis using antibody specific for GFP.
We then asked whether the homeodomain of Pitx2a is essential for its binding to HPV E6. Myc-tagged full-length Pitx2a, Pitx2aΔN, and Pitx2aΔHD were cotransfected into HeLa cells with GFP-tagged HPV E6, respectively. As shown in Fig. 3C, a co-immunoprecipitation experiment indicated that Myc-tagged full-length Pitx2a and Pitx2aΔN (but not Pitx2aΔHD) could bind to GFP-tagged HPV E6. Therefore, deletion of the Pitx2a homeodomain abrogated its ability to bind to HPV E6 and to induce p53 accumulation, suggesting that binding to HPV E6 is likely the cause for Pitx2a to induce p53 accumulation in HeLa cells.
HPV-positive cells, such as HeLa cells, use E6AP as an ubiquitin-protein ligase to ubiquitinate p53, resulting in a low level of p53 protein. However, HPV-negative cells, such as HCT116, U2OS, and C33A, use ubiquitin-protein ligase Hdm2 to ubiquitinate p53 to maintain an appropriate p53 level. Hdm2-mediated p53 degradation is not operating in HPV-positive cervical cancer cells (27). We therefore asked whether Pitx2a could regulate Hdm2-mediated p53 degradation in HPV-negative cells. GFP-tagged full-length Pitx2a, Pitx2aΔN, and Pitx2aΔHD were transfected into HCT116, U2OS, and C33A cells. p53 protein levels were examined 24 h after the transfection. Both full-length and truncated Pitx2a did not affect p53 protein levels in HCT116 (Fig. 4A), U2OS, and C33A (data not shown) cells, indicating that Pitx2a did not regulate Hdm2-mediated p53 degradation. In contrast, nutlin-3, an inhibitor of Hdm2, could significantly induce p53 accumulation in U2OS cells (Fig. 4B).
FIGURE 4. Effect of Pitx2a on Hdm2- and E6-mediated p53 degradation.
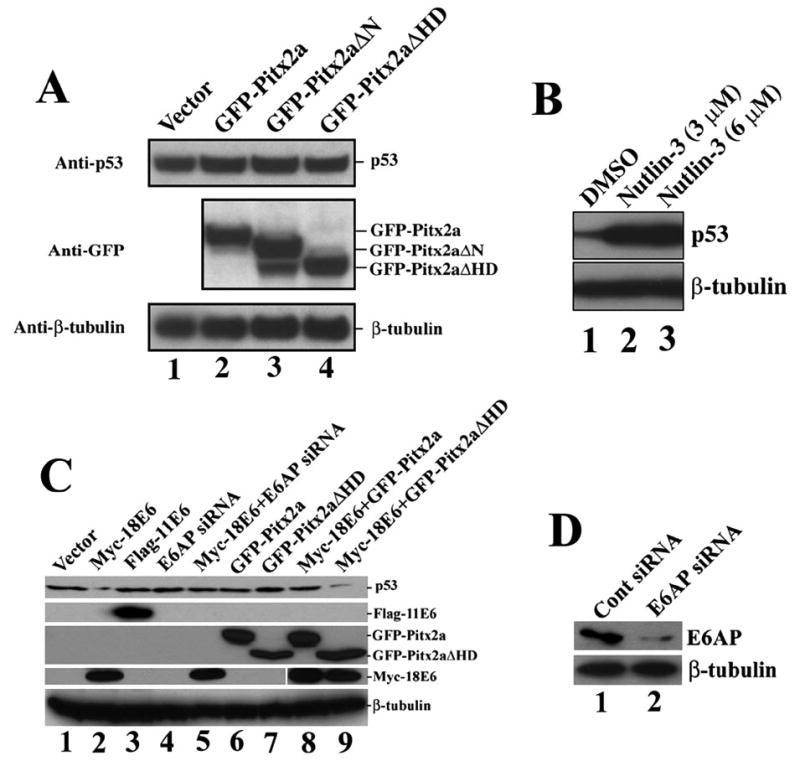
A, Pitx2a did not induce p53 accumulation in HCT116 cells. Vector alone (lane 1) and plasmids encoding GFP-Pitx2a (lane 2), GFP-Pitx2aΔN (lane 3), and GFP-Pitx2aΔHD (lane 4) were transfected into HCT116 cells. Immunoblot analysis was done to assess the p53 protein levels by using an antibody specific for p53. GFP-Pitx2a, GFP-Pitx2aΔN, and GFP-Pitx2aΔHD were detected using an antibody specific for GFP. β-tubulin was detected using antibody specific for -tubulin and was a control to assure equal loading of all lanes. B, Nutlin-3 induced p53 accumulation in U2OS cells. U2OS cells were cultured in the presence of Me2SO (DMSO, lane 1) or nutlin-3 (lanes 2 and 3) for 24 h. The p53 protein level was detected using an antibody specific for p53. C, Pitx2a (but not Pitx2aΔHD) inhibited E6/E6AP-mediated p53 degradation. The indicated plasmids were transfected into U2OS cells. 48 h after transfection, p53 protein level was determined using a specific antibody for p53. Note that expression 18 E6 induced p53 degradation (lane 2), and this p53 degradation was inhibited by E6AP RNAi (lane 5) and Pitx2a (lane 8) but not Pitx2aΔHD (lane 9). The FLAG-11E6, GFP-Pitx2a/Pitx2aΔHD, Myc-18E6, and β-tubulin were detected using antibodies specific for FLAG, GFP, Myc, and β-tubulin, respectively. D, E6AP RNAi reduced E6AP protein in U2OS cells. U2OS cells were transfected with E6AP small interfering RNA. 48 h after transfection, the E6AP protein level was determined using an antibody specific for E6AP.
To further elucidate the relationships between Pitx2a, E6AP, and E6 in the regulation of p53 degradation, we examined whether Pitx2a and its truncated form, Pitx2aΔHD, could inhibit p53 degradation induced by E6 protein in U2OS cells. Consistent with previous reports (28), expression of HPV type 18 (but not type 11) E6 proteins induced p53 degradation in U2OS cells (Fig. 4C, lanes 2 and 3). As expected, E6-mediated p53 degradation was abrogated by reduction of E6AP using RNAi (Fig. 4C, lane 5), confirming E6-mediated p53 degradation was dependent on E6AP. In addition, expression of Pitx2a (but not Pitx2aΔHD) inhibited p53 degradation mediated by E6/E6AP (Fig. 4C, compare lane 8 with lane 9). These experiments further confirmed that Pitx2a could inhibit E6/E6AP-mediated p53 degradation.
Pitx2a is a homeodomain transcription factor, and its homeodomain is essential for p53 accumulation induced by Pitx2a. We therefore asked whether the transcription activity of Pitx2a is required for p53 accumulation in HeLa cells. To answer this question, we performed an in vitro p53 degradation assay. We incubated GST-p53 and in vitro translated HPV 18 E6 in the presence or absence of Pitx2a. It is not necessary to add E6AP, because the rabbit reticulocyte lysate provides functional E6AP (29). As shown in Fig. 5, in the absence of Pitx2a, p53 protein levels significantly decreased after 90 min of incubation at 23 °C (compare lane 1 with lane 3). In contrast, in the presence of Pitx2a, p53 protein levels did not significantly change (Fig. 5, compare lane 2 with lane 4), suggesting that Pitx2a was able to directly inhibit E6/E6AP-mediated p53 degradation. This result also further confirmed that binding of Pitx2a to HPV E6 and the subsequent interference with ubiquitination and degradation of p53 by the E6·E6AP complex is the cause of p53 accumulation induced by Pitx2a in HeLa cells. Importantly, there were no transcriptional and translational events in this assay system, suggesting that inhibition of p53 degradation by Pitx2a was independent of Pitx2a transcriptional activity.
FIGURE 5. Effect of Pitx2a on p53 degradation in vitro.

GST-p53 and in vitro translated Myc-E6 were incubated at 23 °C in the presence (lanes 2 and 4) or absence (lanes 1 and 3) of GST-Pitx2a for 0 or 90 min. Immunoblot analysis was done to assess the p53 protein level. Note that rabbit reticulocyte lysate contains active E6AP (29).
The tumor suppressor function of p53 depends on its ability to activate the transcription of its downstream target genes, such as p21/cip1, GADD45, NOXA, Bax, and 14-3-3 (30). It has been demonstrated that p53-mediated growth suppression is critically dependent on the transcriptional activation of p21/cip1 by p53 (23). We therefore examined whether p21 was up-regulated at the transcription level by Pitx2a. Reverse transcription-PCR revealed that the p21 mRNA level significantly increased when the Pitx2a cells were cultured in the presence of doxycycline (Fig. 1C), indicating that Pitx2a-induced p53 was most likely functional. To further confirm this result, we also examined the transcriptional activity of the accumulated p53 by using a p53-responsive luciferase reporter plasmid, p53-TA-Luc, in which a p53-responsive element was fused to the luciferase reporter gene. Functional p53 protein was expected to activate the transcription of the luciferase reporter gene from p53-TA-Luc. As shown in Fig. 6A, expression of Pitx2a (in the presence of doxycycline) significantly increased the luciferase activity from the p53-responsive luciferase reporter plasmid, p53-TA-Luc, but did not affect the luciferase activity from a control plasmid without the p53-responsive element pTA-Luc. This result suggested that Pitx2a-induced p53 is functional.
FIGURE 6. Pitx2a-induced p53 is functional.
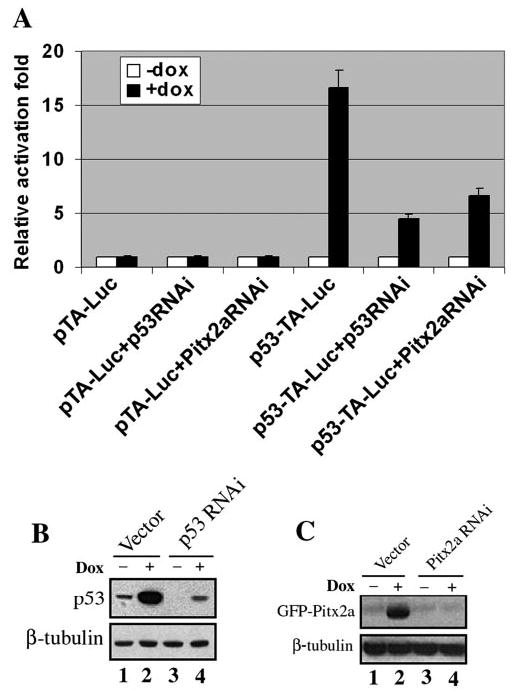
A, pTA-Luc alone, pTA-Luc plus p53 RNAi or Pitx2a RNAi, p53-TA-Luc alone, and p53-TA-Luc plus p53 RNAi or Pitx2a RNAi, were transfected into the Pitx2a cells, respectively. 24 h after the transfection, the cells were split and cultured in the presence or absence of doxycycline for another 24 h. The luciferase activity was measured according to the manufacturer’s instructions. pTA-Luc, control luciferase reporter plasmid; p53-TA-Luc, p53-responsive luciferase reporter plasmid. B, vector alone (lanes 1 and 2) and plasmid expressing p53 RNAi were transfected into Pitx2a cells. 24 h after the transfection, the cells were split and cultured in the presence (lanes 2 and 4) or absence (lanes 1 and 3) of doxycycline for another 24 h. Immunoblot analysis was done to assess the p53 protein level. C, vector alone (lanes 1 and 2) and plasmid expressing Pitx2a RNAi were transfected into Pitx2a cells. 24 h after the transfection, the cells were split and cultured in the presence (lanes 2 and 4) or absence (lanes 1 and 3) of doxycycline for another 24 h. Immunoblot analysis was done to assess the GFP-Pitx2a protein level. β-tubulin was used as a loading control.
These results were further confirmed by RNAi experiments. Both p53 and Pitx2a RNAi significantly reduced activation of the p53-responsive promoter by Pitx2a in the presence of doxycycline (Fig. 6A). To confirm that p53 and Pitx2a RNAi could down-regulate the expression of p53 and Pitx2a, we transfected the Pitx2a cells with the respective RNAi plasmids, followed by immunoblot analysis to detect the protein levels of p53 and Pitx2a. As shown in Fig. 6, B and C, p53 and Pitx2a RNAi significantly knocked down p53 and Pitx2a proteins, respectively.
We then asked whether the accumulated p53 could bind to its consensus DNA binding site. p53 is a transcription factor containing a DNA binding domain, which can bind to its consensus binding site on the promoter region and regulate the transcription of its downstream target genes. We used agarose-conjugated double-stranded DNA oligos containing the p53 consensus binding site to purify p53 from Pitx2a cells cultured in the presence or absence of doxycycline. If the accumulated p53 can bind to its consensus site, the agarose-conjugated p53 oligos should be able to enrich p53 from the cell lysates. As shown in Fig. 7, p53 protein was enriched by the agarose-conjugated double-stranded DNA oligo containing the wild-type p53 consensus binding site. In contrast, the agarose-conjugated oligo containing a mutated p53 binding site was not able to enrich p53 protein induced by Pitx2a (Fig. 7, compare lane 4 with lane 6). This result suggested that Pitx2a-induced p53 maintained its DNA binding activity. This is also consistent with our previous results that indicated that the expression of Pitx2a in HeLa cells induced the accumulation of p21, a p53 downstream target gene, as well as cell cycle arrest at G1/G0 (16).
FIGURE 7. Pitx2a-induced p53 maintains its DNA binding activity.

The addition of doxycycline (Dox) induced p53 accumulation (compare lane 1 with lane 2) in the Pitx2a cells. The accumulated p53 was enriched by an agarose-conjugated double-stranded DNA oligo containing the p53 binding site (lane 4) but was not enriched by an agarose-conjugated double-stranded DNA oligo containing a mutated (mt) p53 binding site (compare lane 4 with lane 6). wt, wild type.
DISCUSSION
We have demonstrated in this article that Pitx2a, a homeodomain transcription factor, can bind to high risk HPV type 18 E6 protein and subsequently interfere with E6/E6AP-mediated p53 degradation, leading to p53 accumulation in HeLa cells. The homeodomain of Pitx2a is essential for both the Pitx2a-HPV E6 interaction and the induction of p53 accumulation in HeLa cells. The accumulation of p53 induced by Pitx2a is independent of Pitx2a transcription activity. In addition, Pitx2a-induced p53 is functional and maintains its DNA binding activity.
HPV E6·E6AP complex formation is required for p53 degradation in cervical cancer cells (11–14). Our results showed that Pitx2a could bind to the E6 protein and interfere with E6/E6AP-mediated p53 degradation in HeLa cells. Deletion of the Pitx2 homeodomain disrupted both the Pitx2a-E6 interaction and the induction of p53 accumulation in HeLa cells. We therefore speculate that there was a link between Pitx2a-E6 interaction and the inhibition of E6/E6AP-mediated p53 degradation, i.e. the Pitx2a-E6 interaction interferes with E6/E6AP-mediated p53 degradation, leading to p53 accumulation in HeLa cells. We do not know at present how the Pitx2a-E6 interaction leads to the inhibition of E6/E6AP-mediated p53 degradation. However, two possibilities must be considered. First, binding of Pitx2a to E6 protein can block the further interactions between HPV E6 and E6AP. Therefore, E6AP, without binding to HPV E6, will not be able to degrade p53 protein. Second, binding of Pitx2a to E6 protein can cause a conformational change of the E6·E6AP complex, which subsequently loses its ability to degrade p53. We favor the first possibility, because we were not able to detect the formation of the trimolecular complex E6·E6AP·Pitx2a (data not shown).
In addition to E6AP, many interacting partners for high risk HPV E6 protein have been identified, such as p300/CBP (31, 32), Bak (33), E6BP/ERC55 (34), E6TP1 (35), interferon regulatory factor-3 (36), Paxillin (37, 38), hDlg (39, 40), MUPP-1 (41), and hScrib (40). Binding of HPV E6 protein to these interacting partners results in different biological effects, such as inhibition of apoptosis (Bak), disruption of the actin cytoskeleton (Paxillin), and inhibition of β-interferon transcription (interferon regulatory factor-3). Only a low level of HPV E6 expression was present in cervical cancer cells, but this amount was sufficient to maintain the transformation phenotype. Do these E6-interacting partners compete with each other for the limited amount of HPV E6 protein in cervical cancer cells? For instance, does binding of p300/CBP to HPV E6 interfere with E6·E6AP·p53 complex formation? This question remains to be answered. At least, however, it has not been previously reported that these E6 interacting partners had an effect on E6/E6AP-mediated p53 degradation in HPV E6-positive cells. We have demonstrated in this article that, not only can Pitx2a bind to HPV E6 protein, but it also can inhibit E6/E6AP-mediated p53 degradation.
High risk HPVs play a critical role in the development of cervical carcinoma. HPV E6 protein is sufficient to induce immortalization of a number of human normal epithelial cell types. In addition, continuous expression of HPV E6 protein is required for the maintenance of the transformed phenotype of cervical cancer cells. Degradation of p53 by E6/E6AP in HPV-positive cells is the underlying mechanism leading to cervical carcinoma associated with high risk HPVs (11–14). Therefore, HPV E6 is an ideal target for molecular anti-cancer strategies to cervical carcinoma. Many approaches, such as antisense oligos and ribozymes directed against HPV E6 mRNA, have been examined to block the expression of HPV E6 protein in HPV-positive cervical cancer cells (42–47). All of these approaches have been shown to inhibit the expression of HPV E6 mRNA and protein, leading to cell growth suppression and a reduction of tumorigenicity in vivo. In addition, blocking E6 activity by using a therapeutic peptide was shown to induce apoptosis of HPV-positive cancer cells and to inhibit E6-induced degradation of its cellular substrates, such as p53, Dlg, and the membrane-associated guanylate kinase inverted family of proteins (48, 49).
Pitx2a, a bicoid-type homeodomain transcription factor, can bind to HPV E6 protein and inhibit p53 degradation in HPV-positive HeLa cells, resulting in p53 accumulation and cell cycle arrest at G1/G0 (16). Pitx2 was also originally isolated as a downstream target for the human acute leukemia ALL1 gene. Loss of function of the ALL1 gene has been implicated in the development of human acute leukemia associated with abnormalities at 11q23 (50). Pitx2 is expressed in normal human bone marrow and leukemic cell lines with a normal ALL1 allele but is not expressed in the leukemic cell lines in which ALL1 is rearranged (8). Additionally, both Pitx2 mRNA and protein are up-regulated by treatment with lithium chloride and expression of constitutively active β-catenin, suggesting that the Pitx2 level is regulated by the Wnt signaling pathway (9). Although the physiological roles for the Pitx2a-E6 interaction remain to be determined, further dissection of the E6-binding domains on the Pitx2a molecule could help in designing strategies for the molecular therapy of cervical carcinoma.
Acknowledgments
I thank Dr. Robert S. Adelstein (NHLBI) for continuous support and critical review of the manuscript. I also thank Drs. Robert Oxford and Di Wu for excellent technical support. I am also grateful to Dr. Peter M. Howley (Harvard Medical School) for the gift of E6AP cDNA, Dr. Cheng-Ming Chiang for 11E6 plasmid, Dr. T. A. Hjalt for Pitx2 antibody, and Dr. Christian Combs (NHLBI Light Microscopy Core) for assistance with confocal microscopy.
Footnotes
This work was supported by National Institutes of Health Grants k22 HL071542-01 (to Q. W.). This is contribution 06-7-J from the Kansas Agricultural Experiment Station, Manhattan, Kansas.
The abbreviations used are: HPV, human papillomavirus; GFP, green fluorescent protein; EGFP, enhanced GFP; E6AP, E6-associated protein; GST, glutathione S-transferase; RNAi, RNA interference; Luc, luciferase.
References
- 1.Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC. Nat Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- 2.Lu MF, Pressman C, Dyer R, Johnson RL, Martin JF. Nature. 1999;401:276–278. doi: 10.1038/45797. [DOI] [PubMed] [Google Scholar]
- 3.Lin CR, Kioussi C, O’Connell S, Briata P, Szeto D, Liu F, Izpisua-Belmonte JC, Rosenfeld MG. Nature. 1999;401:279–282. doi: 10.1038/45803. [DOI] [PubMed] [Google Scholar]
- 4.Kitamura K, Miura H, Miyagawa-Tomita S, Yanazawa M, Katoh-Fukui Y, Suzuki R, Ohuchi H, Suehiro A, Motegi Y, Nakahara Y, Kondo S, Yokoyama M. Development (Camb) 1999;126:5749–5758. doi: 10.1242/dev.126.24.5749. [DOI] [PubMed] [Google Scholar]
- 5.Gage PJ, Suh H, Camper SA. Development (Camb) 1999;126:4643–4651. doi: 10.1242/dev.126.20.4643. [DOI] [PubMed] [Google Scholar]
- 6.Westmoreland JJ, McEwen J, Moore BA, Jin Y, Condie BG. J Neurosci. 2001;21:6810–6819. doi: 10.1523/JNEUROSCI.21-17-06810.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin DM, Skidmore JM, Fox SE, Gage PJ, Camper SA. Dev Biol. 2002;252:84–99. doi: 10.1006/dbio.2002.0835. [DOI] [PubMed] [Google Scholar]
- 8.Arakawa H, Nakamura T, Zhadanov AB, Fidanza V, Yano T, Bullrich F, Shimizu M, Blechman J, Mazo A, Canaani E, Croce CM. Proc Natl Acad Sci U S A. 1998;95:4573–4578. doi: 10.1073/pnas.95.8.4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kioussi C, Briata P, Baek SH, Rose DW, Hamblet NS, Herman T, Ohgi KA, Lin C, Gleiberman A, Wang J, Brault V, Ruiz-Lozano P, Nguyen HD, Kemler R, Glass CK, Wynshaw-Boris A, Rosenfeld MG. Cell. 2002;111:673–685. doi: 10.1016/s0092-8674(02)01084-x. [DOI] [PubMed] [Google Scholar]
- 10.Baek SH, Kioussi C, Briata P, Wang D, Nguyen HD, Ohgi KA, Glass CK, Wynshaw-Boris A, Rose DW, Rosenfeld MG. Proc Natl Acad Sci U S A. 2003;100:3245–3250. doi: 10.1073/pnas.0330217100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huibregtse JM, Beaudenon SL. Semin Cancer Biol. 1996;7:317–326. doi: 10.1006/scbi.1996.0041. [DOI] [PubMed] [Google Scholar]
- 12.Mantovani F, Banks L. Oncogene. 2001;20:7874–7887. doi: 10.1038/sj.onc.1204869. [DOI] [PubMed] [Google Scholar]
- 13.Munger K, Howley PM. Virus Res. 2002;89:213–228. doi: 10.1016/s0168-1702(02)00190-9. [DOI] [PubMed] [Google Scholar]
- 14.Scheffner M, Whitaker NJ. Semin Cancer Biol. 2003;13:59–67. doi: 10.1016/s1044-579x(02)00100-1. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen ML, Nguyen MM, Lee D, Griep AE, Lambert PF. J Virol. 2003;77:6957–6964. doi: 10.1128/JVI.77.12.6957-6964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei Q, Adelstein RS. Mol Biol Cell. 2002;13:683–697. doi: 10.1091/mbc.01-07-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kao WH, Beaudenon SL, Talis AL, Huibregtse JM, Howley PM. J Virol. 2000;74:6408–6417. doi: 10.1128/jvi.74.14.6408-6417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas MC, Chiang CM. Mol Cell. 2005;17:251–264. doi: 10.1016/j.molcel.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 19.Paddison PJ, Caudy AA, Hannon GJ. Proc Natl Acad Sci U S A. 2002;99:1443–1448. doi: 10.1073/pnas.032652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hjalt TA, Semina EV, Amendt BA, Murray JC. Dev Dyn. 2000;218:195–200. doi: 10.1002/(SICI)1097-0177(200005)218:1<195::AID-DVDY17>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 22.Crook T, Tidy JA, Vousden KH. Cell. 1991;67:547–556. doi: 10.1016/0092-8674(91)90529-8. [DOI] [PubMed] [Google Scholar]
- 23.Levine AJ. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 24.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 25.Tao M, Kruhlak M, Xia S, Androphy E, Zheng ZM. J Virol. 2003;77:13232–13247. doi: 10.1128/JVI.77.24.13232-13247.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lechner MS, Mack DH, Finicle AB, Crook T, Vousden KH, Laimins LA. EMBO J. 1992;11:3045–3052. doi: 10.1002/j.1460-2075.1992.tb05375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hengstermann A, Linares LK, Ciechanover A, Whitaker NJ, Scheffner M. Proc Natl Acad Sci U S A. 2001;98:1218–1223. doi: 10.1073/pnas.031470698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Talis AL, Huibregtse JM, Howley PM. J Biol Chem. 1998;273:6439–6445. doi: 10.1074/jbc.273.11.6439. [DOI] [PubMed] [Google Scholar]
- 29.Huibregtse JM, Scheffner M, Howley PM. EMBO J. 1991;10:4129–4135. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogelstein B, Lane D, Levine AJ. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 31.Zimmermann H, Degenkolbe R, Bernard HU, O’Connor MJ. J Virol. 1999;73:6209–6219. doi: 10.1128/jvi.73.8.6209-6219.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel D, Huang SM, Baglia LA, McCance DJ. EMBO J. 1999;18:5061–5072. doi: 10.1093/emboj/18.18.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas M, Banks L. Oncogene. 1998;17:2943–2954. doi: 10.1038/sj.onc.1202223. [DOI] [PubMed] [Google Scholar]
- 34.Chen JJ, Reid CE, Band V, Androphy EJ. Science. 1995;269:529–531. doi: 10.1126/science.7624774. [DOI] [PubMed] [Google Scholar]
- 35.Gao Q, Srinivasan S, Boyer SN, Wazer DE, Band V. Mol Cell Biol. 1999;19:733–744. doi: 10.1128/mcb.19.1.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ronco LV, Karpova AY, Vidal M, Howley PM. Genes Dev. 1998;12:2061–2072. doi: 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tong X, Howley PM. Proc Natl Acad Sci U S A. 1997;94:4412–4417. doi: 10.1073/pnas.94.9.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vande Pol SB, Brown MC, Turner CE. Oncogene. 1998;16:43–52. doi: 10.1038/sj.onc.1201504. [DOI] [PubMed] [Google Scholar]
- 39.Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M. Proc Natl Acad Sci U S A. 1997;94:11612–11616. doi: 10.1073/pnas.94.21.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakagawa S, Huibregtse JM. Mol Cell Biol. 2000;20:8244–8253. doi: 10.1128/mcb.20.21.8244-8253.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SS, Glaunsinger B, Mantovani F, Banks L, Javier RT. J Virol. 2000;74:9680–9693. doi: 10.1128/jvi.74.20.9680-9693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clawson GA, Miranda GQ, Sivarajah A, Xin P, Pan W, Thiboutot D, Christensen ND. Gene Ther. 2004;11:1331–1341. doi: 10.1038/sj.gt.3302303. [DOI] [PubMed] [Google Scholar]
- 43.Cho CW, Poo H, Cho YS, Cho MC, Lee KA, Lee SJ, Park SN, Kim IK, Jung YK, Choe YK, Yeom YI, Choe IS, Yoon DY. Exp Mol Med. 2002;34:159–166. doi: 10.1038/emm.2002.23. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez-Salas LM, Arpawong TE, DiPaolo JA. Antisense Nucleic Acid Drug Dev. 1999;9:441–450. doi: 10.1089/oli.1.1999.9.441. [DOI] [PubMed] [Google Scholar]
- 45.Venturini F, Braspenning J, Homann M, Gissmann L, Sczakiel G. Nucleic Acids Res. 1999;27:1585–1592. doi: 10.1093/nar/27.7.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamada K, Sakaue M, Alemany R, Zhang WW, Horio Y, Roth JA, Mitchell MF. Gynecol Oncol. 1996;63:219–227. doi: 10.1006/gyno.1996.0310. [DOI] [PubMed] [Google Scholar]
- 47.Alvarez-Salas LM, Cullinan AE, Siwkowski A, Hampel A, DiPaolo JA. Proc Natl Acad Sci U S A. 1998;95:1189–1194. doi: 10.1073/pnas.95.3.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Butz K, Denk C, Ullmann A, Scheffner M, Hoppe-Seyler F. Proc Natl Acad Sci U S A. 2000;97:6693–6697. doi: 10.1073/pnas.110538897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sterlinko Grm H, Weber M, Elston R, McIntosh P, Griffin H, Banks L, Doorbar J. J Mol Biol. 2004;335:971–985. doi: 10.1016/j.jmb.2003.10.079. [DOI] [PubMed] [Google Scholar]
- 50.Croce CM. Cancer Res. 1999;59(Suppl 7):1778S–1783S. [PubMed] [Google Scholar]