

Abstract
Numerous soluble proteins convert to insoluble amyloid-like fibrils having common properties. Amyloid fibrils are associated with fatal diseases such as Alzheimer’s, and amyloid-like fibrils can be formed in vitro. For the yeast protein Sup35, conversion to amyloid-like fibrils is associated with a transmissible infection akin to that caused by mammalian prions. A seven-residue peptide segment from Sup35 forms amyloid-like fibrils and closely related microcrystals, which here reveal the atomic structure of the cross-β spine. It is a double β-sheet, with each sheet formed from parallel segments stacked in-register. Sidechains protruding from the two sheets form a dry, tightly self-complementing steric zipper, bonding the sheets. Within each sheet, every segment is bound to its two neighbouring segments via stacks of both backbone and sidechain hydrogen bonds. The structure illuminates the stability of amyloid fibrils, their self-seeding characteristic, and their tendency to form polymorphic structures.
Four decades of research have established that amyloid-like fibrils of different proteins have a common structural ‘cross-β’ spine1. In 1959 Cohen and Calkins2 observed elongated, unbranched fibrils in electron micrographs of diseased tissues, and in 1968 Glenner and Eanes3 discovered that the fibrils exhibit an X-ray diffraction signature known as the cross-β pattern. This pattern shows4 that the strongest repeating feature of the fibril is a set of β-sheets that are parallel to the fibril axis with their strands perpendicular to this axis. The hypothesis of a common molecular organization was supported by the finding5 that amyloid fibrils from 6 different proteins, each associated with its own clinical syndrome, showed similar cross-β diffraction patterns. The degree of similarity pointed to ‘a common core molecular structure.’
Revealing the atomic details of this cross-β spine has been impeded by the limited order of fibrils isolated from diseased tissues, infected cells, and in vitro conversions of proteins to fibrils. There is also evidence for a diversity of crystalline and fibril structures6–8. Nevertheless, an arsenal of biophysical tools has defined important features. These tools include solid-state NMR9–11, model-building constrained by X-ray fiber and powder diffraction6,7,12,13, site-directed spin labeling14,15, cryo-electron microscopy16,17, and proline-scanning mutagenesis18. Despite numerous models suggested by these studies, until now no refined, fully objective atomic model has been available for the common spine structure.
We selected the yeast protein Sup35 for X-ray diffraction analysis because extensive studies have shown that its fibril formation is the basis of protein-based inheritance and prion-like infectivity19–23. Its fibril-forming tendency had been traced to the N-terminus of the ‘prion-determining domain24,25, and from this region we isolated a 7-residue, fibril-forming segment with sequence GNNQQNY6. This peptide dissolves in water and at a concentration of ~400μM, forms amyloid-like fibrils in a few hours. These fibrils display all of the common characteristics of amyloid fibrils, including: elongated, unbranched morphology; the cross-β diffraction pattern; binding of the flat dyes Congo Red and Thioflavin T; the characteristic green-yellow birefringence of Congo Red; lag-dependent cooperative kinetics of formation with self-seeding26; and unusual stability.
GNNQQNY and the related peptide NNQQNY form elongated microcrystals at higher concentrations (~10–100 mM), enabling X-ray diffraction studies. The microcrystals are similar to the fibrils in that the peptide segments are perpendicular to the long dimension of both aggregates and that fibrils and microcrystals have similar diffraction patterns (Fig. S2). In hundreds of crystallization experiments, microcrystals never grew to more than a few micrometers in length, with much narrower cross sections.
Architecture of the GNNQQNY cross-β spine
Three features of the microcrystals made it possible to determine structures for GNNQQNY and NNQQNY. First, the largest microcrystals (Fig.1) are of sufficient size, order, and stability to yield adequate diffraction data on microfocus beamline ID13 at the European Synchrotron Research Facility (ESRF). Second, microcrystals of NNQQNY grow only in the presence of Zn2+ or Cd2+. Anomalous scattering from a crystal of Zn-NNQQNY yielded phases for the structure of Zn-NNQQNY. Third, the structure of GNNQQNY is nearly isomorphous with that of NNQQNY, allowing structure determination from a difference map. Details of data collection and structure determination are listed in Table 1. The NNQQNY structure is described in Supplementary Information. Here we focus on the structure of GNNQQNY.
Figure 1.

The NNQQNY microcrystal used for X-ray diffraction data collection, held to the tip of a glass capillary by cryoprotectant (50% ethylene glycol/water); scale bar shows 10 μm. X-rays were focused on the encircled areas. Separate data sets were collected for each and were merged to provide the final data set. The inset shows an SEM photograph of NNQQNY crystals, suggesting that the ‘large’ microcrystals used for data collection are composed of several aligned, nanometer-sized blocks; scale bar shows 1 μm.
Table 1.
Statistics of data collection, phasing, and atomic refinement
| Data collection | NNQQNY* | GNNQQNY |
| Space group | P21 | P21 |
| Resolution (Å) | 1.3 | 1.8 |
| Unit cell dimensions: a (Å) | 21.15 | 21.94 |
| b (Å) | 4.87 | 4.87 |
| c (Å) | 23.13 | 23.48 |
| ß(°) | 102.93 | 107.08 |
| Measured reflections | 8,241 | 995 |
| Unique reflections | 2,166 | 509 |
| Overall completeness (%) | 97.1 | 89.5 |
| Last shell completeness (%) | 88.8 | 84.2 |
| Overall Rsym† | 0.146 | 0.204 |
| Last shell Rsym | 0.426 | 0.491 |
| Overall I/σ (I) | 9.9 | 3.8 |
| Last shell I/σ (I) | 2.6 | 1.5 |
| Refinement | ||
| Rwork | 0.102 | 0.181 |
| Rfree | 0.152 | 0.190 |
| rmsd bond lengths (Å) | 0.007 | 0.014 |
| rmsd bond angles (°) | 1.1 | 1.2 |
| number of protein atoms | 55 | 59 |
| number of solvent atoms | 12 | 7 |
| average B factor of protein atoms | 5.6 | 13.1 |
| average B factor of solvent atoms | 17.6 | 27.5 |
| PDB ID code | 1yjo | 1yjp |
Friedel pairs are not merged in reported NNQQNY data statistics; the NNQQNY structure was refined with anisotropic B-factors
Rsym(I) = ∑hkl ( (∑i |Ihkl,i − <Ihkl>|) /∑i Ihkl)
GNNQQNY molecules are extended in conformation and are hydrogen-bonded to each other in standard Pauling-Corey parallel β-sheets. Because the strands are perpendicular to the long axis of the microcrystals (Fig. 2a), hydrogen-bonded addition of GNNQQNY molecules to the growing β-sheet accounts for the elongated shape of the crystals as well as the fibrils. As previously suggested from X-ray powder diffraction of the microcrystals6,7, the GNNQQNY β-strands within each sheet are parallel and exactly in register. A parallel, in register arrangement is also seen for Αβ molecules in their fibrils9,10. Each pair of sheets is related by a 21 screw axis: the strands in one sheet are antiparallel to those in the neighbouring sheet, and each sheet is shifted along the screw axis relative to its neighbour by one half the strand-strand separation of 4.87 Å. Thus sidechains extending from a strand in one sheet nestle between sidechains extending from two strands of the neighbouring sheet (Fig. 2b).
Figure 2.

Structure of GNNQQNY. Unless otherwise noted, carbon atoms are coloured in purple or grey/white, oxygen in red, and nitrogen in blue.
a, The pair-of-sheets structure of the fibril-forming peptide GNNQQNY, showing the backbone of each β-strand as an arrow, with ball and stick sidechains protruding. The dry interface is between the two sheets, with the wet interfaces on the outside surfaces. Sidechains Asn2, Gln4 and Asn6 point inwards, forming the dry interface. The 21 screw axis of the crystal is shown as the vertical line. It rotates one of the strands of the near sheet 180° about the axis and moves it up 4.87 Å/2 so that it is superimposed on one of the strands of the far sheet.
b, The steric zipper viewed edge on (down the a axis). Notice the vertical shift of one sheet relative to the other, allowing interdigitation of the sidechains emanating from each sheet. The amide stacks of the dry interface are shaded in grey at the center, and those of the wet interface are shaded in pale red on either side.
c, The GNNQQNY crystal viewed down the sheets (i.e. from the top of panel a, along the b axis). Six rows of β-sheets run horizontally. Peptide molecules are shown in black and water molecules are represented by red +. Notice that the sheets are in pairs, with a closely spaced pair (8.5 Å) alternating with a wider spaced (15 Å) pair. The wide spaces between sheets (wet interfaces) are filled with water molecules, whereas the closely spaced interfaces (dry interfaces) lack waters, other than those hydrating the caroboxylate ions at the C-termini of peptides. The atoms in the lower left unit cell are shown as spheres representing van der Waals radii.
d, The steric zipper. This is a close-up view of a pair of GNNQQNY molecules from the same view as panel c, showing the remarkable shape complementarity of the Asn and Gln sidechains protruding into the dry interface. 2Fo-Fc electron density is shown, and the position of the central screw axis is indicated.
e, Views of the β-sheets from the side (down the c axis), showing 3 β-strands with the inter-strand hydrogen bonds. Sidechain carbon atoms are highlighted in yellow. Backbone hydrogen bonds are shown by purple or black dots and sidechain hydrogen bonds by yellow dots. The length of each hydrogen bond is noted in Å units. The views of the interfaces are close to the views of panel a, where purple or grey/white backbone carbons correspond between equivalent sheets. The left hand set of three strands is viewed from the centre of the dry interface; the right hand set of three strands is viewed from the wet interface. Notice the amide stacks in both interfaces and the tyrosine stack in the wet interface. Also, notice that the wet interface is offset along the vertical axis by one half of the interstrand separation of 4.87 Å
There are two distinctly different interfaces between sheets, which we term the dry and wet interfaces (Fig 2c). The wet interface is lined with water molecules that completely separate GNNQQNY molecules, other than contact between Tyr7 residues in neighbouring sheets. The separation of these sheets is large, about 15 Å. In contrast, the dry interface contains no water, other than two molecules which hydrate the C-terminal carboxylate ions at the ends of the peptide segments. These sheets are closer together, separated by 8.5 Å. Whereas each polar sidechain of the wet interface is hydrated by water molecules, the polar sidechains of the dry interface (Asn 2, Gln 4, and Asn 6) are tightly interdigitated with the same three sidechains of the other sheet (Fig. 2d). These opposing sidechains do not form hydrogen bonds with each other. Rather their shapes complement each other closely, forming van der Waals interactions. Viewed down the sheets (Fig. 2d) the interdigitating sidechains look like the teeth of a zipper, so we call this interaction a steric zipper. The dry interface is a stack of these steric zippers (Fig. 2b).
The shape complementarity of the dry interface is unusually tight when compared to other protein interfaces, as quantified by the SC parameter27. SC measures the shape complementarity of two atomic surfaces by comparing the directions of unit vectors normal to the two surfaces, emanating from nearest points on the opposed surfaces. The average dot product of the pair of vectors approaches 1.0 as the two surfaces follow each other perfectly. The tightly meshing surfaces of proteolytic inhibitor proteins and their cognate proteases have values of SC in the range 0.73 ± 0.03 and SC for protein antigens bound to antibodies are 0.66 ± 0.0227. For the sheets forming the steric zipper in the dry interface SC = 0.86, showing that this interface has unusually high complementarity.
The remarkable complementarity between sheets in the dry interface suggest that the stable structural unit of the cross-β spine is a pair of β-sheets. The wet interface, with only a single peptide-peptide contact, has the features of a crystal contact and may not exist in the fibril structure. A pair-of-sheets organization for the cross-β spine is consistent with several other observations. First, a spine of two sheets is self-limiting in lateral growth, because the same face of both sheets is opposed, exposing a different outward face – in this structure, the wet face. A spine of three or four such stacked sheets would expose a face identical to one of its interior bonding faces, leading to further lateral growth. Second, models of cross-β spines containing three or more sheets sustain distortions in backbone hydrogen bonding that increase as the sheets stack further from the fibril axis. Third, Ivanova et al.28 found that the width of the diffuse equatorial X-ray reflection at ~ 9–11Å resolution in fibrils of β-2-microglobulin corresponds better with a model containing two sheets, than a model containing a single sheet or three sheets. Finally, a pair-of-sheets structure is consistent with studies by cryo-electron microscopy of the amyloid-like protofibrils of SH3 and insulin16,29. In short, the crystal structures of GNNQQNY and NNQQNY suggest that a tight, dry steric fit between a pair of sheets is likely to be a fundamental feature of amyloid-like fibrils. However, it is not yet clear how to reconcile a pair-of-sheets feature with evidence from mass-per-unit-length measurements on Aβ fibrils10 and from EM measurements of GNNQQNY protofibrils7, which are consistent with four sheets.
Another fundamental feature of the cross-β spine of Fig. 2 is that it is built from a short peptide. The self-complimentary steric zipper explains how short segments of proteins are able to form amyloid-like fibrils and raises the question of whether the rest of the protein participates in the spine.
Amide and tyrosine stacks in the cross-β spine
While there are no hydrogen bonds bridging two tightly complementing sheets across the dry interface, each GNNQQNY forms 11 hydrogen bonds to its two neighbouring molecules in the same sheet (Fig. 2e). Five of these are backbone C=O…H-N hydrogen bonds, and four are amide stacks: amide-amide hydrogen bonds between pairs of identical Asn or Gln residues in adjacent molecules within a sheet. It is these hydrogen-bonded amide stacks that force the GNNQQNY and NNQQNY molecules to stack parallel and in register in their respective sheets. This network of backbone and sidechain hydrogen bonds is reminiscent of the polar zipper proposed by Perutz et al30. Amide stacks such as those found here could stabilize the polyglutamine aggregates formed in the CAG expansion diseases and those formed in vitro with polyglutamine-containing peptides. The remaining hydrogen bonds between GNNQQNY molecules in the sheet are from the sidechain amide nitrogen of Gln5 to the hydroxyl of Tyr7 and from the Asn2 backbone nitrogen to the Asn2 sidechain oxygen. Also, the rings of Tyr7 are stacked, but not face-to-face; they pack edge-to-face across the wet interface.
Similarity to other structures
The structure of the GNNQQNY cross-β spine shows limited similarity to β-helices proposed as models for amyloid and prion spines17,31–34. A search for structurally related β–sandwiches in the Protein Data Bank yielded only one significant match to the backbone of GNNQQNY: SufD (PDB entry 1vh4). The search model, which contains six strands of GNNQQNY, three from each sheet forming the dry interface, can be superimposed on the SufD backbone with an RMS deviation of 1.8 Å. The fold of SufD, a member of the β-helix family, resembles GNNQQNY more closely than do canonical right handed β-helices because it has two sheets rather than three, and its sheets abut, rather than having a cylindrical or triangular cross section. SufD’s similarity to GNNQQNY is limited, however, by its lack of a steric zipper; sidechains from opposing sheets contact but do not interdigitate. As a result, the distance between sheets in SufD is nearly 2 Å greater. Hence the complementarity between the two sheets composing SufD (SC=0.70) is significantly lower than in GNNQQNY. In short, the GNNQQNY structure shows only weak similarity to β-helices in general, and differs considerably from the cylindrical and triangular β-helices that have been proposed as models for amyloid-like spines.
Structure-based energetics
The structure of GNNQQNY suggests factors that determine the rate and stability of fibril formation as well as a factor that may underlie amyloid fibril polymorphism and prion strains8,22,23. The structure indicates three levels of organization within the fibrils. The first is the alignment of GNNQQNY molecules to form a β-sheet. The second is the self-complementation of two sheets, forming the pair-of-sheets structure, with a dry interface. Because the self-complementation of two sheets involves van der Waals forces rather than hydrogen bonding, the patterns of bonding are less specific than those of the first level. Alternative interdigitations could give rise to fibril polymorphism and prion strains. In the third level, pair-of-sheets structures interact to form a fibril. For the third level, we note only that the non-covalent forces involved are probably weaker than those driving the formation of the first two levels.
In the alignment of GNNQQNY molecules to form a β-sheet, each GNNQQNY molecule must be extended. Because β-sheets form rapidly35,36 and reversibly, we assume that this level forms more rapidly than the second level. The second level is likely to form more slowly because the amide sidechains must acquire the proper rotamers to permit interdigitation with the mating sheet and must be dehydrated to permit formation of the dry amide-stacking hydrogen bonds. We suggest that the decrease in entropy accompanying this step creates the barrier to fibril formation, which is evident in its lag-dependent cooperative formation. Once a nucleus of the cross-β spine has formed, additional molecules can add more readily, leading to rapid growth. In Supplementary Information we argue from the structure that the nucleus for GNNQQNY fibril formation is ~4 molecules, and that the transition-state complex on the path to the nucleus is ~3 molecules. From energetic considerations we estimate a crude value for the free energy of forming this complex of ~8 kcal/mol-of-GNNQQNY at room temperature. If there are 3 molecules in the transition-state complex, the barrier is ~24 kcal/mole, a substantial barrier to fibril formation.
In the formation of the transition-state complex and of the protofibril itself, there must be enthalpy decreases that compensate for the entropy decreases. Some enthalpy will be released by the van der Waals energy of the tight interdigitation in the steric zipper. The formation of hydrogen bonds between backbone groups and amide stacks will contribute also, but these bonds replace hydrogen bonds between water and the peptide in solution, so there is little net increase in the number of hydrogen bonds37. Conceivably hydrogen bonds in the pair-of-sheets structure are stronger than those in solution. They are in an anhydrous, low dielectric constant environment, and the columns of hydrogen bonds in the amide stacks run antiparallel to neighbouring columns (Fig. 3e), so there could be substantial strengthening of hydrogen bonds through induced dipoles, as is the case in ice38. Though our estimates are crude, the standard free energy change for protofibril formation, ΔG0, the sum of the enthalpic and entropic terms, is unlikely to be strongly negative.
Figure 3.
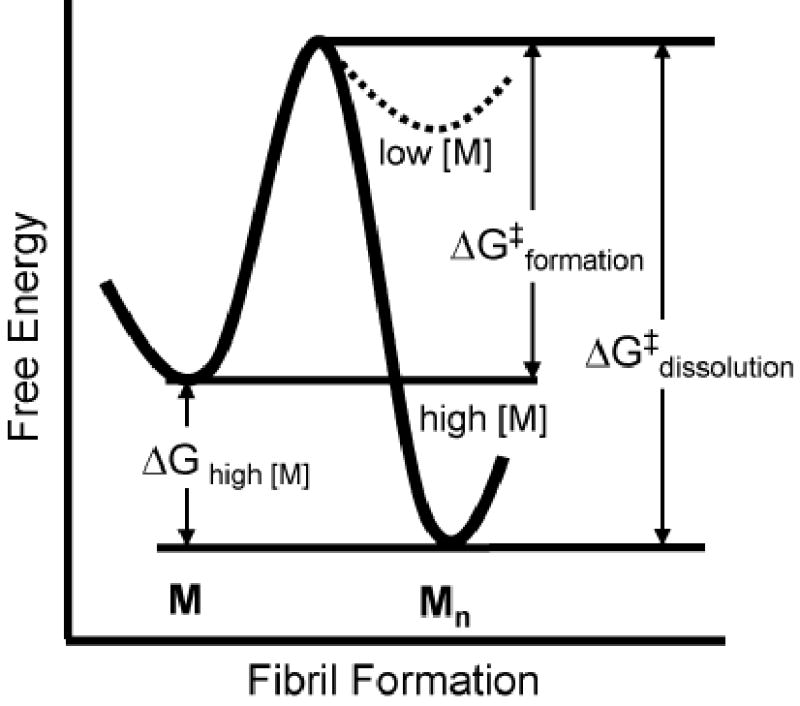
A conjectural plot of the free energy, G, for conversion of monomeric GNNQQNY, M, to the aggregated state, Mn. The standard free energy change ΔG0 for the conversion is small, so that the change in ΔG is controlled mainly by the concentration of monomer. At low concentrations, the monomeric state is favoured over the aggregated state, and the aggregated state is favoured at high concentrations. There is a significant kinetic barrier to formation of the aggregated state, ΔG‡ formation. At high concentrations of protein, the barrier to redissolve fibers, ΔG‡ dissolution, is very large.
Amyloid-like fibrils are stabilized by protein concentration as well as by formation of the steric zipper and the hydrogen bond stacks. For conversion of n peptide monomers, M, to an amyloid spine, Mn , with infinite cooperativity, nM → Mn, the free energy of transition from the dissolved to the aggregated state is given by
in which ΔG0 is the standard free energy, RT is the product of the gas constant with the absolute temperature, and the term on the right is governed by the concentration of monomer. At high concentrations of monomer, this term is strongly negative, favouring transition to the fibrillar state. Thus our structure suggests there is a large entropic barrier to amyloid fibril formation, but once a nucleus is present, high concentrations of protein drive the formation and contribute to an even larger barrier to dissolution of the fibrils (Fig. 3).
Summary and biological implications
The structures of GNNQQNY and NNQQNY determined here by X-ray microcrystallography confirm gross features of the cross-β spine that have been known from other methods: the spine is built from β-strands that are spaced ~4.8 Å apart, perpendicular to the fibril axis, formed into β-sheets with hydrogen bonds parallel to the axis, and exactly in register6,9,10. What is new is the pair-of-sheets organization with the interface between the paired sheets consisting of the closely enmeshed self-complementing sidechains protruding from the two sheets, termed a steric zipper. This interface is dry, in contrast to the highly hydrated external faces of the paired sheets. Disruption or capping of this steric zipper may be a strategy for drug interference of amyloid formation39.
The steric zipper in the structures of GNNQQNY and NNQQNY explains how a fibril can be formed from a short segment of a protein. In fact, fibrils formed from short peptides are well known6,40,41. We suggest that such short segments are capable of self-complementation across a dry intersheet steric zipper, as are the Asn-X-Gln-X-Asn sequences studied here. Similarly, we expect that short segments of low complexity sequences can form steric zippers. The observation that polyamino acids form amyloid-like fibrils42 is consistent with the importance of sidechain interactions in steric zippers, notably size and shape complementarity.
The self-complementing GNNQQNY sequence is a segment of the yeast prion Sup35, a protein known to convert copies of itself to an amyloid fibril-like state. This fibrillar state has been shown to be at the basis of the transition to the [PSI+] prion state of Sup3520,21,24,25. Presumably, self-complementation by a steric zipper is a preliminary step in the process of molecular self-recognition that leads to conversion. Because the steric zipper involves non-specific van der Waals forces, a given sequence may form more than one self-complementing steric zipper, possibly leading to amyloid polymorphism and prion strains.
Regulation of protein concentration within cells and tissues takes on significance in preventing fibril formation, in light of the structure-based arguments presented here that the standard free energy of fibril formation is not strongly negative. If in fact the dissolved and fibrillar forms of proteins are nearly iso-energetic in the biological milieu of an organism, there are two factors that influence the formation of amyloid-like fibrils. The first is the concentration of a protein in a given tissue. Breakdown in the cellular machinery that regulates protein synthesis or protein degradation could raise the concentration of protein monomers to the point of favouring an aggregated state. If the protein in question contains self-complementing segments of sequence, the aggregate could be the amyloid-like state. Chaperones that isolate proteins as they fold would be of critical importance when those sequences contain self-complementing segments. The second factor is the energetic barrier on the reaction pathway. The GNNQQNY structure suggests that several self-complementary segments must be properly arranged to act as a nucleus for fibril growth, presenting a significant barrier to fibril formation. However, once fibrils form at high protein concentration, the barrier to the reverse reaction – dissolution of the fibril – is even higher, rendering fibril formation difficult to reverse.
Methods
Peptide crystallization
Lyophilized, synthetic GNNQQNY (AnaSpec, San Jose, CA, www.anaspec.com; CS Bio Company, Inc., Menlo Park, CA, www.csbio.com) and NNQQNY (AnaSpec) peptides dissolve easily in water and aqueous solutions. Because of residual trifluoroacetic acid in the lyophilized peptide, dissolving the material in water results in a low pH solution; this low pH solution was used for crystallization.
GNNQQNY crystals were grown from a solution of 10 mg/mL peptide in water (pH ~2.0) at ~20ºC. An orthorhombic crystal polymorph was previously grown6,7 from this condition. In later preparations, seed crystals from previous batches were used to promote faster and more reliable crystallization.
NNQQNY crystals were grown using the hanging-drop vapour diffusion method by mixing a 5:4:1 ratio of peptide solution, reservoir solution, and additive solution, respectively. The peptide solution contained 30 mg/mL NNQQNY in water. The reservoir solution contained 100 mM HEPES (pH 7.5) and 1 M sodium acetate. The additive solution contained 0.1 M zinc sulphate. The final pH of the drop was ~7.5, and crystals were grown at ~20ºC. GNNQQNY and NNQQNY crystals were transferred to a cryoprotectant, either 50% ethylene glycol/water or 50% glycerol/water, prior to data collection.
X-ray data collection and processing
X-ray diffraction data sets were collected from the GNNQQNY and NNQQNY crystals at the European Synchrotron Radiation Facility (ESRF) beamline ID13, equipped with a MAR CCD detector43. Data were collected in 5° wedges at a wavelength of 0.975 Å using a 5 μm beam size. The crystals were cryo-cooled (100K) for data collection. Due to the extremely small focal size of the X-ray beam, the effect of localized radiation damage could be minimized by illuminating three different portions of the NNQQNY crystal during data collection (Fig. 1). All data were processed and reduced using Denzo/Scalepack from the HKL suite of programs44.
Structure solution and refinement
An initial set of phases for the NNQQNY structure could be derived by the method of single wavelength anomalous dispersion (SAD) using the anomalous scattering signal from a well-ordered zinc ion. The location of the zinc ion was readily deduced from the presence of a 5 σ peak in an anomalous difference Patterson map (Fig. S1). SAD phases were calculated with the program MLPHARE45. Density modification with the program DM45 significantly improved the interpretability of the electron density map, despite an extremely low solvent content (18%). A six residue long β-strand could be immediately recognized and modelled in the electron density with no ambiguity in orientation or position. Side chain torsion angles were adjusted using the graphics program “O”46. Coordinates were refined with the program REFMAC47. Refinement statistics are reported in Table 1. The geometric quality of the model was assessed with the programs PROCHECK48 and WHATIF49. All residues were found in the most favoured region of the Ramachandran plot. The GNNQQNY structure could be refined by difference Fourier methods since its unit cell was nearly isomorphous with that of the NNQQNY crystal. Protein structures were illustrated using the program PyMOL50.
Supplementary Material
Acknowledgments
We thank the late Carl Branden for initiating the UCLA-ESRF collaboration; D.L.D. Caspar, R. Diaz-Avalos, Y.Fujiyoshi, R. G. Griffin, Sine Larsen, K. Mitsuoka, P. W, Stevens, and T.O. Yeates for discussions, Dr. Suzanna Horvath of Caltech for peptide synthesis, and NIH, NSF, HHMI, and USPHS National Research Service Award GM07185 for support.
Footnotes
Competing Interests Statement The authors declare that they have no competing financial interests.
Supplementary Information accompanies the paper on www.nature.com/nature.
References
- 1.Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struct Biol. 2000;130:88–98. doi: 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- 2.Cohen AS, Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature. 1959;183:1202–3. doi: 10.1038/1831202a0. [DOI] [PubMed] [Google Scholar]
- 3.Eanes ED, Glenner GG. X-ray diffraction studies on amyloid filaments. J Histochem Cytochem. 1968;16:673–7. doi: 10.1177/16.11.673. [DOI] [PubMed] [Google Scholar]
- 4.Geddes AJ, Parker KD, Atkins ED, Beighton E. “Cross-beta” conformation in proteins. J Mol Biol. 1968;32:343–58. doi: 10.1016/0022-2836(68)90014-4. [DOI] [PubMed] [Google Scholar]
- 5.Sunde M, et al. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997;273:729–39. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 6.Balbirnie M, Grothe R, Eisenberg DS. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated beta-sheet structure for amyloid. Proc Natl Acad Sci U S A. 2001;98:2375–80. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diaz-Avalos R, et al. Cross-beta order and diversity in nanocrystals of an amyloid-forming peptide. J Mol Biol. 2003;330:1165–75. doi: 10.1016/s0022-2836(03)00659-4. [DOI] [PubMed] [Google Scholar]
- 8.Petkova AT, et al. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 2005;307:262–5. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 9.Benzinger TL, et al. Propagating structure of Alzheimer's beta-amyloid(10–35) is parallel beta-sheet with residues in exact register. Proc Natl Acad Sci U S A. 1998;95:13407–12. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petkova AT, et al. A structural model for Alzheimer's beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci U S A. 2002;99:16742–7. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaroniec CP, MacPhee CE, Astrof NS, Dobson CM, Griffin RG. Molecular conformation of a peptide fragment of transthyretin in an amyloid fibril. Proc Natl Acad Sci U S A. 2002;99:16748–53. doi: 10.1073/pnas.252625999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sunde M, Blake CC. From the globular to the fibrous state: protein structure and structural conversion in amyloid formation. Q Rev Biophys. 1998;31:1–39. doi: 10.1017/s0033583598003400. [DOI] [PubMed] [Google Scholar]
- 13.Sumner Makin, O., Atkins, E., Sikorski, P., Johansson, J. & Serpell, L. C. Molecular basis for amyloid fibril formation and stability. Proc Natl Acad Sci U S A (2005). [DOI] [PMC free article] [PubMed]
- 14.Serag AA, Altenbach C, Gingery M, Hubbell WL, Yeates TO. Identification of a subunit interface in transthyretin amyloid fibrils: evidence for self-assembly from oligomeric building blocks. Biochemistry. 2001;40:9089–96. doi: 10.1021/bi010655s. [DOI] [PubMed] [Google Scholar]
- 15.Torok M, et al. Structural and dynamic features of Alzheimer's Abeta peptide in amyloid fibrils studied by site-directed spin labeling. J Biol Chem. 2002;277:40810–5. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 16.Jimenez JL, et al. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. Embo J. 1999;18:815–21. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kishimoto A, et al. beta-Helix is a likely core structure of yeast prion Sup35 amyloid fibers. Biochem Biophys Res Commun. 2004;315:739–45. doi: 10.1016/j.bbrc.2004.01.117. [DOI] [PubMed] [Google Scholar]
- 18.Williams AD, et al. Mapping abeta amyloid fibril secondary structure using scanning proline mutagenesis. J Mol Biol. 2004;335:833–42. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 19.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–9. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 20.Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–6. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 21.Serio TR, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–21. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 22.King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319–23. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–8. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 24.DePace AH, Santoso A, Hillner P, Weissman JS. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell. 1998;93:1241–52. doi: 10.1016/s0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- 25.Santoso A, Chien P, Osherovich LZ, Weissman JS. Molecular basis of a yeast prion species barrier. Cell. 2000;100:277–88. doi: 10.1016/s0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- 26.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–8. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 27.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–50. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 28.Ivanova MI, Sawaya MR, Gingery M, Attinger A, Eisenberg D. An amyloid-forming segment of beta2-microglobulin suggests a molecular model for the fibril. Proc Natl Acad Sci U S A. 2004;101:10584–9. doi: 10.1073/pnas.0403756101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jimenez JL, et al. The protofilament structure of insulin amyloid fibrils. Proc Natl Acad Sci U S A. 2002;99:9196–201. doi: 10.1073/pnas.142459399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91:5355–8. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pickersgill RW. A primordial structure underlying amyloid. Structure (Camb) 2003;11:137–8. doi: 10.1016/s0969-2126(03)00009-1. [DOI] [PubMed] [Google Scholar]
- 32.Wetzel R. Ideas of order for amyloid fibril structure. Structure (Camb) 2002;10:1031–6. doi: 10.1016/s0969-2126(02)00809-2. [DOI] [PubMed] [Google Scholar]
- 33.Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc Natl Acad Sci U S A. 2002;99:5591–5. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc Natl Acad Sci U S A. 2004;101:8342–7. doi: 10.1073/pnas.0402254101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varley P, et al. Kinetics of folding of the all-beta sheet protein interleukin-1 beta. Science. 1993;260:1110–3. doi: 10.1126/science.8493553. [DOI] [PubMed] [Google Scholar]
- 36.Sivaraman T, Kumar TK, Chang DK, Lin WY, Yu C. Events in the kinetic folding pathway of a small, all beta-sheet protein. J Biol Chem. 1998;273:10181–9. doi: 10.1074/jbc.273.17.10181. [DOI] [PubMed] [Google Scholar]
- 37.Eisenberg D, Wesson M, Yamashita M. Interpretation of Protein Folding and Binding with Atomic Solvation Parameters. Chemica Scripta. 1989;29A:217–221. [Google Scholar]
- 38.Coulson CA, Eisenberg D. Interactions of H2O Molecules in Ice. Proc Royal Society. 1966;291:445–453. [Google Scholar]
- 39.Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci U S A. 2002;99:2754–9. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de la Paz ML, Serrano L. Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci U S A. 2004;101:87–92. doi: 10.1073/pnas.2634884100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tjernberg L, Hosia W, Bark N, Thyberg J, Johansson J. Charge attraction and beta propensity are necessary for amyloid fibril formation from tetrapeptides. J Biol Chem. 2002;277:43243–6. doi: 10.1074/jbc.M205570200. [DOI] [PubMed] [Google Scholar]
- 42.Fandrich M, Dobson CM. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. Embo J. 2002;21:5682–90. doi: 10.1093/emboj/cdf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riekel C. Recent developments in micro-diffraction on protein crystals. J Synchrotron Radiat. 2004;11:4–6. doi: 10.1107/s090904950302541x. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 45.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 46.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47 (Pt 2):110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 47.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 48.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK - a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–91. [Google Scholar]
- 49.Vriend G, Sander C. Quality control of protein models: directional atomic contact analysis. J Appl Cryst. 1993;26:47–60. [Google Scholar]
- 50.DeLano, W. L. The PyMOL user's manual (DeLano Scientific, San Carlos, CA, USA, 2002).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.